
Abstract
The Lrp and MarR families are two groups of transcriptional regulators widely distributed among prokaryotes. However, the hierarchical-regulatory relationship between the Lrp family and the MarR family remains unknown. Our previous study found that an Lrp (SACE_Lrp) from Saccharopolyspora erythraea indirectly repressed the biosynthesis of erythromycin. In this study, we characterized a novel MarR family protein (SACE_6745) from S. erythraea, which is controlled by SACE_Lrp and plays a direct regulatory role in erythromycin biosynthesis and export. SACE_Lrp directly regulated the expression of marR by specifically binding a precise site OM (5’-CTCCGGGAACCATT-3’). Gene disruption of marR increased the production of erythromycin by 45% in S. erythraea A226. We found that MarR has direct DNA-binding activity for the promoter regions of the erythromycin biosynthetic genes, as well as an ABC exporter SACE_2701-2702 which was genetically proved to be responsible for erythromycin efflux. Disruption of SACE_Lrp in industrial S. erythraea WB was an efficient strategy to enhance erythromycin production. Herein, we jointly engineered SACE_Lrp and its target MarR by deleting marR in WBΔSACE_Lrp, resulting in 20% increase in erythromycin yield in mutant WBΔLrpΔmarR compared to WBΔSACE_Lrp, and 39% to WB. Overall, our findings provide new insights into the hierarchical-regulatory relationship of Lrp and MarR proteins and new avenues for coordinating antibiotic biosynthesis and export by joint engineering regulators in actinomycetes.
Key points
• The hierarchical-regulatory relationship between SACE_Lrp and MarR was identified.
• MarR directly controlled the expression of erythromycin biosynthesis and export genes.
• Joint engineering of SACE_Lrp-MarR regulatory element enhanced erythromycin production.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Transcription regulators can respond to alterations of environmental and physiological signals by tuning the expression of relevant genes (Liu et al. 2013). As typical representatives, the Lrp (leucine-responsive regulatory protein) and the MarR (multiple antibiotic resistance regulator) regulator families are both widely distributed and extremely well-characterized regulatory transcriptional factors among prokaryotes (Grove 2013; Peeters and Charlier 2010). Lrp family regulators (LFRs) are involved in diverse biological processes, especially in amino acid metabolism and transport (Brinkman et al. 2003; Peeters and Charlier 2010). LFRs are identified by an N-terminal helix-turn-helix (HTH) domain for DNA binding and a C-terminal αβ sandwich domain for ligand response (Deng et al. 2011; Peeters and Charlier 2010). MarR family regulators (MFRs) also modulate a variety of biological processes, mainly including antibiotic resistance, virulence, and stress responses (Grove 2013; Gupta et al. 2018). Similar to LFRs, MFRs are characterized by a winged HTH DNA-binding domain at the N-terminus, and the DNA-binding activity of MFRs is altered by conformational changes in response to structural ligands (Deochand and Grove 2017; Grove 2013).
Actinobacteria are well known for their ability to produce abundant antibiotics and related compounds (Bérdy 2005). Biosynthesis of these antibiotics involves sophisticated regulatory networks, which are finely adjusted by extracellular and intracellular signals (Liu et al. 2013; Martín and Liras 2010). Recently, studies on LFRs have made significant progress in providing insight into the molecular mechanism involved in the regulation of antibiotic biosynthesis by SACE_Lrp (Liu et al. 2017) and SACE_5717 (Liu et al. 2019) from S. erythraea, SCO3361 from Streptomyces coelicolor (Liu et al. 2017), SLCG_Lrp from S. lincolnensis (Xu et al. 2020) in our laboratory, and SSP_Lrp from S. spiramyceticus (Lu et al. 2019). MFRs are one of the most abundant groups of transcriptional regulators in antibiotic-producing actinobacteria; however, only a few members have been identified being involved in antibiotic biosynthesis, such as OhrR and PcaV from S. coelicolor (Davis et al. 2013; Oh et al. 2007), DptR3 from S. roseosporus (Zhang et al. 2015), SAV4189 and OhrR from S. avermitilis (Guo et al. 2018; Sun et al. 2018), and CtcS from S. aureofaciens (Kong et al. 2019).
In addition to antibiotic biosynthesis, antibiotic export is an important factor affecting antibiotic production. However, transcription regulators controlling antibiotic export in natural producers such as actinobacteria remain underexplored. TetR family regulator (TFR) GouR from S. graminearus modulates gougerotin export by directly controlling the major facilitator superfamily transporter gene gouM (Wei et al. 2014). TFR ActR from S. coelicolor regulates actinorhodin export and resistance by directly acting on the two putative export pumps actAB operon (Tahlan et al. 2008; Xu et al. 2012). Another TFR RifQ from Amycolatopsis mediterranei directly represses the expression of the rifamycin efflux pump RifP for regulation of rifamycin export (Lei et al. 2018).
S. erythraea is a model representative of actinomycetes that are used for industrial production of erythromycin A (Butler 2008). Erythromycin and its derivatives are widely used as clinical broad-spectrum polyketide antibiotics (Butler 2008; McDaniel et al. 2001). In S. erythraea, massive prediction of transcriptional regulators has offered the opportunity to elucidate specialized regulatory mechanisms for the biosynthesis and export of erythromycin, but for most of them, the specific biological functions have never been examined (Oliynyk et al. 2007). Bioinformatics analysis revealed 30 MFR genes in the S. erythraea genome (Marcellin et al. 2013; Oliynyk et al. 2007). However, the biological function of MFR links with erythromycin production in S. erythraea is unclear. Besides, it is also unknown whether the export of erythromycin is regulated. In this study, we characterized a novel MarR family protein (SACE_6745) from S. erythraea, which is regulated by SACE_Lrp and plays a significant role in the biosynthesis and export of erythromycin (Liu et al. 2017; Oliynyk et al. 2007). Furthermore, we successfully demonstrated a notable enhancement in erythromycin production through combinatorial engineering of SACE_Lrp and MarR regulatory elements in the S. erythraea industrial strain.
Materials and methods
Strains, plasmids, and primers
All bacterial strains and plasmids used in the present study are listed in Table 1, and all primers are listed in Supplementary Table S1.
Escherichia coli DH5α was used to construct plasmids, and E. coli BL21 (DE3) was used to overexpress proteins SACE_Lrp and MarR S. erythraea, and its derivatives were grown at 30°C on R5 solid medium for sporulation or in liquid TSBY medium for growth of mycelia with appropriate antibiotics as previously described (Wu et al. 2014). TSBY medium was also used for protoplast preparation and seed culture, and R5 agar was also used for protoplast regeneration and selection of transformants. R5 liquid medium was used for erythromycin fermentation (Wu et al. 2014).
Protein overexpression and purification
To express the MarR protein in E. coli, marR (SACE_6745) gene was amplified from S. erythraea A226 genome by PCR with the 6745-32a-F/R primers and inserted into the corresponding EcoRI/HindIII sites of pET32a obtaining the constructed pET32a-marR plasmid. pET32a-marR and pET28a-Lrp (Liu et al. 2017) was respectively introduced into E. coli BL21 (DE3) to express MarR and SACE_Lrp. Overexpression and purification of His6-tagged proteins were performed as previously described (Liu et al. 2017).
Electrophoretic mobility shift assay (EMSA)
The EMSAs were performed as previously described (Hellman and Fried 2007). The putative promoter regions of marR, SACE_2701-2702, and the erythromycin biosynthetic gene cluster (ery cluster) genes were amplified from the A226 genome by PCR with appropriate primers (Table S1). The PCR products were purified for EMSA probes, and the concentration of probes was determined by the microplate reader (BioTek). The above probes and the PLrp-5387 probe were individually incubated with His6-tagged protein as described previously (Liu et al. 2017).
DNase I footprinting assay
To determine the SACE_Lrp binding site within SACE_Lrp-SACE_5387-int, DNase I footprinting assays were performed as previously described (Liu et al. 2019). For each experiment, 100 ng of labeled probe (5’-FAM and 3’-HEX) was incubated with different concentrations of His6-tagged SACE_Lrp in 40 μL of binding buffer. After incubation at room temperature for 20 min, DNase I digestion was performed at room temperature for 0.5 min. All reactions were stopped by adding 10 μL DNase I stop solution at 65°C for 10 min. All samples were extracted with phenol/chloroform and precipitated with ethanol for further sequencing. Electropherograms were then analyzed with GeneMarker v2.2 (Applied Biosystems).
GFP reporter assay in E. coli
To construct the GFP reporter plasmids, the enhanced green fluorescent protein gene (egfp) fragment was amplified from pUPW-EGFP (Liu et al. 2017) by PCR with DE-F/R primers (Xu et al. 2020) and digested with XbaI/BamHI. In addition, a fragment containing the putative promoter region of marR was amplified from the A226 genome by PCR with P6745-F/R primers (Table S1) and digested with HindIII/XbaI. The two fragments were ligated into pKC1139 to create the reporter plasmid pmarR-EGFP. To evaluate the regulatory effect of SACE_Lrp on the marR gene, the Paac(3)IV promoter with EcoRV/NdeI and SACE_Lrp with NdeI/EcoRI were amplified from pIB139 (Wilkinson et al. 2002) and A226 genome using primers Papr-F/R (Xu et al. 2020) and 5388P-F/R (Table S1), respectively. The two fragments were together joined with pmarR-EGFP to create the reporter plasmid pLrp-marR-EGFP. The above method was also used to obtain the reporter plasmids p6744-EGFP and pLrp-6744-EGFP with the corresponding primers listed in Table S1. These plasmids were transformed into DH5α, detecting green fluorescence (excitation at 485 nm; emission at 510 nm, Molecular Devices). All fluorescence values were normalized to the growth rate (OD600).
Gene disruption and complementation
To construct the marR disruption mutant in S. erythraea A226, two 1.5-kb DNA flanking fragments of marR were amplified by PCR with the 6745-up-F/R and 6745-down-F/R primers using A226 genomic DNA as a template (Table S1). The obtained fragments of PCR products were treated with HindIII/XbaI and KpnI/EcoRI, and then inserted into the corresponding sites of plasmid pUCTSR (Han et al. 2011) to obtain pUC-ΔmarR, as confirmed by DNA sequencing. Plasmid pUC-ΔmarR was introduced into the parental strain A226 by PEG-mediated protoplast transformation. A 225-nt DNA fragment of the marR gene was replaced by the thiostrepton resistance gene (tsr) by homologous chromosomic recombination. The desired mutant ΔmarR with thiostrepton-resistant was further determined by PCR with the 6745-C-F/R primers (Table S1). Using the same procedures, plasmid pUC-Δ2701-2702 was constructed and introduced into A226 by PEG-mediated protoplast transformation. An 1800-nt DNA fragment of the SACE_2701-2702 gene was replaced with the tsr gene, generating the deletion mutant Δ2701-2702 in S. erythraea.
To construct the complementation strains ΔmarR/pIB-marR and Δ2701-2702/pIB-2701-2702, the full-length marR gene of 384-bp and SACE_2701-2702 gene of 2456-bp were amplified by PCR using the 6745-C-F/R and 2701-C-F/R primers (Table S1) with A226 genomic DNA as a template, respectively. The PCR products were cloned into the corresponding NdeI/XbaI sites of plasmid pIB139 to construct plasmids pIB-marR and pIB-2701-2702. These two plasmids were respectively introduced into ΔmarR and Δ2701-2702, generating complemented ΔmarR/pIB-marR and Δ2701-2702/pIB-2701-2702 by screening with apramycin and PCR using the apr-test-F/R primers (Table S1). pIB-2701-2702 was also introduced into A226 to generate the overexpressed strain A226/pIB-2701-2702 using the same method.
In the industrial high-yield S. erythraea WB, the marR gene was also disrupted by the tsr gene through the above procedures, generating the WBΔmarR strain. To further knock out marR in WBΔLrp (Liu et al. 2017), a 1.38-kb DNA fragment of the apramycin resistance gene was prepared by PCR from pIB139 with the apr-F/R primers, treated with XbaI/KpnI, and then replaced the tsr gene of pUC-ΔmarR to obtain the pUC-apr-ΔmarR. Similarly, the marR gene was displaced by the apramycin resistance gene using the pUC-apr-ΔmarR by the above method. The desired mutant WBΔLrpΔmarR with apramycin-resistant was further determined by PCR with the 6745-C-F/R primers (Table S1).
Fermentation and erythromycin determination
Flask fermentation of S. erythraea A226, S. erythraea WB, and their derivatives was carried out as described previously (Wu et al. 2014). Isolation of erythromycin A and HPLC analysis of erythromycin A yield were performed as described previously (Wu et al. 2014).
RNA preparation and qRT-PCR assay
Cells of S. erythraea A226 and its derivatives were harvested by centrifugation, and total RNA was collected using the RNA extraction/purification kit (SBS) as described previously (Liu et al. 2017). All operation procedures were carried out following the manufacturer’s instructions (MBI Fermentas). The specific primers used for the qRT-PCR assay are listed in Supplementary Table S1. The hrdB gene in S. erythraea served as an internal control to normalize samples.
Erythromycin resistance test
To determine the resistance of S. erythraea A226 and its derivatives against erythromycin, 10 μL spore suspension of A226, A/2701-2702, and ΔmarR was inoculated into 5 mL of liquid TSBY with various concentrations of erythromycin A (0, 5, 10, 25, 50, and 100 mg/L) and cultured at 30°C for 24 h to compare their growth with OD600.
Statistical analysis
The data of erythromycin production in the present study were stated as means ± standard error of mean (SEM) and analyzed by Student’s t-test, with *p < 0.05, **p < 0.01, and ***p < 0.001 representing significant differences.
Results
Determination of the SACE_Lrp binding sequence in the SACE_Lrp-SACE_5387 intergenic region
In our previous report, SACE_Lrp specifically interacted with the intergenic region between SACE_Lrp and SACE_5387 (Liu et al. 2017). In order to elucidate the precise DNA-binding site of SACE_Lrp with its target, DNase I footprinting experiments were performed with the PLrp-5387 probe (Fig. 1a). The results showed that only one protected region (named site OL: 5’-CTCCGGGCAACATT-3’) was identified from nucleotide position −39 to −26 relative to the putative SACE_5387-5386 transcriptional start site (TSS) (Fig. 1b). The site OL-deletion analysis showed that the DNA-binding activity of SACE_Lrp to the mutated probe Pd(site OL) completely disappeared as compared with the original PLrp-5387 probe by EMSAs (Fig. 1c). Taken together, these results demonstrated that the 14 bp asymmetric sequence (5’-CTCCGGGCAACATT-3’) within the intergenic region between SACE_Lrp and SACE_5387 is indispensable for SACE_Lrp binding activity.

DNase I footprinting for determination of the SACE_Lrp binding site. a The fluorograms correspond to the control DNA and to the protection reactions (with increasing concentrations of 0.9, 1.2, and 1.5 μM of SACE_Lrp respectively). b Nucleotide sequences of the intergenic segment between SACE_Lrp and SACE_5387-5386. The transcriptional start site (TSS) is indicated by a bent arrow. Sequence protected from DNase I digestion is indicated with box and labeled with Site OL. Presumptive −10 and −35 regions of the SACE_Lrp and SACE_5387-5386 promoters are underlined. c EMSA of binding of SACE_Lrp to the probes PLrp-5387 and Pd(site OL). The probe PLrp-5387 represents the intergenic segment between SACE_Lrp and SACE_5387-5386, and the Pd(site OL) is derived from PLrp-5387 with deleting the site OL. The amounts of SACE_Lrp used were 0, 250, and 500 nM, respectively
SACE_Lrp directly interacts with the promoter region of a novel MarR homologue
The 14 bp consensus sequence (5’-CTCCGGGCAACATT-3’) of site OL was used to search for SACE_Lrp putative target genes by scanning the genomic sequence of S. erythraea using PREDetector software (Hiard et al. 2007). A total of 31 upstream regions containing potential SACE_Lrp binding sites with high similarity to the site OL were identified (data not shown). Interestingly, PREDetector identified a 14 bp sequence (5’-CTCCGGGAACCATT-3’) that was highly similar to site OL, which lies in the putative promoter region of the marR gene (SACE_6745), encoding a MarR family regulator, so named as site OM (Fig. 2a). The site OM sequence is located at nucleotide positions −57 to −44 relative to the putative marR TSS (Fig. 2b).

SACE_Lrp directly controls the transcription of marR gene. a Blast analysis of site OL and site OM. b Nucleotide sequences of the promoter region of marR gene. The transcriptional start site (TSS) is indicated by a bent arrow. Presumptive sequence protected by SACE_Lrp is indicated with box and labeled with Site OM. Presumptive −10 and −35 regions of the marR promoter are underlined. c EMSA of binding of SACE_Lrp to the probes PmarR and Pd(site OM). The probe PmarR represents the promoter region of marR gene, and the Pd(site OM) is derived from PmarR with deleting the site OM. The amounts of SACE_Lrp used were 0, 500, and 1000 nM, respectively. d An illustration of the reporter plasmids in biosensor system. e Detection of the interaction of SACE_Lrp with the promoters of marR and its divergently transcribed SACE_6744 using the relative fluorescence units (RFUs) in E. coli. The mean values of three replicates are shown, with the standard deviation indicated by error bars
In order to determine whether SACE_Lrp directly interacts with the site OM from the marR promoter, EMSAs were performed to evaluate the binding ability of SACE_Lrp to the original probe PmarR and the mutated probe Pd(site OM). Results from EMSAs showed that a SACE_Lrp-PmarR complex specifically formed in the presence of probe PmarR but completely disappeared in the presence of probe Pd(site OM) (Fig. 2c). We also utilized a biosensor system with green fluorescence in E. coli to verify the interaction of SACE_Lrp with the probe PmarR in vivo. As shown in Fig. 2d, plasmids pmarR-EGFP and p6744-EGFP, in which the egfp gene was directly controlled by the promoters of marR and SACE_6744, were transformed into E. coli DH5α as controls. The SACE_Lrp gene driven by the promoter of the aac(3)IV gene (Paac(3)IV) was inserted into the control plasmids and transformed into E. coli DH5α. When the SACE_Lrp gene was inserted into the pmarR-EGFP, the green fluorescence was enhanced by 64% compared to that without SACE_Lrp (Fig. 2e). However, when the SACE_Lrp gene was inserted into p6744-EGFP, the green fluorescence was almost unchanged compared to that without SACE_Lrp (Fig. 2e). Taken together, these findings indicate that SACE_Lrp specifically binds to the marR promoter by directly interacting with the site OM and indeed activates the expression of marR promoter in a heterologous E. coli host.
MarR negatively regulates biosynthesis of erythromycin
To further clarify the correlation between SACE_Lrp and marR, the transcription profile of marR in the deletion strain ΔSACE_Lrp (Liu et al. 2017) and the parent strain A226 was measured during the process of growth by qRT-PCR. The results showed that the transcription level of marR in ΔSACE_Lrp was reduced by 36% after 24 h and 50% after 48 h compared with A226 (Fig. 3a), suggesting that SACE_Lrp positively regulates the expression of marR in S. erythraea.

Effects of marR deletion on erythromycin biosynthesis in S. erythraea. a Effects of SACE_ Lrp disruption on transcriptional levels of marR. qRT-PCR was used to quantify the amounts of transcripts in A226 and ΔmarR cultured for 24 h and 48 h in liquid R5 medium. Mean values of three replicates are shown, with the standard deviation indicated by error bars. b Schematic deletion of marR by homologous recombination in S. erythraea A226. c PCR confirmation of the marR deletion mutant by the primers 6745-C-F/R. Lanes: M, 5000-bp DNA ladder, lane 1 of the size of 1520 bp was detected in pUC-ΔmarR as a positive control, lane 2 of the size of 384 bp for the PCR amplified band was observed in A226 as a negative control, and lane 3 of the size 1520 bp was detected in mutant ΔmarR. d Erythromycin A production in S. erythraea A226 and its derivatives by HPLC analysis. Mean values of three replicates are shown, with the standard deviation indicated by error bars. e Growth curves of A226 and ΔmarR. The two strains were cultured in the R5 liquid medium, and their dry weights of mycelia were measured. f Effects of marR disruption on transcriptional levels of ery cluster. qRT-PCR was used to quantify the amounts of transcripts in A226 and ΔmarR cultured for 24 h and 48 h in liquid R5 medium. Mean values of three replicates are shown, with the standard deviation indicated by error bars
Given that the transcription of marR was decreased in ΔSACE_Lrp (Fig. 3a) and ΔSACE_Lrp resulted in a 25% increase in erythromycin production (Liu et al. 2017), it was reasonable to predict that marR deletion would increase erythromycin production. To test this concept, the marR gene was disrupted with tsr replacement (Han et al. 2011) in the parent strain A226 (Fig. 3b), and the resulting mutant ΔmarR was confirmed by PCR (Fig. 3c) and tested for erythromycin production. In comparison with A226, the level of erythromycin production in ΔmarR was improved by 45% (p < 0.01) (Fig. 3d). The complemented strain ΔmarR/pIB-marR with a single copy of marR restored the production of erythromycin to the level of the parent strain A226 (Fig. 3d). ΔmarR and A226 showed similar dry weight of mycelia cultured in R5 liquid medium (Fig. 3e), demonstrating that the increased erythromycin yield in ΔmarR did not result from changes in cell growth. Taken together, these results indicate that the MarR from S. erythraea plays a significant repressor role in erythromycin production.
To investigate the relationship between MarR and erythromycin biosynthesis, we measured the transcription profile of ΔmarR and A226 during the erythromycin production process. The qRT-PCR results showed that transcript levels of erythromycin biosynthetic genes in the ery cluster in ΔmarR significantly increased by 1.7–4.6 fold compared with that in A226 (Fig. 3f), including eryAI (4.6 fold), eryBI (4.4 fold), eryBIII (2.0 fold), eryBIV (3.2 fold), eryBVI (1.9 fold), eryCI (2.1 fold), and eryK (1.7 fold). These results suggest that MarR negatively controls the expression of erythromycin biosynthetic genes in S. erythraea.
MarR negatively regulates export and resistance of erythromycin
Control of genes encoding antibiotic efflux pumps is a vital, well-documented role of MarR family regulators (Beggs et al. 2020). We scanned the genomic data of S. erythraea and found that only SACE_2701-2702 encoded an ABC exporter of polyketide antibiotics, probably responsible for erythromycin export (Oliynyk et al. 2007). The qRT-PCR experiments showed that the transcripts of SACE_2701-2702 in ΔmarR increased by 3.0 fold at 24 h and 2.3 fold at 48 h compared with that in A226, respectively (Fig. 4a), suggesting that MarR indeed negatively controls the expression of the antibiotic efflux gene SACE_2701-2702. To confirm the physiological role of SACE_2701-2702 on erythromycin export, a SACE_2701-2702 disruption mutant Δ2701-2702 was constructed with tsr replacement in S. erythraea A226 (Fig. S1). We therefore assessed the extracellular amount of erythromycin in the mutant Δ2701-2702 and the parent strain A226. The final extracellular accumulation of erythromycin was decreased by 52% (p <0.001) in Δ2701-2702 compared with the parent strain A226 (Fig. 4b). The results were consisted with that the accumulation of extracellular erythromycin in ΔmarR, and A226/pIB-2701-2702 were enhanced by 55% (p <0.01) and 23% (p <0.01), respectively, due to the increased expression of the antibiotic efflux gene SACE_2701-2702 (Fig. 4b). We also assessed its intracellular erythromycin content and found that there was little erythromycin in the cells and no obvious difference between the above strains (Fig. S2). These findings confirmed that the ABC exporter SACE_2701-2702 is responsible for the secretion of erythromycin outside the cell in S. erythraea and the MarR modulates erythromycin production by coordinating its biosynthesis in the cell and export from the cell to the medium.

Effects of marR deletion on erythromycin export in S. erythraea. a Effects of marR disruption on transcriptional levels of SACE_2701-2702. qRT-PCR was used to quantify the amounts of transcripts in A226 and ΔmarR cultured for 24 h and 48 h in liquid R5 medium. Mean values of three replicates are shown, with the standard deviation indicated by error bars. b Concentrations of the extracellular erythromycin production in A226 and its derivatives by HPLC analysis. c Optical densities (OD600) of A226 and ΔmarR with different concentration of erythromycin. The two strains were cultured in the R5 liquid medium for 24 h, and their optical densities of mycelia were measured. d Effects of marR disruption on transcriptional levels of ermE. qRT-PCR was used to quantify the amounts of transcripts in A226 and ΔmarR cultured for 24 h and 48 h in liquid R5 medium. Mean values of three replicates are shown, with the standard deviation indicated by error bars
Given the above fact that gene disruption of marR in S. erythraea A226 notably increased the extracellular production of erythromycin (Fig. 4b), it was reasonable to explore whether disruption of marR would affect the resistance against erythromycin of S. erythraea. Therefore, we tested the resistance of ΔmarR and A226 against erythromycin. As shown in Fig. 4c, when the erythromycin concentration reached 10 mg/L, the growth state of ΔmarR was obviously superior to that of the parent strain A226. These results suggested that deletion of marR effectively improved the resistance against erythromycin of S. erythraea, consistent with the qRT-PCR experiments, which showed that the transcripts of the resistance gene ermE in the ery cluster in ΔmarR increased by 1.5 and 3.9 fold compared with that in A226 at 24 h and 48 h, respectively (Fig. 4d).
MarR directly controls genes of erythromycin biosynthesis, export, and resistance
There are five regulatory regions in the ery cluster, including the promoter regions of eryBVI and eryK and the intergenic regions of eryAI-ermBIV, eryBI-ermBIII, and eryCI-ermE (Fig. 5a). To determine whether MarR directly binds these regulatory regions of the ery cluster, we performed EMSA to evaluate the ability of MarR binding to the corresponding probes using purified His6-tagged MarR protein (Fig. 5b). The results showed that MarR protein can bind to the intergenic regions of eryAI-ermBIV, eryBI-ermBIII, and ermE-eryCI (Fig. 5c), but did not bind to the promoter regions of eryBVI and eryK (Fig. 5d). With an increase in the protein concentration (0, 0.5, 1.0, and 2.0 μM), we observed a decrease in the abundance of the lower band and an increase in the abundance of the upper band (Fig. 5c). Similarly, MarR protein can bind to the promoter DNA of the erythromycin efflux gene SACE_2701-2702 (Fig. 5e). These results demonstrate that MarR plays a direct regulatory role in erythromycin biosynthesis, export and resistance in S. erythraea.

Binding analyses of purified MarR to the putative targets. a Organization of the erythromycin biosynthetic gene cluster. b SDS-PAGE analysis of purified His6-MarR. M, molecular mass marker. c EMSA assays of binding of MarR to PeryAI-eryBIV, PeryBI-eryBIII, and PermE-eryCI. The probe PeryAI-eryBIV represents the intergenic segment between eryAI and eryBIV, PeryBI-eryBIII represents the intergenic segment between eryBI and eryBIII, and PermE-eryCI represents the intergenic segment between ermE and eryCI. d EMSA assays of binding of MarR to PeryBVI and PeryK. PeryBVI and PeryK represent the promoter regions of eryBVI and eryK, respectively. e EMSA assays of binding of MarR to P2701-2702. P2701-2702 represents the promoter region of SACE_2701-2702
Joint engineering of SACE_Lrp and MarR for industrial erythromycin overproduction
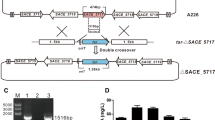
The above findings indicated that erythromycin production was distinctly improved by marR disruption in S. erythraea A226 (Fig. 3), so its practical application was explored in an industrial erythromycin high-yield strain. The marR gene was disrupted in industrial S. erythraea WB, and erythromycin production in shake-flask fermentation was increased by 18% (p < 0.05) in WBΔmarR in industrial fermentation medium (Fig. 6b).

Combinatorial deletion of SACE_Lrp and marR in the industrial S. erythraea. a Erythromycin A production of S. erythraea WB and its derivatives in flask fermentation by HPLC analysis. Mean values of three replicates are shown, with the standard deviation indicated by error bars. b Schematic deletion of marR by homologous recombination in the engineered industrial S. erythraea WBΔSACE_Lrp
In our previous study, gene deletion of SACE_Lrp in WB resulted in a 19% increase in erythromycin production (Liu et al. 2017). To evaluate the potential application of joint engineering of these two regulators for erythromycin overproduction, we further inactivated marR with aac(3)IV replacement in WBΔSACE_Lrp (Fig. 6a). As expected, the engineered strain WBΔLrpΔmarR enhanced the yield of erythromycin A by 20% (p <0.01) and 39% (p < 0.001) relative to WBΔSACE_Lrp and WB, respectively (Fig. 6b). Taken together, the above results indicate that this strategy of joint engineering the Lrp regulator and its target MarR regulator was effective in improving erythromycin production in industrial high-yield strains.
Discussion
It is vitally important to maintain the balance between the biosynthetic process and export process of antibiotics through fine-tuning (Severi and Thomas 2019). As shown in Fig. 7, we previously reported that SACE_Lrp is an efficient regulator for transporting branched-chain amino acids (BCAAs), playing an important role in regulating erythromycin production (Liu et al. 2017). In this study, we further identified the significant regulatory function of SACE_Lrp on a novel MarR protein, highlighting the key role of MarR as a repressor of erythromycin biosynthesis and export in S. erythraea as briefly epitomized in Fig. 7. Lrp proteins are widely distributed among prokaryotes and regulate various biological processes (Peeters and Charlier 2010). However, it is little known that the cascade regulation of Lrp family regulators controls other family regulators to form secondary- or hierarchical-regulatory networks. In S. coelicolor, our reported SCO3361 of the Lrp family protein directly regulates the cluster-situated regulator (CSR) gene actII-ORF4, controlling the biosynthesis of actinorhodin (Act) (Liu et al. 2017). In S. spiramyceticus, SSP_Lrp modulates the expression of three positive regulatory genes bsm42, bsm23, and acyB2, affecting the biosynthesis of spiramycin (SP) and bitespiramycin (BT) (Lu et al. 2019). For MarR family regulators, the cascade regulatory phenomenon is also limited. In Neisseria gonorrhoeae, FadR of MarR family protein was identified as being regulated by MtrR of TetR family members by directly binding to the promoter of FarR (Lee et al. 2003). As stated above, our study builds a previously unknown hierarchical-regulatory relationship between the LFR and MFR.

Proposed model of the regulatory roles of SACE_Lrp and MarR in control of erythromycin biosynthesis and export in S. erythraea. ery cluster: the erythromycin biosynthetic gene cluster; SACE_2701-2702, encodes the ABC exporter of erythromycin (little circle); SACE_5386-5387, encodes the ABC exporter of branched-chain amino acid (BCAA) (triangle); SACE_5388, encodes the SACE_Lrp protein; SACE_6745, encodes the MarR protein; SACE_Lrp directly repressed (blue flat-headed arrow) the expression of SACE_5386-5387 while directly activated (blue pointed arrow) the expression of SACE_6745. MarR directly repressed (red flat-headed arrow) the expressions of the erythromycin biosynthetic genes and SACE_2701-2702. Intracellular BCAA catabolism provides the precursors for erythromycin biosynthesis in S. erythraea (black dashed arrow)
To our knowledge, MarR from S. erythraea is the first reported MFR showing explicit regulatory role on the export of antibiotic through a direct manner in antibiotic-producing actinobacteria. In previous researches, there were only three transcription regulators from the TetR family: S. graminearus GouR, S. coelicolor ActR, and Amycolatopsis mediterranei RifQ could modulate antibiotic export in actinobacteria (Lei et al. 2018; Wei et al. 2014; Xu et al. 2012). The TetR family is a large group of transcriptional regulators widely present in bacteria and controls a number of diverse processes (Cuthbertson and Nodwell 2013). The most common targets of TetR proteins are genes encoding the antibiotic efflux pumps, which are often situated right next to the tetR gene (Lei et al. 2018; Wei et al. 2014; Xu et al. 2012). Differently, S. erythraea MarR modulated the export of erythromycin by controlling a faraway ABC exporter, SACE_2701-2702 (Fig. 4a). Our work indicates that there may be various complex regulatory mechanisms of the role of transcription regulators in antibiotic efflux in entire antibiotic-producing actinobacteria.
Most MarR regulators have been shown to control antibiotic biosynthesis through indirectly, such as AbsC of S. coelicolor, DptR3 of S. roseosporus, SAV4189 of S. avermitilis, and CtcS of S. aureofaciens (Deng et al. 2011; Guo et al. 2018; Hesketh et al. 2010; Kong et al. 2019). However, MarR of S. erythraea controlled erythromycin production by directly controlling the expression of erythromycin biosynthetic genes in the ery cluster (Fig. 4a and 5a). Similarly, OhrR of S. avermitilis controls avermectin production by directly repressing the expression of the pathway-specific activator gene aveR (Sun et al. 2018). In addition, in this study, we found that deletion of marR did not affect morphological differentiation in S. erythraea (Fig. S3). Similarly, MarR proteins CtcS from S. aureofaciens, SAV4189, and OhrR from S. avermitilis had no obvious effect on morphological differentiation (Guo et al. 2018; Kong et al. 2019; Sun et al. 2018). However, S. roseosporus MarR protein DptR3 deletion delayed aerial mycelium formation and sporulation (Zhang et al. 2015). These findings indicate that the regulatory roles and mechanisms of MFRs from different actinobacteria may not be entirely the same.
A common regulatory function of MarR proteins is directly regulation of its own gene’s expression, such as PcaV and TamR of S. coelicolor, SAV4189 and OhrR of S. avermitilis, DptR3 of S. roseosporus, and CtcS of S. aureofaciens (Davis et al. 2013; Guo et al. 2018; Huang and Grove 2013; Kong et al. 2019; Sun et al. 2018; Zhang et al. 2015). In S. erythraea, the marR transcription level in ΔmarR significantly decreased compared with A226 by qRT-PCR (Fig. S4a). Unexpectedly, MarR did not bind to PmarR including the entire marR promoter region by EMSA (Fig. S4b). This case is different from most MarR proteins, implying that the binding affinity of S. erythraea MarR be induced by an unknown cofactor or signal.
In this work, although we demonstrated the direct control of erythromycin biosynthesis, export, and resistance by MarR in S. erythraea (Figs. 4a and 5a), erythromycin is not the ligand of MarR protein (data not shown). In S. aureofaciens, the antibiotics CTC and TC could weaken the DNA-binding activity of CtcS (Kong et al. 2019). Pentalenolactone and two intermediates, pentalenolactones D and F, act as ligands of PenR of S. exfoliatus and its homologue PntR of S. arenae (Zhu et al. 2013). SAV4189 of S. avermitilis did not respond to antibiotics AveB1 and Oli but response to antibiotics HygB and Thi, which are produced by other Streptomyces species, to weaken its DNA-binding activity (Guo et al. 2018), implying that the MarR of S. erythraea can sense some other antibiotic, and it will be of interest to solve this question in the future. Overall, these findings reveal the relatively high variability in the ligand-binding domain of MarR proteins and their complex mechanisms in response to different ligands.
Currently, genetic engineering of regulatory systems provides a potential approach for relevant antibiotic improvement (Li et al. 2015). Based on the regulatory properties of these transcription regulators, they have been employed for the overproduction of target metabolites by modulating transcription repression or activation (Niu and Tan 2013). For example, disruption of LFR SACE_Lrp combined with overexpression of its target SACE_5387-5386 in industrial S. erythraea WB (Liu et al. 2017) and overexpression of MFR SAV4189 combined with disruption of its target gene sav_4190 in industrial S. avermitilis A-144 (Guo et al. 2018) were efficient approaches to enhance the relevant antibiotic production. In this study, we found that deletion of marR in S. erythraea A226 reduced the expression of its own gene but promoted erythromycin production (Fig. 3d), implying that reduction of the marR expression level is an effective strategy for erythromycin overproduction. As expected, deletion of SACE_Lrp combined with deletion of its target marR improved erythromycin production of industrial S. erythraea strain by 39% in industrial strain WB (Fig. 6b). On the one hand, this strategy can improve the production erythromycin precursor by deleting SACE_Lrp; on the other hand, it can promote erythromycin biosynthesis and efflux by deleting marR. Based on bioinformatics analysis with SACE_Lrp and MarR, we found that LFRs and MFRs are widely present among typical antibiotic-producing actinomycetes (Table 2). To summarized, our present findings raise the knowledge of the molecular regulation of antibiotic biosynthesis and export by LFRs and MFRs and provide a potential use in large-scale industrial applications for target metabolite overproduction by joint engineering of these regulators.
To summarize, in our previous report, SACE_Lrp indirectly regulated the erythromycin production through directly controlling the BCAAs transport, and a question remains whether SACE_Lrp can control the erythromycin biosynthesis by secondary-or hierarchical-regulatory networks. Here in this study, our work revealed that the novel regulator MarR (SACE_6745) from S. erythraea, which is controlled by SACE_Lrp, plays a negative role in erythromycin biosynthesis and export. Dissection of the regulatory mechanism of MarR revealed that MarR directly controlled the genes that are responsible for erythromycin biosynthesis, export, and resistance. Moreover, joint engineering of the SACE_Lrp-MarR regulatory system for practical application on previously constructed high-yield WBΔSACE_Lrp resulted in notable overproduction of erythromycin by further deletion of marR. Based on the above findings, we present new insights into the hierarchical-regulatory relationship of Lrp and MarR for the first time, and new avenues for coordinating antibiotic biosynthesis and export with combinatorial engineering regulators in actinobacteria.
Data availability
All data generated or analyzed during this study are available from the corresponding author on reasonable request.
References
Beggs GA, Brennan RG, Arshad M (2020) MarR family proteins are important regulators of clinically relevant antibiotic resistance. Protein Sci 29:647–653. https://doi.org/10.1002/pro.3769
Bérdy J (2005) Bioactive microbial metabolites. J Antibiot 58(1):1–26. https://doi.org/10.1038/ja.2005.1
Brinkman AB, Ettema TJ, de Vos WM, van der Oost J (2003) The Lrp family of transcriptional regulators. Mol Microbiol 48(2):287–294. https://doi.org/10.1046/j.1365-2958.2003.03442.x
Butler MS (2008) Natural products to drugs: natural product-derived compounds in clinical trials. Nat Prod Rep 25(3):475–516. https://doi.org/10.1002/chin.200839259
Cuthbertson L, Nodwell JR (2013) The TetR family of regulators. Microbiol Mol Biol Rev 77(3):440–475. https://doi.org/10.1128/MMBR.00018-13
Davis JR, Brown BL, Rebecca P, Sello JK (2013) Study of PcaV from Streptomyces coelicolor yields new insights into ligand-responsive MarR family transcription factors. Nucleic Acids Res 6:3888–3900. https://doi.org/10.1093/nar/gkt009
Deng W, Wang H, Xie J (2011) Regulatory and pathogenesis roles of Mycobacterium Lrp/AsnC family transcriptional factors. J Cell Biochem 112(10):2655–2662. https://doi.org/10.1002/jcb.23193
Deochand DK, Grove A (2017) MarR family transcription factors: dynamic variations on a common scaffold. Crit Rev Biochem Mol Biol 52:595–613. https://doi.org/10.1080/10409238.2017.1344612
Grove A (2013) MarR family transcription factors. Curr Biol 23(4):142–143. https://doi.org/10.1016/j.cub.2013.01.013
Guo J, Zhang X, Lu X, Liu W, Chen Z, Li J, Deng L, Wen Y (2018) SAV4189, a MarR-family regulator in Streptomyces avermitilis, activates avermectin biosynthesis. Front Microbiol 9:1358. https://doi.org/10.3389/fmicb.2018.01358
Gupta A, Pande A, Sabrin A, Thapa SS, Gioe BW, Grove A (2018) MarR family transcription factors from Burkholderia species: hidden clues to control of virulence-associated genes. Microbiol Mol Biol Rev 83(1). https://doi.org/10.1128/MMBR.00039-18
Han S, Song P, Ren T, Huang X, Cao C, Zhang B (2011) Identification of SACE_7040, a member of TetR family related to the morphological differentiation of Saccharopolyspora erythraea. Curr Microbiol 63(2):121–125. https://doi.org/10.1007/s00284-011-9943-z
Hellman LM, Fried MG (2007) Electrophoretic mobility shift assay (EMSA) for detecting protein-nucleic acid interactions. Nat Protoc 2(8):1849–1861. https://doi.org/10.1038/nprot.2007.249
Hesketh A, Kock H, Mootien S, Bibb M (2010) The role of absC, a novel regulatory gene for secondary metabolism, in zinc-dependent antibiotic production in Streptomyces coelicolor A3(2). Mol Microbiol 74(6):1427–1444. https://doi.org/10.1111/j.1365-2958.2009.06941.x
Hiard S, Rl M, Colson S, Hoskisson PA, Titgemeyer F, Wezel GPV, Joris B, Wehenkel L, Rigali S (2007) PREDetector: a new tool to identify regulatory elements in bacterial genomes. Biochem Biophys Res Commun 357(4):861–864. https://doi.org/10.1016/j.bbrc.2007.03.180
Huang H, Grove A (2013) The transcriptional regulator TamR from Streptomyces coelicolor controls a key step in central metabolism during oxidative stress. Mol Microbiol 87(6):1151–1166. https://doi.org/10.1111/mmi.12156
Kong L, Liu J, Zheng X, Deng Z, You D (2019) CtcS, a MarR family regulator, regulates chlortetracycline biosynthesis. BMC Microbiol 19(1):1–11. https://doi.org/10.1186/s12866-019-1670-9
Lee EH, Rouquette-Loughlin C, Folster JP, Shafer WM (2003) FarR regulates the farAB-encoded efflux pump of Neisseria gonorrhoeae via an MtrR regulatory mechanism. J Bacteriol 185(24):7145–7152. https://doi.org/10.1128/JB.185.24.7145-7152.2003
Lei C, Wang J, Liu Y, Liu X, Zhao G, Wang J (2018) A feedback regulatory model for RifQ-mediated repression of rifamycin export in Amycolatopsis mediterranei. Microb Cell Fact 17(1):14. https://doi.org/10.1186/s12934-018-0863-5
Li L, Zhao Y, Ruan L, Yang S, Mei G, Jiang W, Lu Y (2015) A stepwise increase in pristinamycin II biosynthesis by Streptomyces pristinaespiralis through combinatorial metabolic engineering. Metab Eng 29:12–25. https://doi.org/10.1016/j.ymben.2015.02.001
Liu G, Chater KF, Chandra G, Niu G, Tan H (2013) Molecular regulation of antibiotic biosynthesis in Streptomyces. Microbiol Mol Biol Rev 77(1):112–143. https://doi.org/10.1128/MMBR.00054-12
Liu J, Chen Y, Li L, Yang E, Zhang B (2019) Characterization and engineering of the Lrp/AsnC family regulator SACE_5717 for erythromycin overproduction in Saccharopolyspora erythraea. J Ind Microbiol Biotechnol 46(7):1013–1024. https://doi.org/10.1007/s10295-019-02178-2
Liu J, Chen Y, Wang W, Ren M, Wu P, Wang Y, Li C, Zhang L, Wu H, Weaver DT, Zhang B (2017) Engineering of an Lrp family regulator SACE_Lrp improves erythromycin production in Saccharopolyspora erythraea. Metab Eng 39:29–37. https://doi.org/10.1016/j.ymben.2016.10.012
Liu J, Li J, Dong H, Chen Y, Wang Y, Wu H, Li C, Weaver DT, Zhang L, Zhang B (2017) Characterization of an Lrp/AsnC family regulator SCO3361, controlling actinorhodin production and morphological development in Streptomyces coelicolor. Appl Microbiol Biotechnol 101(14):5773–5783. https://doi.org/10.1007/s00253-017-8339-9
Lu Z, Zhang X, Dai J, Wang Y, He W (2019) Engineering of leucine-responsive regulatory protein improves spiramycin and bitespiramycin biosynthesis. Microb Cell Fact 18(1):38. https://doi.org/10.1186/s12934-019-1086-0
Marcellin E, Licona-Cassani C, Mercer TR, Palfreyman RW, Nielsen LK (2013) Re-annotation of the Saccharopolyspora erythraea genome using a systems biology approach. BMC Genomics 14:699. https://doi.org/10.1186/1471-2164-14-699
Martín JF, Liras P (2010) Engineering of regulatory cascades and networks controlling antibiotic biosynthesis in Streptomyces. Curr Opin Microbiol 13(3):263–273. https://doi.org/10.1016/j.mib.2010.02.008
McDaniel R, Licari P, Khosla C (2001) Process development and metabolic engineering for the overproduction of natural and unnatural polyketides. Adv Biochem Eng Biotechnol 73:31–52. https://doi.org/10.1007/3-540-45300-8_3
Niu G, Tan H (2013) Biosynthesis and regulation of secondary metabolites in microorganisms. Sci China Life Sci 56(7):581–583. https://doi.org/10.1007/s11427-013-4501-5
Oh SY, Shin JH, Roe JH (2007) Dual role of OhrR as a repressor and an activator in response to organic hydroperoxides in Streptomyces coelicolor. J Bacteriol 189(17):6284–6292. https://doi.org/10.1128/JB.00632-07
Oliynyk M, Samborskyy M, Lester JB, Mironenko T, Scott N, Dickens S, Haydock SF, Leadlay PF (2007) Complete genome sequence of the erythromycin-producing bacterium Saccharopolyspora erythraea NRRL23338. Nat Biotechnol 25(4):447–453. https://doi.org/10.1038/nbt1297
Peeters E, Charlier D (2010) The Lrp family of transcription regulators in archaea. Archaea 2010:750457–750410. https://doi.org/10.1155/2010/750457
Severi E, Thomas GH (2019) Antibiotic export: transporters involved in the final step of natural product production. Microbiology 165(8):805–818. https://doi.org/10.1099/mic.0.000794
Sun M, Mengya L, Wen Y, Song Y, Li J, Chen Z (2018) Organic peroxide-sensing repressor OhrR regulates organic hydroperoxide stress resistance and avermectin production in Streptomyces avermitilis. Front Microbiol 9:1398. https://doi.org/10.3389/fmicb.2018.01398
Tahlan K, Yu Z, Xu Y, Davidson AR, Nodwell JR (2008) Ligand recognition by ActR, a TetR-like regulator of actinorhodin export. J Mol Biol 383(4):753–761. https://doi.org/10.1016/j.jmb.2008.08.081
Wei J, Tian Y, Niu G, Tan H (2014) GouR, a TetR family transcriptional regulator, coordinates the biosynthesis and export of gougerotin in Streptomyces graminearus. Appl Environ Microbiol 80(2):714–722. https://doi.org/10.1128/aem.03003-13
Wilkinson CJ, Hughes-Thomas ZA, Martin CJ, Bohm I, Mironenko T, Deacon M, Wheatcroft M, Wirtz G, Staunton J, Leadlay PF (2002) Increasing the efficiency of heterologous promoters in actinomycetes. J Mol Microbiol Biotechnol 4(4):417–426. https://doi.org/10.1016/S0167-7012(02)00036-2
Wu H, Chen M, Mao Y, Li W, Liu J, Huang X, Zhou Y, Ye BC, Zhang L, Weaver DT, Zhang B (2014) Dissecting and engineering of the TetR family regulator SACE_7301 for enhanced erythromycin production in Saccharopolyspora erythraea. Microb Cell Fact 13:158. https://doi.org/10.1186/s12934-014-0158-4
Xu Y, Tang Y, Wang N, Liu J, Zhang B (2020) Transcriptional regulation of a leucine-responsive regulatory protein for directly controlling lincomycin biosynthesis in Streptomyces lincolnensis. Appl Microbiol Biotechnol 104(6):2575–2587. https://doi.org/10.1007/s00253-020-10381-w
Xu Y, Willems A, Au-Yeung C, Tahlan K, Nodwell JR (2012) A two-step mechanism for the activation of actinorhodin export and resistance in Streptomyces coelicolor. mBio 3(5). https://doi.org/10.1128/mbio.00191-12
Zhang Q, Chen Q, Zhuang S, Chen Z, Wen Y, Li J, Liu SJ (2015) A MarR family transcriptional regulator, DptR3, activates daptomycin biosynthesis and morphological differentiation in Streptomyces roseosporus. App Environ Microbiol 81(11):3753–3765. https://doi.org/10.1128/AEM.00057-15
Zhu D, Wang Y, Zhang M, Ikeda H, Deng Z, Cane DE (2013) Product-mediated regulation of pentalenolactone biosynthesis in Streptomyces species by the MarR/SlyA family activators PenR and PntR. J Bacteriol 195(6):1255–1266. https://doi.org/10.1128/JB.02079-12
Funding
This work was supported by the National Natural Science Foundation of China (grant numbers 31800057, 31972930, 31570074), the Initial Foundation of Scientific Research in Anhui Agricultural University (grant number yj2018-08), and the University Synergy Innovation Program of Anhui Province (grant number GXXT-2019-035).
Author information
Authors and Affiliations
Contributions
JL, HW, and BZ conceived the study. JL designed the study. JL, LL, YW, BL, and XC performed the experiments. JL, LT, SD, EY, and HW analyzed the data. JL wrote the manuscript. HW and BZ checked the final version. All authors have read and approved the manuscript.
Corresponding authors
Ethics declarations
Ethics approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
ESM 1
(PDF 634 kb)
Rights and permissions
About this article

Cite this article
Liu, J., Li, L., Wang, Y. et al. Joint engineering of SACE_Lrp and its target MarR enhances the biosynthesis and export of erythromycin in Saccharopolyspora erythraea. Appl Microbiol Biotechnol 105, 2911–2924 (2021). https://doi.org/10.1007/s00253-021-11228-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-021-11228-8