
Abstract
Aspartic proteases exhibit optimum enzyme activity under acidic conditions and have been extensively used in food, fermentation, and leather industries. In this study, a novel aspartic protease precursor (proTlAPA1) from Talaromyces leycettanus was identified and successfully expressed in Pichia pastoris. Subsequently, the auto-activation processing of the zymogen proTlAPA1 was studied by SDS-PAGE and N-terminal sequencing, under different processing conditions. TlAPA1 shared the highest identity of 70.3% with the aspartic endopeptidase from Byssochlamys spectabilis (GAD91729) and was classified into a new subgroup of the aspartic protease A1 family, based on evolutionary analysis. Mature TlAPA1 protein displayed an optimal activity at 60 °C and remained stable at temperatures of 55 °C and below, indicating the thermostable nature of TlAPA1 aspartic protease. During the auto-activation processing of proTlAPA1, a 45-kDa intermediate was identified that divided the processing mechanism into two steps: formation of intermediates and activation of the mature protein (TlAPA1). The former step can be processed without proteolytic activity, while the latter process depended on protease activity completely. The discovery of the novel aspartic protease TlAPA1 and the study of its activation process will contribute to a better understanding of the mechanism of aspartic protease auto-activation.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Proteases (EC3.4.11-24) make up a large share of the total global industrial enzymes (Raveendran et al. 2018). They have extensive applications in the dairy, baking, beverages, brewing, meat, and functional food industries (Raveendran et al. 2018; Zhang et al. 2018). On the basis of the optimal pH of hydrolysis, proteases have been grouped into three categories—acidic, neutral, and alkaline proteases. They can also be classified into serine, cysteine, metallo, and aspartic proteases, depending upon their catalytic residues. Aspartic proteases (EC 3.4.23) have two aspartic residues at their catalytic center, which are vital for hydrolytic cleavage of peptide bonds (Yegin et al. 2011). The activity of aspartic proteases can be specifically inhibited by pepstatin A. Molecular weights of aspartic proteases commonly range between 30 to 50 kDa, while some can weigh up to 55 kDa. Aspartic proteases are generally considered as acidic proteases, because they have isoelectric points of 3.0–4.5 and show optimal activity at pH 3.0–5.0. Most aspartic proteases have an optimal temperature in the range of 30–50 °C, while some exhibit maximum activity at 55 °C. Most aspartic proteases are also sensitive to high temperatures and show poor thermostability, limiting their applications to mesophilic conditions. Thus, the thermal stability of aspartic proteases has been the subject of attention in many recent studies (Silva et al. 2016). The discovery of novel thermostable enzymes, especially from extremophiles, is a potential method to tackle the aforesaid problem (Zhang et al. 2018; Kangwa et al. 2018).
Aspartic proteases are widespread in many organisms—vertebrates, insects, plants, fungi, and even viruses have been widely reported as sources of aspartic proteases (Mandujano-González et al. 2016). The production of aspartic proteases from fungi has several advantages including a short productive cycle, simple late purification, and low costs (Souza et al. 2015). Most commercial aspartic proteases used currently in industrial production are derived from filamentous fungi. Aspartic proteases from fungi are mainly categorized into two groups—pepsin-like and rennin-like enzymes (Rao et al. 1998). The pepsin-like enzymes include aspergillopepsin (Vishwanatha et al. 2009), penicillopepsin (Fraser et al. 1992), trichodermapepsin (Nascimento et al. 2008), and rhizopuspepsin (Kumar et al. 2005), while the rennin-like enzymes are mainly produced by Mucor, Rhizomucor, and Chryphonectria (Yegin et al. 2011). However, the yield of fungal aspartic proteases by industrial fermentation is usually low. An aspartic protease from Aspergillus foetidus was extracellularly produced with an activity of only 63.7 U/mL (Souza et al. 2017). Pichia pastoris is an excellent expression system that has been effectively used to solve the problems of low yield of proteases. Many aspartic proteases have been heterogeneously expressed in P. pastoris (Yang et al. 2013; Yegin and Fernandez-Lahore 2013; Gama Salgado et al. 2013). An aspartic protease from Rhizomucor miehei was produced in P. pastoris with the activity of 3480.4 U/mL (Sun et al. 2018).
Typical aspartic proteases are initially synthesized in the form of inactive precursors (zymogens), which protect host cells from proteolysis (Mandujano-González et al. 2016). The functions of the N-terminal prosegments of aspartic proteases have been studied extensively and include facilitating correct folding, blocking the active site, and stabilizing the protein (Koelsch et al. 1994; Horimoto et al. 2009). It is generally accepted that the propeptides are autocatalytically cleaved at acidic pH (Richter et al. 1998; Khan et al. 1999; Dunn 2002), and their further processing by other peptidases is important for the activation of aspartic proteases from Candida parapsilosis (Dostal et al. 2005). Crystal structures of some aspartic protease zymogens and their activation intermediates have been reported (Richter et al. 1998; Khan et al. 1999; Morales et al. 2012; Lee et al. 1998; Ostermann et al. 2004), which contribute to the understanding of propeptide interactions with catalytic proteins. Current studies on zymogen activation have mainly concentrated on aspartic proteases associated with diseases (Dunn 2002; Hanova et al. 2018), yet little is known about this process among aspartic proteases from fungi. There is evidence that the predicted zymogens vary in length depending on each fungus, suggesting their unique activation processes (Mandujano-González et al. 2016). Therefore, studying the activation processing of fungal aspartic proteases is of great significance to understand the mode of activation of the whole family.
Given the importance of novel and thermostable aspartic proteases in industrial processes, a gene coding for a novel thermostable aspartic protease, Tlapa1, was found and cloned from the thermophilic filamentous fungus Talaromyces leycettanus. Phylogenetic analysis indicated that TlAPA1 belonged to a new subgroup of aspartic proteases A1 family. Moreover, TlAPA1 was expressed in P. pastoris with a zymogen form and its auto-activation was studied in detail. The auto-activation process of TlAPA1 was affected by pH and enzymatic activity and occurred in two stages distinguished by the presence of a processing intermediate. The mature proteases with activity were subsequently purified and characterized biochemically. In this study, a novel thermostable aspartic protease was discovered and synthesized as a zymogen in P. pastoris, and its autocatalytic activation was studied.
Materials and methods
Strains, vectors, and substrates
The gene donor strain Talaromyces leycettanus JCM12802 was purchased from Japan Collection of Microorganisms RIKEN BioResource Center. Escherichia coli Trans1-T1 (TransGen) was used for gene cloning and sequencing. The target gene was expressed in P. pastoris GS115 (Invitrogen). Cloning and expression vectors used were pEASy-T3 (TransGen, Beijing, China) and pPIC9 (Invitrogen, Carlsbad, CA), respectively. Casein sodium salt from bovine milk (C8654, Sigma-Aldrich, St. Louis, MO) was used as a substrate, and other chemicals of analytical grade were commercially available.
Gene cloning and mutagenesis
Talaromyces leycettanus JCM12802, with optimal growth at pH 5.0–6.0, was cultivated as described previously (Zhang et al. 2017). DNA and total RNA were extracted from the mycelia of T. leycettanus JCM12802 after 3 days of growth at 40 °C, and the cDNA was prepared according to the manufacturer’s instructions (TOYOBO, Osaka, Japan). On the basis of the whole genome sequence of T. leycettanus JCM12802 (Wang et al. 2017), the primer pairs used for amplification of Tlapa1 gene are designed and listed in Table 1. The gene that encodes aspartic protease TlAPA1 was amplified from DNA and cDNA of Talaromyces leycettanus, respectively, by the polymerase chain reaction (PCR) method. The PCR reaction program was as follows: a predegeneration at 94 °C for 3 min, followed by 34 cycles of 94 °C for 30 s, 55 °C for 30 s, 72 °C for 1 min, and a final step of 72 °C for 10 min. Finally, the purified PCR products were cloned into the pEASY-T3 and sequenced. A single-point mutation D103N was used to study the effects of proteolytic activity on auto-activation processing. Mutant D103N was performed using the Fast Mutagenesis System Kit (TransGen). Sequences of primer pairs used in these mutations are listed in Table 1.
Bioinformatic analysis of Tlapa1 gene
The sequence results were assembled using DNA Star 7.1 software. The amino acid sequences obtained by the Vector NTI Advance 10.0 software (Invitrogen) were searched with BLASTp programs (http://www.ncbi.nlm.nih.gov/BLAST/) to analyze the homologous sequences. The signal peptide sequence of TlAPA1 was predicted with SignalP (http://www.cbs.dtu.dk/services/SignalP/). The potential N-glycosylation sites were predicted using NetNGlyc 1.0 Server (http://www.cbs.dtu.dk/services/NetNGlyc/). Alignment of multiple protein sequences was accomplished using Clustal W software (http://www.clustal.org/) and rendered using the ESPript3.0 program (http://espript.ibcp.fr/ESPript/cgi-bin/ESPriptcgi).
Phylogenetic analysis
The full amino acid sequence of TlAPA1 was used as the query sequence in BLASTp searches in NCBI (http://blast.ncbi.nlm.nih.gov/Blast.cgi). In total, 22 sequences of A1 family aspartic proteases were obtained. Multiple sequence alignments of TlAPA1 with other representative aspartic protease enzymes, characterized enzymes, and enzymes with determined three-dimensional (3D) structures, were performed as described previously (Revuelta et al. 2014). Sequence information for the A1 family of aspartic proteases was obtained from the MEROPS database (https://www.ebi.ac.uk/merops/cgi-bin/family_index?type=P#A). Phylogenetic analyses of TlAPA1 and A1 family of aspartic proteases were performed as described in previous studies (Revuelta et al. 2014). The distance matrix for nucleotides was calculated by Kimura’s two-parameter model. The phylogenetic tree was constructed with the neighbor-joining method using MEGA 7.0 and assessed using 1000 bootstrap replications (Kumar et al. 2016).
Expression and purification of zymogens
Recombinant proteins were expressed in P. pastoris GS115, as described previously (Zhang et al. 2017). Briefly, the gene fragment coding for the zymogen (proTlAPA1) without the signal peptide was amplified using the PCR method. PCR products were digested with EcoRI and NotI and ligated into the pPIC9 plasmid using T4 DNA ligase (New England Laboratory). The recombinant plasmid pPIC9-proTlapa1, linearized by BglII, was transformed into P. pastoris GS115 competent cells by electroporation. Positive transformants were screened based on the transparent zone on skim milk plates as described below. The transformants showing the largest transparent zones were inoculated into 30 mL YPD and incubated at 30 °C. The seed medium containing the positive transformant was inoculated into 1-L conical flasks containing 300 mL of BMGY for fermentation. Conical flasks containing 200 mL of BMMY and 0.5% (v/v) methanol were prepared.
The cells were harvested by centrifugation for 10 min at 12,000g and resuspended in BMMY medium and for the next subsequent fermentation at 30 °C. Methanol was added every 24 h to obtain a final concentration of 0.5% (v/v). After 48 h of cultivation, cell-free cultures were centrifugated at 12,000g, 4 °C for 10 min, and the fermentation broth was collected. The crude pro-enzymes were concentrated using an ultrafiltration membrane with a molecular weight cut-off of 10 kDa (Vivascience, Hannover, Germany). A HiTrap Q Sepharose XL 5 mL FPLC column (GE Healthcare, Sweden) was used for purification. Protein binding and equilibration was performed using buffer A (10 mM sodium phosphate, pH 6.0), and a linear gradient of NaCl (0–1.0 M) was used to elute the proteins.
Activation of purified zymogen proTlAPA1
To determine the processing of zymogen conversion, pH values of the proTlAPA1 samples were adjusted to 3.0 using 0.5 M lactic acid-sodium lactate buffer. proTlAPA1 was autocatalytically activated at 37 °C for 0, 15, 30, 45, 60, 75, and 90 min, respectively. Processed polypeptides were detected by SDS-PAGE and N-terminal sequencing. To investigate whether the maturation of proTlAPA1 is protein concentration-dependent or not, zymogen proteins of wild-type and mutants with two concentrations (0.1 mg/mL and 0.2 mg/mL) were incubated at 37 °C for 5 and 10 min and analyzed with SDS-PAGE. To study the effects of pepstatin A on the zymogen conversion of proTlAPA1, 5 μM pepstatin A was added into the conversion system before the autocatalytic processing and incubated at 37 °C for 15, 30, 45, and 60 min, respectively. Zymogen conversion systems were subjected to SDS-PAGE analysis. Samples without pepstatin A were also treated similarly, as a control group. To determine the optimum pH of zymogen conversion, the pH values of the proTlAPA1 samples were adjusted to 2.0, 3.0, 4.0, 5.0, and 6.0 using 0.5 M lactic acid-sodium lactate buffer. The samples at different pH conditions were incubated at 37 °C; and the proteolytic activities were analyzed with casein (1%, w/v) at different incubation times. Finally, the samples were analyzed using SDS-PAGE.
Enzyme activity assay
The activity of aspartic proteases was assayed in a 1000 μl reaction mixture containing 500 μl of 1% (w/v) casein sodium salt and 500 μl enzyme sample in buffer at pH 3.0. After incubation at 60 °C for 10 min, 1000 μl of 40% (w/v) trichloroacetic acid (TCA) was added to terminate the reaction. A total of 500 μL of the supernatant was obtained from the mixture using centrifugation after 12,000g for 3 min. 2.5 mL of 0.4 M sodium carbonate and 500 μL Folin-phenol was added into supernatant in turn before incubating at 40 °C for 20 min. The amount of released tyrosine was measured at 680 nm. One unit of proteolytic activity was defined as the amount of enzyme that released 1 μmol of tyrosine equivalent per minute under the conditions described above (pH 3.5, 60 °C and 10 min).
Properties of recombinant aspartic protease TlAPA1
Optimal conditions for purified TlAPA1 activity were measured in lactic acid-sodium lactate buffer under the following conditions–temperatures ranging from 30 to 80 °C (at a constant pH 3.5); and pH ranging from 2.0 to 4.5 (at a constant temperature of 60 °C). Thermostability of TlAPA1 was assessed by preincubating the purified enzyme for 1 h in lactic acid-sodium lactate buffer under varying temperature conditions: 55 °C, 60 °C and 65 °C (at constant pH of 3.5). The pH stability was tested after preincubating at a constant temperature of 37 °C and varying pH conditions: 100 mM glycine-HCl for pH 1.0–3.0, 100 mM citric acid–Na2HPO4 for pH 3.0–8.0, 100 mM Tris-HCl for pH 8.0–9.0, and 100 mM glycine-NaOH for pH 9.0–10.0. And then the residual enzyme activities were measured under optimum conditions (60 °C, pH 3.5).
The proteolytic activities of TlAPA1 were measured under standard conditions (pH 3.5, 60 °C, 10 min) with 0.5–10 mg/mL casein sodium salt, and the constants were determined by linear regression fitting using GraphPad Prism version 7.01. All experiments were performed in three biological and technical replicates.
N-terminal sequencing
Purified proteins were separated by SDS-PAGE and electro transferred onto a polyvinylidene difluoride (PVDF) membrane. Stained with Coomassie Brilliant Blue R-250, the target protein bands were excised and subjected to N-terminal amino acid sequence analysis using a PPSQ-33 automatic sequence analysis system (Shimadzu, Kyoto, Japan).
Results
Gene cloning and sequence analysis
The gene encoding a novel aspartic protease zymogen was identified from the genome of Talaromyces leycettanus and named as proTlapa1. The DNA sequence (MK108371 in the GenBank database) had 1396 bp consisting of 3 introns and 4 exons. proTlapa1 encodes a polypeptide of 424 amino acids including a putative signal peptide of 19 residues and a propeptide of 61 residues at the N-terminus. The molecular mass and pI value were estimated to be 42.5 kDa and 4.7, respectively. Three N-glycosylation sites (N144, N253, and N357) were predicted using the NetNGlyc 1.0 Server. The deduced amino acid sequence of proTlapa1 shared the highest identity of 70.3% with the aspergillopepsin A-like aspartic endopeptidase from Byssochlamys spectabilis (GAD91729, Oka et al. 2014), and of only 41.4% with a functionally characterized aspergillopepsin-1 from Aspergillus oryzae RIB40 (Q06902, Gomi et al. 1993). Among aspartic proteases with determined three-dimensional structures, proTlapa1 (residues 86 to 405) showed the highest identity (41.7%) with the corresponding domain of the mature aspartic protease (PDB 1IZD) from Aspergillus oryzae (Kamitori et al. 2003).
Phylogenetic analysis of TlAPA1
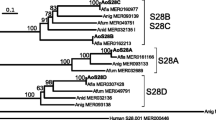
The results of BLASTP analysis showed that TlAPA1 belonged to the A1 family of aspartic proteases. However, this family of proteases comprises of many subgroups with complex evolutionary relationships. To obtain a clear evolutionary relationship between TlAPA1 and other homologs of the A1 family, a phylogenetic analysis based on the amino acid sequence alignment was performed using MEGA 7.0. These results indicated that aspartic proteases from different microorganisms were separated from each other in the evolutionary tree (Fig. 1). Seven subgroups that had been reported in previous studies emerged in the process of evolution in the following order: aspergillopepsin, penicillopepsin, trichoderpepsin, podosporapepsin, endothiapepsin, rhizopuspepsin, and murcorpepsin subgroups. However, TlAPA1 did not belong to any of these subgroups. TlAPA1 along with Q4WZS3 from Aspergillus fumigates belonged to a new clade in the evolutionary tree (Fig. 1). This clade emerged after the formation of the rhizopuspepsin subgroup in the evolutionary tree. The evolutionary position of TlAPA1 suggested that TlAPA1 might have unique characteristics distinct from other aspartic peptidases.

Phylogenetic analysis of the subgroups of aspartic proteases A1 family. The amino acid sequences of A1 family were obtained by BLAST analysis using the TlAPA1 protein (GenBank accession number MK108371) as the query sequence. The evolutionary tree was constructed by the neighbor-joining method. The sequences are labeled with their GenBank accession numbers and host fungi. Numbers indicated in the tree branches are the bootstrap values (%) based on 1000 replications. The subgroups of A1 family are classified using gray shadow and names are indicated in overstriking. TlAPA1 is shown in red which, together with Q4WZS3, belonged to a new unknown clade
Heterologous expression and purification of proenzyme
The zymogen proTlAPA1, consisting of the N-terminal propeptide and the mature domain, was expressed in P. pastoris GS115. Expression of the recombinant protein was confirmed by electrophoresis. As shown by SDS-PAGE (Fig. 2), a specific protein band corresponding to a molecular mass of approximately 53 kDa was obtained, which was higher than the calculated value (45 kDa) of proTlAPA1. Upon treating the samples with Endo H, the target band appeared at approximately 45 kDa. The first five residues of purified proTlAPA1 were V-P-A-P-S, as identified by N-terminal amino acid sequence analysis. These results indicated that TlAPA1 was produced in the form of a zymogen in P. pastoris GS115.

SDS-PAGE analysis of the recombinant proTlAPA1.Lane M, molecular mass standard; Lane 1, crude zymogen proTlAPA1; Lane 2, purified recombinant proTlAPA1; and Lane 3, deglycosylated zymogen proTlAPA1 treated with Endo H
Process of auto-activation
The inactive precursors of aspartic proteases were usually autocatalytically activated under acidic conditions. In this study, the processing of the zymogen conversion was determined by SDS-PAGE and N-terminal amino acid sequencing. As shown in Fig. 3, the activation process had been already initiated at 0 min of incubation at room temperature (about 25 °C), indicating the rapidity of the process, potentially due to disintegration of zymogens in the reaction whose pH had been adjusted to 3.5 before incubation. The appearance of two new bands was accompanied by the weakening of the zymogen band (50 kDa) before 30 min of incubation (Fig. 3). The molecular weights of the two new products were 45 kDa and 40 kDa, and their first 5 residues were identified as L-D-F-E-P and V-A-Q-P-A, respectively. After 90 min of incubation at room temperature at pH 3.5, the proTlAPA1 was completely converted to a 40-kDa band (Fig. 3). We suspected that the 45-kDa product was an intermediate in the conversion process of proTlAPA1 to mature TlAPA1, and the processing sites on this 45-kDa product were confirmed by N-terminal sequencing. The above results indicated that the auto-activation processing of proTlAPA1 proceeded in two stages, and two processing sites (L67-L68, D85-V86) of proTlAPA1 auto-activation were identified.

Analysis of proTlAPA1 auto-activation for 90 min. The time course of processing at room temperature (~ 20 °C) was analyzed using 12% SDS-PAGE. ProTlAPA1, recombinant precursor without the signal peptide; Int-TlAPA1, intermediate produced during auto-activation processing; Mat-TlAPA1, mature protein after auto-activation
Effects of proteolytic activity on auto-activation processing
As previous studies have illustrated, processing induced auto-activation was related to its own proteolytic activity. The relationship between auto-processing of proTlAPA1 and protein concentration was determined by SDS-PAGE after incubation at 37 °C for 5 and 10 min. Although the auto-activation processing of proTlAPA1 with two different concentrations both occurs after the incubation at pH 3.5 for 5 and 10 min, differences in activation rates were observed. Compared with the 0.1 mg/mL concentration group, protein samples with a concentration of 0.2 mg/mL have a faster activation rate (Fig. 4a). This result indicates that the auto-processing of proTlAPA1 is protein concentration-dependent, which means the activation of TlAPA1 involves an intermolecular event.

SDS-PAGE analysis of auto-activation processing of proTlAPA1 and its variant D103N. a Effect of the protein concentration on proTlAPA1 auto-processing. b Effect of the inhibitor pepstain A on proTlAPA1 auto-processing. CK, the auto-activation processing of recombinant zymogens without pepstain A; + pepstain A, the auto-activation processing of recombinant zymogens with pepstatin A. c Effect of active mature TlAPA1 on variant D103N zymogens processing. D103N, the auto-activation processing of variant D103N zymogens; D103N+TlAPA1, the auto-activation processing of variant D103N zymogens with 0.2 μg/ml active TlAPA1. M, molecular mass standard; Pro, purified zymogens before auto-activation; 5, 10, 15, 30, 45, and 60, the processing time of the auto-activation
Pepstatin A is a specific inhibitor of aspartic protease, which can effectively inhibit its protease activity. Hence, the effect of pepstatin A on the zymogen conversion of TlAPA1 was examined at a concentration of 5 μM of pepstatin A. Electrophoretic analyses revealed that upon pepstatin A treatment, the apparent molecular mass of the proTlAPA1 decreased from 50 to 45 kDa (Fig. 4b). The first 5 amino acid residues of processed proteins (45 kDa) were determined as L-D-F-E-P, which were identical to the cleavage intermediate of proTlAPA1. Although the proTlAPA1 can be auto-processed to intermediates (45-kDa products), the mature protein (40 kDa) was not obtained even upon prolonged the incubation time up to 3 h in the presence of pepstatin A (Fig. 4b). This demonstrated that the auto-processing from intermediates to mature proteins was inhibited by pepstatin A. Hence, we conclude that the activation process from zymogen proteins to 45-kDa intermediates could be completed when aspartic protease PsAPA was inhibited by pepstatin A, while the conversion of 45-kDa intermediates to mature proteases was blocked by pepstatin A.
To further study the effects of proteolytic activity on proTlAPA1 auto-activation processing, the catalytic Asp103 residue of TlAPA1 was replaced with Asn; and it was confirmed that the variant D103N lost its proteolytic activity completely (Fig. S2). The variant D103N did not prevent processing of the precursor into the 45 kD intermediates, while it terminated the auto-activation of proTlAPA1 in the intermediate stage (Fig. 4c). This showed that the auto-processing of proTlAPA1 into a 45-kDa intermediate still can be performed after the catalytic residue Asp103 was mutated, and the process that turned intermediates into mature form failed to proceed to the variant D103N. To further investigate whether the 45-kD intermediates could be processed into mature form in an intermolecular manner, the zymogen proteins of variant D103N were treated with 0.2 μg/ml mature TlAPA1. In the presence of active mature TlAPA1, the zymogen proteins of variant D103N were processed into the mature forms after incubation of 60 min (Fig. 4c). The above results show that the auto-processing of proTlAPA1 is susceptible to active mature TlAPA1 and this process occurs in intermolecular manners.
Effect of pH on auto-activation processing
The auto-activation processing of aspartic proteases is often triggered by low pH. To determine the optimum pH of zymogen conversion, this processing was executed in the range of pH 3.0–6.0, and the proteolytic activities of treated samples were detected using casein (1%, w/v) as a substrate, during the processing of the aspartic protease zymogen. During prolonged incubation at pH 3.0, the proteolytic activities were increased and the enzyme activity peaked after 60 min (Fig. 5a). We speculated that the precursor was almost completely processed after 60 min of incubation at pH 3.0, which was confirmed by SDS-PAGE (Fig. 5b, pH 3.0). The processing induced auto-activation at pH 4.0 and pH 3.0 were comparable, although complete activation at pH 4.0 required longer incubation time (about 90 min). As shown in Fig. 5a, the increase in proteolytic activities was absent at pH 6.0, and no differences in protein bands were observed (Fig. 5b, pH 6.0). This indicated that zymogen proTlAPA1 was not converted at pH 6.0. Interestingly, the precursor band was cleaved at pH 5.0 (Fig. 5b), although the final activity was dramatically lower than at pH 3.0 and pH 4.0. In order to understand the reason for the lowered activity at pH 5.0, the N-terminal amino acid sequences of mature proteins at pH 3.0 and pH 5.0 were determined and found to be V-A-Q-P-A and A-V-Q-G-G. This demonstrated that a degradation product M2-TlAPA1 was generated at pH 5.0 because its activity was not detected at any aforementioned temperature and pH conditions. In conclusion, precursors of aspartic proteases could be activated at the range of pH 3.0–4.0, while auto-activation occurred most efficiently at pH 3.0, which is closest to the optimum pH of mature aspartic protease.

Analysis of proTlAPA1 auto-activation at varying pH 3.0–6.0. a Proteolytic activities measured at different times with the range of pH 3.0–6.0 at 60 °C. b SDS-PAGE analysis of auto-activation processing at 37 °C with pH 3.0, 5.0, and 6.0
Biochemical characterization of TlAPA1
The enzymatic characteristics of mature aspartic protease TlAPA1 were assessed using casein (1%, w/v) as a substrate. TlAPA1 showed the highest activity at pH 3.5 (Fig. 6a), similar to that that seen in most fungal aspartic proteases. As shown in Fig. 6b, TlAPA1 had an optimal temperature of 60 °C, which was higher than aspartic proteases obtained from most other fungi. We further measured stabilities of aspartic protease TlAPA1 under different pH and temperature conditions. TlAPA1 retained greater than 80% of its initial activity after 60 min of incubation at 37 °C over a range of pH 2.0–6.0 (Fig. 6c). The stability at acidic pH makes TlAPA1 favorable for applications in food, beverages, and brewing industries. Figure 6d shows that TlAPA1 was extremely stable below 55 °C, retaining almost all of its initial activity after 1 h of incubation. At higher temperatures, half-life of TlAPA1 was 30 min at 60 °C and 5 min at 65 °C. The thermostability of TlAPA1 was higher than that of highly homologous aspartic proteases from other fungi. Purified recombinant TlAPA1 had a specific activity of 2187.4 ± 67.3 U·mg−1, while the Km, Vmax, kcat, and kcat/Km values were determined as 1.9 mg mL−1, 2321 μmoL min−1 mg−1, 1410 s−1, and 723.5 mL s−1 mg−1, respectively.

Characterization of purified mature enzymeTlAPA1. a pH-activity profile. b Temperature-activity profile. c pH stability profile after 1 h-incubation at 37 °C at different pH values. d Temperature-stability profile after incubation at pH 3.0 and different temperatures for various durations
Discussion
Talaromyces leycettanus JCM12802, a thermophilic fungus that has an optimal growth temperature of 42 °C, is the source of various thermostable hydrolases including β-mannanase (Wang et al. 2015), xylanase (Wang et al. 2017), β-glucanase (You et al. 2016), and α-Amylase (Zhang et al. 2017). In this study, a novel aspartic protease precursor comprising of 424 amino acid residues was identified from Talaromyces leycettanus JCM12802 and expressed in Pichia pastoris successfully. Another aspartic protease named TlAP has been detailedly characterized from the same strain in our previous study (Guo et al. 2019). By comparing the homology between TlAPA1 and TlAP, we observed that there was only 37.6% identity in terms of the amino acid sequence. The molecular mass of TlAPA1 without the signal peptide (mature protease) was predicted to be 42.5 kDa, which was larger than that of TlAP (39.2 kDa). The predicted pI values of the two putative proteins from Talaromyces leycettanus JCM12802 were 4.7 and 4.1, which were in line with what has been reported in the literature. In addition, the properties of aspartic protease TlAPA1 were analyzed in detail. Purified TlAPA1 showed a peak activity at pH 3.5, which was higher than that of TlAP (pH 3.0). Similar to the reported aspartic protease TlAP, TlAPA1 was stable over the pH range of 3.0 to 6.0. These characteristics were similar to the homologs from other fungi, including Monascus pilosus (Lakshman et al. 2010), Trichoderma asperellum (Yang et al. 2013), and A. foetidus (Souza et al. 2017). The acidic adaptation of TlAPA1 makes it a promising candidate in many industries including cheese manufacturing, juice clarification, and leather softening. TlAPA1 also hydrolyzed proteins at higher temperatures. Its optimal activity was recorded at 60 °C and greater than 80% of the maximal activity remained at 65 °C. The temperature optimum of TlAPA1 is higher than most reported homologs including the TlAP (Table 2). TlAPA1 retains greater than 80% of its original activity after incubation at 55 °C for 30 min, indicating superior thermostability of TlAPA1 compared with most other aspartic proteases that are commonly stable at 50 °C and below (Guo et al. 2019; Takenaka et al. 2017; Rao et al. 2011). The thermostability of TlAPA1 is a favorable characteristic for its potential application in many areas. For example, during proteolysis, most substrate proteins are resistant to proteases because of their structural stability at moderate temperatures. At higher temperatures, substrate proteins become unfold and their cleavage sites become exposed and accessible to the catalytic enzyme. This suggests a more efficient hydrolysis of TlAPA1 substrates at high temperatures, due to the thermostable nature of TlAPA1.
The properties of enzymes are ultimately determined by the sequence of amino acids, so we performed a phylogenetic analysis of aspartic protease TlAPA1. Pfam protein family prediction indicated TlAPA1 belongs to the A1 family of aspartic proteases, and these aspartic proteases have undergone large sequence diversification leading to their evolutionary complexity (Revuelta et al. 2014; Rawlings et al. 2018). When full sequences of the A1 family proteins were used for phylogenetic analysis, the phylogenetic tree indicated TlAPA1 as a sister to an uncharacterized aspartic protease Q4WZS3 from Aspergillus fumigates, whose clade has not yet been discovered. These results suggested that TlAPA1 was a potential evolutionary intermediate linking rhizopuspepsins and other subgroups. A1 family of aspartic proteases contains many biochemically-characterized enzymes including pepsin, chymosin, rennin, and cathepsin D (Revuelta et al. 2014; Rawlings and Barrett 2013). Phylogenetic analysis has contributed to an in-depth understanding of aspartic protease evolution. The phylogenetic tree showed that TlAPA1 largely differed with other homologs in terms of the amino acid sequence, indicating that TlAPA1 potentially had distinct enzyme characteristics and special applications like the mucorpepsin subgroup, a unique clade in the evolutionary process and an important class of proteases, widely used as milk-coagulating agents (Yegin and Dekker 2013). Our analysis suggested that phylogenetic analysis is able to provide deeper insights into the evolution of a new clade and it has contributed to an in-depth understanding of fungal aspartic protease evolution.
It is well known that all aspartic proteases are synthesized in forms of zymogens, and they are processed into the mature aspartic proteases with proteolytic activity. At present, there have been many researches about the activation processing of aspartic protease zymogens, and some mechanisms have been studied in depth (Richter et al. 1998; Dunn 2002). Interestingly, the auto-activation processing of proTlAPA1 proceeded in two stages: formation of intermediates and activation of the mature protein (TlAPA1). This varies from other aspartic proteases, such as the cathepsins D and E (Ostermann et al. 2004), HIV protease (Lee et al. 1998), yeast proteinase A (Dostal et al. 2005), aspergillopepsinogen I (Dunn 2002), and rhizopuspepsin (Chen et al. 1991).
The autocatalytic activation of zymogens is a complex process, involving a dramatic conformational rearrangement and the proteolytic cleavage of the prosegment (Richter et al. 1998). The auto-activation of most aspartic protease zymogens depended on the decrease of pH values (Khan et al. 1999). At low pH, the protonated acidic residues (the two catalytic aspartic residues) disrupt the electrostatic interactions between the prosegment and the mature enzyme, initiating the activation reaction through the conformational changes (Dunn, 2002). We further studied the conversion processing of proTlAPA1 at different pH conditions and determined that the optimum pH of conversion was similar to that of its peak activity, suggesting that the auto-activation process was related to aspartic protease activity. Interestingly, when we activated the zymogens at pH 5.0, a minor protein band was generated in SDS-PAGE. This auto-processing site (S98-A99) was also confirmed by N-terminal sequencing and was located at 13 residues downstream of the mature site. However, a degradation product was generated at pH 5.0 because of the activation with mistakes (Fig. 7). In the N-terminal propeptide, the segment around the processing site underwent inaccurate conformational changes at pH 5.0 and the S98-A99 site became sensitive to proteolysis. Similar results also have been reported for other zymogens such as phytepsin, cardosin, and rhizopuspepsin (Chen et al. 1991).

Schematic representation of the signal peptide, the propeptide region and the mature protein in TlAPA1. ProTlAPA1, the recombinant precursor without the signal peptide; Int-TlAPA1, the intermediate produced during auto-activation processing; TlAPA1, mature protein after auto-activation at pH 3.0; M2-TlAPA1, mature protein after auto-activation at pH 5.0
The prosegment cleavage often occurs by either an intramolecular or intermolecular reaction (Dunn 2002). Three models of activation were proposed: complete self-processing, self-processing assisted with protease, and fully assisted processing (Richter et al. 1998). In order to understand better the autocatalytic activation mechanism of proTlAPA1, a few experiments were carried out in vitro. Our result indicates that the auto-processing of proTlAPA1 is positively correlated with protein concentration, which is in agreement with gastric aspartic proteases (Richter et al. 1998), and this means that the prosegment can be auto-processed intermolecularly. Generally, the activation of aspartic protease zymogens was prevented by pepstatin A (Glathe et al. 1998; Domingos et al. 2000), while proTlAPA1 still can be auto-processed to a 45-kDa intermediate at the presence of 5 μM pepstatin A. We predicted that the formation of 45-kDa intermediates maybe occur independent of catalytic residues and this hypothesis has been confirmed by the activation process of mutant D103N. However, the conversion of 45-kDa intermediates to mature TlAPA1 was dependent on the catalytic residues of TlAPA1. As for the zymogen proteins of mutant D103N, the auto-activation processing terminated in the intermediate stage without activated TlAPA1. After the addition of the activated mature TlAPA1, zymogen proteins of mutant D103N can be processed into mature forms. These showed that the conversion of 45-kDa intermediates occurs in an intermolecular manner. The above results show that there are quite differences from other aspartic proteases for the auto-activation mechanism, but the activation mechanisms still remain to be proven in future work based on the tertiary structure.
This large difference in the processing mechanism of proTlAPA1 appears to be attributable to its primary structure of the prosegment. Compared the N-terminal propeptide of TlAPA1 precursor to other homologs, the N-terminal propeptide of TlAPA1 precursor showed low sequence identity (< 30%) to other known aspartic proteases (Fig. S1). In addition, the N-terminal propeptide of TlAPA1 precursor possessed an additional 61 residues with a greater abundance of arginine residues, compared with other homologs (Fig. S1). We thus speculated that the sequence peculiarity of N-terminal prosegment lead to the special auto-activation processing of the TlAPA1 precursor.
In summary, a novel aspartic protease, TlAPA1, was identified, that belonged to a new clade in the phylogenetic tree. The sequence analysis of the propeptide region showed that proTlAPA1 could have a unique mechanism of auto-activation processing. The results indicated that there are two steps in the processing of auto-activation, and a 45-kDa intermediate was confirmed. Moreover, the characteristics of mature protein TlAPA1 demonstrated that it is excellent in terms of specific activity and thermostability.
References
Chen Z, Koelsch G, Han HP, Wang XJ, Lin XL, Hartsuck JA, Tang J (1991) Recombinant rhizopuspepsinogen: expression, purification, and activation properties of recombinant rhizopuspepsinogens. J Biol Chem 266:11718–11725
Domingos A, Cardoso PC, Xue ZT, Clemente A, Brodelius PE, Pais MS (2000) Purification, cloning and autoproteolytic processing of an aspartic proteinase from Centaurea calcitrapa. Eur J Biochem 267:6824–6831
Dostal J, Dlouha H, Malon P, Pichova I, Hruskova-Heidingsfeldova O (2005) The precursor of secreted aspartic proteinase sapp1p from Candida parapsilosis can be activated both autocatalytically and by a membrane-bound processing proteinase. J Biol Chem 386:791–799
Dunn BM (2002) Structure and mechanism of the pepsin-like family of aspartic peptidases. Chem Rev 102:4431–4458
Fraser ME, Strynadka NC, Bartlett PA, Hanson JE, James MN (1992) Crystallographic analysis of transition-statemimics bound to penicillopepsin: phosphorus-containing peptide analogues. Biochemistry 31:5201–5214
Gama Salgado JA, Kangwa M, Fernandez-Lahore M (2013) Cloning and expression of an active aspartic proteinase from Mucor circinelloides in Pichia pastoris. BMC Microbiol 13:250–260
Glathe S, Kervinen J, Nimtz M, Li GH, Tobin GJ, Copeland TD, Ashford DA, Wlodawer A, Costa J (1998) Transport and activation of vacuolar aspartic proteinase phytepsin in barley (Hordeum vulgare L.). J Biol Chem 273:31230–31236
Gomi K, Arikawa K, Kamiya N, Kitamoto K, Kumagai C (1993) Cloning and nucleotide sequence of the acid protease-encoding gene (pepA) from Aspergillus oryzae. Biosci Biotechnol Biochem 57:1095–1100
Guo Y, Tu T, Yuan P, Wang Y, Ren Y, Yao B, Luo H (2019) High-level expression and characterization of a novel aspartic protease from Talaromyces leycettanus JCM12802 and its potential application in juice clarification. Food Chem 281:197–203
Hanova I, Brynda J, Houstecka R, Alam N, Sojka D, Kopacek P, Maresova L, Vondrasek J, Horn M, Schueler-Furman O, Mares M (2018) Novel structural mechanism of allosteric regulation of aspartic peptidases via an evolutionarily conserved exosite. Cell Chem Biol 25:318–329
Horimoto Y, Dee DR, Yada RY (2009) Multifunctional aspartic peptidase prosegments. New Biotechnol 25:318–324
Kamitori S, Ohtaki A, Ino H, Takeuchi M (2003) Crystal structures of Aspergillus oryzae aspartic proteinase and its complex with an inhibitor pepstatin at 1.9A resolution. J Mol Biol 326:1503–1511
Kangwa M, Salgado JAG, Fernandez-Lahore HM (2018) Identification and characterization of N-glycosylation site on a Mucor circinelloides aspartic protease expressed in Pichia pastoris: effect on secretion, activity and thermo-stability. AMB Express 8:157–170
Khan AR, Khazanovich-Bernstein N, Bergmann EM, James MNG (1999) Structural aspects of activation pathways of aspartic protease zymogens and viral 3C protease precursors. Proc Natl Acad Sci U S A 96:10968–10975
Koelsch G, Mareš M, Metcalf P, Fusek M (1994) Multiple functions of pro-parts of aspartic proteinase zymogens. FEBS Lett 343:6–10
Kumar S, Sharma NS, Saharan MR, Singh R (2005) Extracellular acid protease from Rhizopus oryzae: purification and characterization. Process Biochem 40:1701–1705
Kumar S, Stecher G, Tamura K (2016) Mega 7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol 33:1870–1874
Lakshman PLN, Toyokawa Y, Toyama H, Taira T, Yasuda M (2010) Purification and characterisation of two extracellular acid proteinases from Monascus pilosus. Food Chem 121:1216–1224
Lee AY, Gulnik SV, Erickson JW (1998) Conformational switching in an aspartic proteinase. Nat Struct Biol 5:866–871
Mandujano-González V, Villa-Tanaca L, Anducho-Reyes MA, Mercado-Flores Y (2016) Secreted fungal aspartic proteases: a review. Rev Iberoam Micol 33:76–82
Morales R, Watier Y, Bocskei Z (2012) Human prorenin structure sheds light on a novel mechanism of its autoinhibition and on its non-proteolytic activation by the (pro)renin receptor. J Mol Biol 421:100–111
Nascimento AS, Krauchenco S, Golubev AM, Gustchina A, Wlodawer A, Polikarpov I (2008) Statistical coupling analysis of aspartic proteinases based on crystal structures of the Trichoderma reesei enzyme and its complex with pepstatin a. J Mol Biol 382:763–778
Oka T, Ekino K, Fukud K, Nomura Y (2014) Draft genome sequence of the formaldehyde-resistant fungus Byssochlamys spectabilis No. 5. Genome Announc 2:e01162–e01113
Ostermann N, Gerhartz B, Worpenberg S, Trappe J, Eder J (2004) Crystal structure of an activation intermediate of cathepsin E. J Mol Biol 342:889–899
Rao MB, Tanksale AM, Ghatge MS, Deshpande VV (1998) Molecular and biotechnological aspects of microbial proteases. Microbiol Mol Biol Rev 62:597–635
Rao S, Mizutani O, Hirano T, Masaki K, Iefuji H (2011) Purification and characterization of a novel aspartic protease from basidiomycetous yeast Cryptococcus sp. S-2. J Biosci Bioeng 112:441–446
Raveendran S, Parameswaran B, Ummalyma SB, Abraham A, Mathew AK, MadhavanA RS, Pandey A (2018) Applications of microbial enzymes in food industry. Food Technol Biotechnol 56:16–30
Rawlings ND, Barrett AJ (2013) Introduction: aspartic and glutamic peptidases and their clans. In: Barrett AJ, Rawlings ND, Woessner JF (eds) Handbook of proteolytic enzymes, vol 2, 3rd edn. Elsevier, Amsterdam, pp 3–19
Rawlings ND, Barrett AJ, Thomas PD, Huang X, Bateman A, Finn RD (2018) The MEROPS database of proteolytic enzymes, their substrates and inhibitors in 2017 and a comparison with peptidases in the PANTHER database. Nucleic Acids Res 46:624–632
Revuelta MV, Kan JALV, Kay J, Have AT (2014) Extensive expansion of A1 family aspartic proteinases in fungi revealed by evolutionary analyses of 107 complete eukaryotic proteomes. Genome Biol Evol 6:1480–1494
Richter C, Tanaka T, Yada RY (1998) Mechanism of activation of the gastric aspartic proteinases: pepsinogen, progastricsin and prochymosin. Biochem J 335:481–490
Silva RR, Suoto TB, Oliveira TB, Oliveira LCG, Karcher D, Juliano MA, Juliano L, Oliveira AHC, Rodrigues A, Rosa JC (2016) Evaluation of the catalytic specificity, biochemical properties, and milk clotting abilities of an aspartic peptidase from Rhizomucor miehei. J Ind Microbiol Biotechnol 43:1059–1069
Souza PM, Bittencourt MLD, Caprara CC, Freitas M, Almeida RPC, Silveira D, Fonseca YM, Ferreira EX, Pessoa A, Magalhaes PO (2015) A biotechnology perspective of fungal proteases. Braz J Microbiol 46:337–346
Souza PMD, Werneck G, Aliakbarian B, Siqueira F, Ferreira FEX, Perego P, Converti A, Magalhaes PO, Junior AP (2017) Production, purification and characterization of an aspartic protease from Aspergillus foetidus. Food Chem Toxicol 109:1103–1110
Sun Q, Chen F, Geng F, Luo Y, Gong S, Jiang Z (2018) A novel aspartic protease from Rhizomucor miehei expressed in Pichia pastoris and its application on meat tenderization and preparation of turtle peptides. Food Chem 245:570–577
Takenaka S, Umeda M, Senba H, Koyama D, Tanaka K, Yoshida KI, Doi M (2017) Heterologous expression and characterisation of the Aspergillus aspartic protease involved in the hydrolysis and decolorisation of red-pigmented proteins. J Sci Food Agric 97:95–101
Vishwanatha KS, Rao AGA, Singh SA (2009) Characterisation of acid protease expressed from Aspergillus oryzae MTCC 5341. Food Chem 114:402–407
Wang C, Luo H, Niu C, Shi P, Huang H, Meng K, Bai Y, Wang K, Hua H, Yao B (2015) Biochemical characterization of a thermophilic β-mannanase from Talaromyces leycettanus JCM12802 with high specific activity. Appl Microbiol Biotechnol 99:1217–1228
Wang X, Ma R, Xie X, Liu W, Tu T, Zheng F, You S, Ge J, Xie H, Yao B, Luo H (2017) Thermostability improvement of a Talaromyces leycettanus xylanase by rational protein engineering. Sci Rep 7:152–187
Yang X, Cong H, Song J, Zhang J (2013) Heterologous expression of an aspartic protease gene from biocontrol fungus Trichoderma asperellum in Pichia pastoris. World J Microbiol Biotechnol 29:2087–2094
Yegin S, Dekker P (2013) Progress in the field of aspartic proteinases in cheese manufacturing: structures, functions, catalytic mechanism, inhibition, and engineering. Dairy Sci Technol 93:565–594
Yegin S, Fernandez-Lahore M (2013) A thermostable aspartic proteinase from Mucor mucedo DSM 809: gene identification, cloning, and functional expression in Pichia pastoris. Mol Biotechnol 54:661–672
Yegin S, Fernandez-Lahore M, Salgado AJG, Guvenc U, Goksungur Y, Tari C (2011) Aspartic proteinases from Mucor spp. in cheese manufacturing. Appl Microbiol Biotechnol 89:949–960
You S, Tu T, Zhang L, Wang Y, Huang H, Ma R, Shi PJ, Bai YG, Su XY, Lin ZM, Luo HY, Yao B (2016) Improvement of the thermostability and catalytic efficiency of a highly active β-glucanase from Talaromyces leycettanus JCM12802 by optimizing residual charge–charge interactions. Biotechnol Biofuels 9:124–135
Zhang D, Tu T, Wang Y, Li Y, Luo X, Zheng F, Wang X, Bai Y, Huang H, Su X, Yao B, Zhang T, Luo H (2017) Improvement of the catalytic performance of a Talaromyces leycettanusα-amylase by changing the linker length. J Agric Food Chem 65:5041–5048
Zhang Y, He S, Simpson BK (2018) Enzymes in food bioprocessing–novel food enzymes, applications, and related techniques. Curr Opin in Food Sci19:30–35
Funding
This work was financially supported by the National Key Research and Development Program of China (2016YFD0501409-02), the Fundamental Research Funds for Central Non-profit Scientific Institution (Y2017JC31), and the China Modern Agriculture Research System (CARS-41).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical statement
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(PDF 254 kb)
Rights and permissions
About this article

Cite this article
Guo, Y., Tu, T., Zheng, J. et al. A novel thermostable aspartic protease from Talaromyces leycettanus and its specific autocatalytic activation through an intermediate transition state. Appl Microbiol Biotechnol 104, 4915–4926 (2020). https://doi.org/10.1007/s00253-020-10569-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-020-10569-0