
Abstract
Corynebacterium glutamicum is a versatile workhorse for producing industrially important commodities. The design of an optimal strain often requires the manipulation of metabolic and regulatory genes to different levels, such as overexpression, downregulation, and deletion. Unfortunately, few tools to achieve multiple functions simultaneously have been reported. Here, a dual-functional clustered regularly interspaced short palindromic repeats (CRISPR) (RE-CRISPR) system that combined genome editing and transcriptional repression was designed using a catalytically active Cas12a (a.k.a. Cpf1) in C. glutamicum. Firstly, gene deletion was achieved using Cas12a under a constitutive promoter. Then, via engineering of the guide RNA sequences, transcriptional repression was successfully achieved using a catalytically active Cas12a with crRNAs containing 15 or 16 bp spacer sequences, whose gene repression efficiency was comparable to that of the canonical system (deactivated Cas12a with full-length crRNAs). Finally, RE-CRISPR was developed to achieve genome editing and transcriptional repression simultaneously by transforming a single crRNA plasmid and Cas12a plasmid. The application of RE-CRISPR was demonstrated to increase the production of cysteine and serine for ~ 3.7-fold and 2.5-fold, respectively, in a single step. This study expands the application of CRISPR/Cas12a–based genome engineering and provides a powerful synthetic biology tool for multiplex metabolic engineering of C. glutamicum.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Corynebacterium glutamicum is an important microorganism for industrial production of various amino acids, widely used in medicine, food, and feed industry (Becker et al. 2018; Hermann 2003; Zhao et al. 2018). As a cell factory, it possesses several advantages, such as rapid growth, good adaptability, and the generally regarded as safe status (Heider and Wendisch 2015; Ikeda and Nakagawa 2003). At the early stage, random mutagenesis with phenotypic resistance selection was most commonly used to improve the productivity of amino acids (Vertès et al. 2005). Later, genetic manipulation for strain improvement was based on allelic exchange method, which was achieved via two rounds of recombination events with low efficiency (Schäfer et al. 1994). Generally, to maximize the yield of the desired product, multiple metabolic engineering targets with different modes of regulation should be manipulated simultaneously (Lian et al. 2017; Nielsen and Keasling Jay 2016). Although short oligonucleotides mediated heterologous recombination system has been introduced, the application of MAGE (multiplex automated genome engineering) (Pu et al. 2016; Wang et al. 2009) targeting multiple loci in C. glutamicum was less successful due to low homologous recombination efficiency (Binder et al. 2013). Therefore, the development of a combinatorial metabolic engineering strategy to modify the genome of C. glutamicum in a high-throughput manner is highly demanded.
To tackle these problems, the recently developed CRISPR (clustered regularly interspaced short palindromic repeats) systems (CRISPR/Cas9 and CRISPR/Cas12a), widely used for genome editing in many species (Pickar-Oliver and Gersbach 2019), were employed. Cas9 from Streptococcus pyogenes (spCas9) was firstly evaluated in C. glutamicum and showed toxicity under constitutive expression (Cleto et al. 2016), while inducible or weak expression resulted in decent genome editing efficiency (Cho et al. 2017; Liu et al. 2017; Peng et al. 2017). Subsequently, Cas12a from Francisella novicida was found to be non-toxic and showed comparable genome editing efficiency as Cas9 (Jiang et al. 2017). However, due to the low transformation and recombination efficiency, simultaneous genome editing of more than two loci is still difficult to achieve (Liu et al. 2017). CRISPRi (CRISPR interference) was also developed for blocking gene transcription by using deactivated spCas9 (dCas9) in C. glutamicum (Cleto et al. 2016; Park et al. 2018). CRISPRi was widely harnessed not only to fine tune metabolic pathways (Zhang et al. 2016; Yoon and Woo 2018; Park et al. 2019) but also to identify unknown genes (Lee et al. 2018). Due to the cytotoxicity of dCas9 overexpression in C. glutamicum, all the aforementioned studies were performed using inducible expression systems. Nevertheless, constitutive expression of a non-toxic Cas protein, such as Cas12a, will be preferred for persistent transcription inhibition.
Notably, different modes of genomic alterations were required for cell factory development, while the currently available mono-functional CRISPR systems could not fully accommodate the metabolic engineering needs in C. glutamicum. Fortunately, there have been a few reports on the development of multi-functional CRISPR systems beyond C. glutamicum. Recently, Cas9 and Cas12a were found to lose the cleavage activity while maintain the binding capability when truncated gRNAs or crRNAs were used (Breinig et al. 2019; Dahlman et al. 2015; Kiani et al. 2015). Thus, the catalytically active Cas proteins were further used for simultaneous multiple genome manipulation in mammal cells, where truncated gRNAs or crRNAs were developed for transcriptional regulation and full-length gRNAs or crRNAs for genome editing (Breinig et al. 2019; Dahlman et al. 2015; Kiani et al. 2015). A tri-functional orthogonal CRISPR-AID system in yeast was developed by using three different Cas proteins, where the catalytically inactive Cas12a from Lachnospiraceae bacterium fused with an activation domain was used for CRISPRa (CRISPR activation), catalytically dead Cas9 from Streptococcus pyogenes for CRISPRi, and catalytic Cas9 from Staphylococcus aureus for CRISPRd (CRISPR deletion) (Lian et al. 2017).
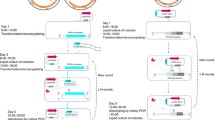
This study aimed to develop a multi-functional CRISPR system for metabolic engineering of C. glutamicum. First of all, CRISPR/Cas12a–based genome engineering system was developed. Then, deactivated Cas12a with full-length crRNA and active Cas12a with truncated crRNA was evaluated for transcriptional repression in C. glutamicum for the first time. Based on these results, a dual-functional CRISPR system (RE-CRISPR) using a single Cas12a protein was developed by using truncated crRNA for transcriptional repression and full-length crRNA for genome editing (Fig. 1). Finally, the metabolic engineering application of the RE-CRISPR system was demonstrated by increasing cysteine and serine production in a single step.

Schematic diagram of RE-CRISPR system for targeting gene A (transcriptional repression) and gene B (gene deletion). RNAP represented RNA polymerase
Materials and methods
Strains and growth conditions
All the strains used in this work are listed in Table 1. E. coli was grown in LB medium containing 10 g/L tryptone, 5 g/L yeast extract, and 10 g/L NaCl at 37 °C. Unless specifically mentioned, C. glutamicum was grown in LB medium supplemented with 5 g/L glucose at 30 °C. Modified Epo medium (LB medium supplemented with 30 g/L glycine and 0.1% Tween 80) was used for the preparation of C. glutamicum competent cells. Brain heart infusion-supplemented (BHIS) medium containing 37 g/L brain heart infusion powder and 91 g/L sorbitol was used for the transformation of C. glutamicum. CGXII medium (40 g/L glucose, 5 g/L urea, 20 g/L (NH4)2SO4, 1 g/L K2HPO4, 1 g/L KH2PO4, 0.25 g/L MgSO4·7H2O, 10 mg/L CaCl2, 10 mg/L FeSO4·7H2O, 10 mg/L MnSO4·H2O, 1 mg/L ZnSO4·7H2O, 0.2 mg/L CuSO4, 0.02 mg/L NiCl2, 0.2 mg/L biotin, 42 g/L 3-morpholinopropanesulfonic acid, and 30 mg/L protocatechuic acid) was used for fluorescence measurement of C. glutamicum. Seed medium for cysteine production contained 30 g/L glucose, 10 g/L corn steep liquor, 10 g/L (NH4)2SO4, 0.5 g/L K2HPO4, 0.5 g/L KH2PO4, 0.5 g/L MgSO4·7H2O, 0.2 mg/L biotin, and 0.2 mg/L thiamine. Cysteine fermentation medium (MC medium) contained 50 g/L glucose, 25 g/L corn steep liquor, 15 g/L (NH4)2SO4, 1 g/L K2HPO4, 1 g/L KH2PO4, 1 g/L MgSO4·7H2O, 5 g/L Na2S2O3, 10 mg/L FeSO4·7H2O, 10 mg/L MnSO4·H2O, 1 mg/L ZnSO4·7H2O, 0.2 mg/L CuSO4, 0.02 mg/L NiCl2, 0.2 mg/L biotin, 0.2 mg/L thiamine, and 20 g/L CaCO3. When necessary, 50 μg/mL kanamycin and 34 μg/mL chloramphenicol were supplemented.
Plasmids construction
All the plasmids and primers used in this study are listed in Tables S1 and S2. The Francisella novicida cas12a and Arabidopsis thaliana cysE (ATcysE) were codon optimized for expression in C. glutamicum (GenBank MN062361 and MN062362). The crRNA fragment without the spacer sequence (Fig. S1) was chemically synthesized by Generay Biotech (Shanghai, China). Plasmids used in this study were constructed via the restriction/ligation method or the Gibson Assembly method using a pEASY-Uni Seamless Cloning and Assembly Kit (TransGen Biotech, Beijing, China). The detailed procedures were described in Supplementary Materials.
Genome editing and transcriptional repression in C. glutamicum
The traditional pK19mobsacB–based gene deletion and insertion were performed as previously described (van der Rest et al. 1999). For CRISPR-based genome editing and transcriptional repression, pCas12atrc, pCas12atuf, or pdCas12atuf were firstly transformed to the wild-type strain, Cg-ΔaecD, Cg-ΔaecDΔsdaA, or the reporter strains Cg-GFP, Cg-RFP, Cg-LacZ, and Cg-RL. Then, the corresponding crRNA plasmids were further introduced, i.e., psg-ldh-HR for the deletion of ldhA and psg-aecD-HR for the deletion of aecD. The colonies were selected on the kanamycin and chloramphenicol containing BHIS plates and verified by diagnostic PCR, DNA sequencing, and fluorescence detection.
For simultaneous genome editing and transcriptional repression (RE-CRISPR), psg-gfp16-aecD20, psg-aecD20-mcbR16, psg-sdaA20-glyA15, and psg-sdaA20-glyA15-ATcysE were transformed to strain Cg-GFP/pCas12atuf, Cg/pCas12atuf, and Cg-ΔaecD/pCas12atuf, respectively. The colonies grown on the kanamycin and chloramphenicol containing plates were randomly picked and verified for aecD or sdaA deletion. To evaluate the repression of gfp expression, the aecD disrupted strains were cultured in CGXII medium to measure GFP fluorescence intensities. For the repression of mcbR and glyA, the aecD or sdaA disrupted strains were cultured in MC medium for quantifying the mRNA level as well as cysteine production.
Cysteine and serine production
A single colony of recombinant C. glutamicum strain was aerobically cultured at 30 °C in 30 mL BHIS medium containing 0.5% glucose for 18 h and inoculated at a ratio of 5% (v/v) into a 30-mL seed medium for 16 h. Then, seed culture was inoculated into 1 L shake flask containing 100 mL MC medium with the initial OD600 of ~ 1. The cultures were incubated at 30 °C and 120 rpm for cysteine and serine production.
Detection of GFP and RFP fluorescence and β-galactosidase activity
To measure the fluorescence of GFP or RFP, single colonies of recombinant C. glutamicum were cultured in 5-mL BHIS medium containing 0.5% glucose for 18 h and inoculated at a ratio of 5% into 30 mL CGXII medium. After incubation at 30 °C and 200 rpm for 24 h, OD600 and fluorescence intensity were measured by a Varioskan Flash Multimode Reader (Thermo Scientific, Waltham, USA). The excitation of GFP and RFP was set at 485 and 560 nm as well as emission at 510 and 607 nm, respectively. Fluorescence intensities normalized against culture OD600 were used to indicate the expression level of GFP and RFP. The β-galactosidase assay was carried out as previously described (Shang et al. 2018).
Analytical methods
Cell growth was monitored by measuring the optical density at 600 nm (OD600) with a spectrophotometer. Cysteine concentration in the supernatant of C. glutamicum culture was determined using the colorimetric method (Gaitonde 1967). Serine concentration was measured by high-performance liquid chromatography (HPLC) using phenylisothiocyanate as a precolumn derivatization agent (Zhang et al. 2014).
Results
Genome editing of C. glutamicum using CRISPR/Cas12a
In order to establish a system in C. glutamicum for simultaneous genome editing and transcriptional regulation using a single CRISPR protein, suitable Cas system in C. glutamicum should be chosen. As Cas9 showed toxicity in C. glutamicum (Fig. S2), which was consistent with previous reports (Cho et al. 2017; Cleto et al. 2016), Cas12a from Francisella novicida was employed. To test the genome editing efficiency of Cas12a, ldhA was chosen as the deletion target. The expression of crRNA was driven by the constitutive promoter Ptuf followed by insertion with two flanking regions as homologous recombination donor in the same plasmid to delete 614 bp of the coding sequence (Fig. 2a). As shown in Fig. 2, the editing efficiency using Cas12a under tuf promoter reached up to 58.6% (Fig. 2b, d; Fig. S3). On the contrary, only 25.8% correct colonies were obtained when the inducible promoter Ptrc was used (Fig. 2b, c; Fig. S3), although the same crRNA plasmid (psg-ldh-HR) was used. Thus, it was supposed that constitutive expression of the non-toxic Cas12a protein was beneficial for genome editing of C. glutamicum. In addition, the deletion of aecD was also carried out using constitutively expressed Cas12a, with a comparable efficiency (50%) achieved (Fig. 2b, e; Fig. S3).

CRISPR/Cas12a–based genome editing. a Schematic diagram of CRISPR/Cas12a–based ldhA gene deletion using two plasmid CRISPR/Cas12a system. b A summary of deletion efficiency targeting ldhA and aecD. c PCR verification of ldhA deletion using Cas12a under trc promoter in replicate 1. d PCR verification of ldhA deletion using Cas12a under tuf promoter in replicate 1. e PCR verification of aecD deletion using Cas12a under tuf promoter in replicate 1. M represented marker, C represented the wild-type control strain, and numbers represented the randomly picked colonies. c–e The result of PCR verification of replicate 1 in b, and the corresponding diagnostic PCR results for replicate 2 and replicate 3 were provided in Fig. S3
Transcriptional repression using deactivated Cas12a (dCas12a) and catalytically active Cas12a
To test whether truncated crRNA could still recruit Cas12a to the desired locus but without introducing a double strand break, a series of gfp targeting crRNA plasmids with different lengths of spacer sequences (13, 14, 15, 16, 18, and 20 nt) targeting g1 site (Fig. 3a) were constructed and introduced into the reporter strain Cg-GFP/pCas12atuf. As expected (Fig. 3b), no colonies appeared with 18- and 20-nt guides due to the introduction of a double strand break and the lack of a repair template. Fifteen- or 16-nt crRNAs decreased the expression of gfp for about 60%, while crRNAs with 13- or 14-nt spacers dramatically impaired repression efficiency (Fig. 3b). The effect of crRNA length on transcriptional repression efficiency was also evaluated using chromosomally integrated rfp and lacZ as reporter genes. As shown in Fig. 4a, crRNAs with 15 and 16 nt still showed much higher repression efficiency than the shorter ones. Therefore, catalytically active Cas12a with a 15-nt or16-nt crRNA was used for transcriptional repression in the following studies.

Transcriptional repression of gfp using dCas12a and Cas12a. a Schematic diagram of four different crRNAs targeting different locations of the template (g1, g3, and g4) and non-template (g2) strand of gfp gene integrated into the chromosome of C. glutamicum. b Comparison of gfp expression level repressed by Cas12a with crRNA of different length ranging from 13 to 20 bp. Asterisk represented that no colonies were obtained. c Comparison of gfp expression level repressed by dCas12a with 20-nt crRNA (full-length) and Cas12a with 16-nt (truncated) crRNA. d Comparison of gfp expression level repressed by Cas12a with 16-nt guide RNA targeting different locations of the template and non-template strand. g1, g2, g3, and g4 represented strain Cg-GFP containing plasmid pCas12atuf and different crRNAs with16-nt spacer (psg-gfp16, psg-gfp16-2, psg-gfp16-3, and psg-gfp16-4). Control represented strain Cg-GFP containing plasmid pCas12atuf and empty crRNA plasmid psg-E. For each experiment, three colonies were randomly picked and cultured with technical duplicates. Data represent the mean values of all the replicates and error bars represent the standard deviations

Transcriptional repression of rfp and lacZ using Cas12a. a Comparison of rfp and lacZ expression level repressed by Cas12a with crRNA of different lengths ranging from 13 to 20 bp. Asterisk represented that no colonies were obtained. C-rfp and C-lacZ represented strain Cg-RFP and Cg-LacZ containing plasmid pCas12atuf and the empty crRNA plasmid psg-E, respectively. b Transcriptional repression of rfp and lacZ simultaneously using pCas12atuf. Control represented strain Cg-RL containing pCas12atuf and psg-E-E2. For each experiment, three colonies were randomly picked and cultured with technical duplicates. Data represent the mean values of all the replicates and error bars represent the standard deviations
Next, more targeting sites were tested for transcriptional repression when 16-nt crRNAs were used (Fig. 3a, d). Plasmids with crRNAs targeting front, middle, and end part of the template strand (g1, g3, and g4) and targeting non-template strand (g2) of gfp coding sequence were constructed. As shown in Fig. 3d, all the template strand targeting crRNAs effectively repressed gfp expression, with the efficiency was slightly decreased with longer distance from the start codon. On the contrary, only 20% repression was achieved when targeting the front part of the non-template strand, which showed a different pattern with that of dCas9 (Cleto et al. 2016; Park et al. 2018; Zhang et al. 2016), and was consistent with EedCas12a (Kim et al. 2017). To confirm that gfp repression was caused by CRISPRi instead of gene mutation, the gfp expression cassette of the target strains was sequenced and found to be intact (Fig. S4).
As Cas12a could mediate gene repression with a truncated crRNA, its simultaneous regulation of multiple loci was further tested, with the simultaneous repression of rfp and lacZ chosen as a case study. After introducing pCas12atuf and psg-2rfp16-lacZ15 containing 16-nt rfp crRNA cassette and 15-nt lacZ crRNA cassette, rfp and lacZ expression levels were decreased by 51.2% and 54.7%, respectively (Fig. 4b). The multiplex CRISPRi efficiency was comparable with that using individual crRNA (Fig. 4; Fig. S5). Taken together, these results demonstrated that the design of a catalytically active Cas12a with a 15- or 16-nt crRNA could reliably mediate multiplex transcriptional repression in C. glutamicum.
Construction of the RE-CRISPR system using a catalytically active Cas12a protein
As Cas12a has been demonstrated to mediate genome editing and transcriptional repression, these two functions were combined by using Cas12a with two kinds of crRNAs (Fig. 1): a truncated crRNA for transcriptional repression and a full-length crRNA for genome editing. As a proof of concept, simultaneous repression of gfp and deletion of aceD gene was attempted first. The two crRNA expression cassettes (16-nt for gfp and 20-nt for aecD) together with the flanking regions of aecD were assembled to construct plasmid psg-gfp16-aecD20. After transformation into strain Cg-GFP/pCas12atuf with the dual-crRNA plasmid, aecD deletion efficiency and gfp repression efficiency were evaluated. As shown in Fig. 5a, 5 out of 10 randomly picked colonies were found to harbor the desired deletion, comparable to that with an individual aecD deletion crRNA (Fig. 2b). Then, gfp expression level of all the five correct colonies was measured and found to be around 44% of the control strain (Fig. 5b). In addition, the expression of gfp was decreased to a similar level in the other five un-edited colonies (Fig. 5b), while aecD showed no mutation and no obvious repression (Figs. S6 and S7), indicating that the CRISPRi crRNA and CRISPRd crRNA should function independently. Notably, the genome editing and transcriptional repression efficiency of the RE-CRISPR system were comparable to those when performed separately, indicating that multiplex and multi-functional CRISPR system could be developed with a single Cas12a protein by crRNA engineering.

RE-CRISPR system for simultaneously genome editing and transcriptional repression using active Cas12a. a PCR verification of aecD deletion. M represented marker, C represented the wild-type control strain and 1–10 represented the randomly picked colonies on BHIS plates containing kanamycin and chloramphenicol. b Expression level of gfp in strains targeting gene aecD and gfp double loci created by RE-CRISPR. The double loci targeted recombinants were first verified for aecD deletion, and then the correct strains were picked and cultured for fluorescence intensity assay. Control represented strain Cg-GFP containing plasmids pCas12atuf and psg-E. gfp-2, gfp-4, gfp-6, gfp-9, and gfp-10 were the five colonies which had been verified for aecD deletion. gfp-1, gfp-3, gfp-5, gfp-7, and gfp-8 were the five colonies in which aecD was not deleted. Data represent the mean values of biological triplicates and error bars represent the standard deviations
Application of RE-CRISPR for metabolic engineering of cysteine and serine production
Finally, the application of the RE-CRISPR system was demonstrated for metabolic engineering of C. glutamicum for cysteine and serine overproduction. As a proof-of-concept, aecD involved in the degradation of cysteine (Wada et al. 2002) and mcbR encoding a transcriptional repressor in cysteine biosynthetic pathways (Rey et al. 2003) were chosen as gene deletion and transcription repression targets, respectively, to improve cysteine production (Fig. 6). The aecD deletion crRNA (20-nt) together with the 1000 bp flanking regions and the mcbR repression crRNA (16-nt) were assembled to construct plasmid psg-aecD20-mcbR16. After transformation and selection, recombinant strain Cg-aecDd-mcbRi was obtained with an aecD deletion efficiency of 55% (Fig. S8A) and a mcbR repression efficiency of 51.4% (Fig. S8B). As for cysteine production (Fig. 7a), the deletion of aecD alone increased the production for about 2.5-fold (28.7 mg/L), while the combined aecD deletion and mcbR repression using RE-CRISPR system further increased the production to 3.7-fold (42.8 mg/L), as compared to the wild-type strain. In addition, RE-CRISPR-mediated metabolic engineering was compared with the traditional metabolic engineering strategy, where aecD-deficient strain Cg-ΔaecD was firstly constructed and followed by mcbR repression with the crRNA plasmid psg-mcbR16. The resultant strain Cg-ΔaecD-mcbRi showed similar performance with that of Cg-aecDd-mcbRi in cysteine production (Fig. 7a, b), suggesting the feasibility of targeting two genes simultaneously using the RE-CRISPR system.

Schematic diagram of the main metabolic pathways in C. glutamicum for cysteine and serine production. Genes in purple were the targets for transcriptional repression, in red for deletion, and in green for overexpression. aecD and mcbR were chosen as the targets for gene deletion and repression in strain B253. sdaA, glyA, and cysE were chosen as the targets for deletion, repression, and overexpression in strain Cg-ΔaecD, respectively

Application of RE-CRISPR for increasing cysteine production by targeting aecD and mcbR in strain B253. a Cysteine concentration of different recombinant strains. Strains were cultured in MC medium, and cysteine production was compared at 35 h. b Growth curves of different recombinant strains. WT represented the wild-type strain B253. Data represent the mean values of biological triplicates, and error bars represent the standard deviations
For practical metabolic engineering applications, the transcriptional repression of essential genes should be demonstrated for CRISPRi and RE-CRISPR system. Therefore, sdaA and glyA involved in serine biosynthesis were chosen as the targets for genome editing and transcriptional repression, respectively. Serine is an important precursor of cysteine, which could be degraded by serine dehydratases encoded by sdaA and cleaved into glycine and 5,10-methylene tetrahydrofolate by serine hydroxymethyltransferase encoded by glyA (Simic et al. 2002) (Fig. 6). As an important reaction involved in C1 metabolism (Fig. 6), glyA-deficient strain could not be obtained even on complex medium supplemented with glycine, confirming glyA as an essential gene (Peters-Wendisch et al. 2005; Schweitzer et al. 2009). To further improve cysteine production by enhancing the supply of precursor serine, Cg-ΔaecD was chosen as the parent strain. The sdaA deletion crRNA (20-nt) together with the 1000 bp flanking regions and the glyA repression crRNA (15-nt) were assembled to construct plasmid psg-sdaA20-glyA15. After transformation and selection, recombinant strain Cg-ΔaecD-sdaAd-glyAi was obtained with an sdaA deletion efficiency of ~ 40% (Fig. S8C) and a glyA repression efficiency of ~ 50% (Fig. S8E). As shown in Fig. 8a, the deletion of sdaA resulted in 1.3-fold increase in serine accumulation (175 mg/L), while simultaneous deletion of sdaA and repression of glyA (strain Cg-ΔaecD-sdaAd-glyAi) using RE-CRISPR system increased serine production by 2.5-fold (329 mg/L). Interestingly, cysteine production in strain Cg-ΔaecDΔsdaA and Cg-ΔaecD-sdaAd-glyAi was not significantly improved. It was supposed that the native l-serine acetyltransferase encoded by cysE was feedback inhibited by cysteine (Wei et al. 2019). To address this problem, a feedback inhibition insensitive cysE from Arabidopsis thaliana (ATcysE) was integrated into the plasmids of RE-CRISPR system targeting gene sdaA and glyA to enhance the cysteine formation from serine (Fig. 6; Fig. S8d, e). As expected, the resultant strain Cg-ΔaecD-sdaAd-glyAi-ATcysE accumulated 5.5-fold (158.3 mg/L) more cysteine than the parent strain Cg-ΔaecD (28.7 mg/L) and 1.34-fold higher than strain Cg-ΔaecD-ATcysE (118.5 mg/L). In addition, plasmid psg-ATcysE-glyA15–containing glyA repression crRNA cassette and ATcysE overexpression cassette were introduced to strain Cg-ΔaecDΔsdaA containing pCas12atuf. The resultant strain Cg-ΔaecDΔsdaA-ATcysE-glyAi showed similar performance with that of Cg-ΔaecD-sdaAd-glyAi-ATcysE in growth rate and cysteine production (Fig. 8a, b).

Application of RE-CRISPR for increasing cysteine and serine production by targeting sdaA and glyA in strain Cg-ΔaecD. a Cysteine and serine concentration of different recombinant strains. Strains were cultured in MC medium and cysteine and serine production were compared at 35 h. b Growth curves of different recombinant strains. Data represent the mean values of biological triplicates, and error bars represent the standard deviations
Discussion
To maximize the microbial product titers and yields, the metabolic and regulatory networks of the microorganism should be rewired. The development of a multiplex and multi-functional genome engineering tools in C. glutamicum is highly demanded for metabolic engineering applications. As most of the current studies focus on a mono-function CRISPR in C. glutamicum, the RE-CRISPR system realized the combination of two functions (genome editing and transcriptional repression) using a single catalytically active Cas12a protein. In addition, RE-CRISPR was successfully used for improving serine and cysteine production for 2.7-fold and 5.5-fold, respectively, by targeting two targets in a single step (Figs. 5, 7, and 8), which would be a powerful genome engineering and synthetic biology tool for multiplex genome engineering of C. glutamicum, particularly when essential genes should be manipulated. Compared with the reported inducible Cas9 system (Cho et al. 2017; Liu et al. 2017; Peng et al. 2017) and all-in-one plasmid based Cas12a system (Jiang et al. 2017), the RE-CRISPR system developed in this study showed comparable efficiencies (Fig. 2; Figs. S3 and S9). However, due to the relative low homologous recombination efficiency of C. glutamicum, gene deletion efficiency was still lower than eukaryotes, such as yeast (DiCarlo et al. 2013). As reported, Cas9 or Cas12a combined with heterologous recombinase could mediate genome editing with an efficiency approaching 100% (Cho et al. 2017; Liu et al. 2017; Jiang et al. 2017). Therefore, heterologous recombinase, such as RecT, should be introduced to further improve the genome editing efficiency of the RE-CRISPR system.
As for transcriptional repression, the deactivated and catalytically active Cas12a were explored for transcriptional repression using full-length and truncated crRNA in C. glutamicum for the first time, respectively. Decent efficiency was obtained for transcriptional repression of one or two genes using active Cas12a (Figs. 3 and 4). Notably, the repression efficiency was slightly lower than that of the canonical system (dCas12a with a 20-nt crRNA) (Fig. 3c). As reported (Singh et al. 2018), ultrastable binding of Cas12a to the target DNA required a minimum of 17-nt sequence with perfect match and the stability was decreased with shorter pairing bases. Thus, the slightly decreased repression efficiency observed in this study might be caused by the relatively lower stability between Cas12a and the truncated crRNA. Interestingly, it was found that dithiothreitol (DTT) enhanced the binding affinity of LbCas12a (Cas12a form Lachnospiraceae bacterium) and truncated crRNA (16-nt) for ~ 20-fold, which was comparable to that with a full-length crRNA (Singh et al. 2018). Therefore, other Cas12a orthologues (i.e., LbCas12a) should be evaluated for the RE-CRISPR system to further improve the transcriptional repression efficiency using a truncated crRNA.
For metabolic engineering applications, the original design of this study was to develop a tri-functional system using the catalytically active Cas12a fused with an activation domain (i.e., ω subunit of the RNA polymerase) (Table 1; Tables S1 and S2). In such a system, the full-length crRNA mediated genome editing, the truncated crRNA bound to the coding sequences blocked transcription elongation, and the truncated crRNA bound to the upstream sequences resulted in ω subunit-dependent transcriptional activation. The fusion of ω subunit to the C-terminus of Cas12a and dCas12a protein showed no negative effects on genome editing and transcriptional repression efficiencies (Figs. S10 and S11). Then, CRISPRa was investigated by designing crRNAs targeting the upstream of Psod promoter with gfp as the reporter using dCas12a-ω with a 20-nt length crRNA. As shown in Fig. S12, about 2- and 1.8-fold increase in gfp expression was observed when targeting 328 and 183 bp upstream of the transcriptional start site (TSS) of Psod-gfp expression cassette, respectively. Unfortunately, the activation fold is still limited and Cas12a-dependent CRISPRa was not further explored for metabolic engineering applications. CRISPRa has been well developed in E. coli and the fusion of dCas9 with various activators resulted in dramatically different gene activation efficiency (Dong et al. 2018). Therefore, the activation domain for RNA polymerase recruitment should be further engineered with optimal CRISPRa efficiency and then integrated into the RE-CRISPR system to develop a tri-functional CRISPR system in C. glutamicum.
In summary, a dual-functional RE-CRISPR system was developed by combining genome editing and transcriptional repression in a single system and its metabolic engineering application was demonstrated by improving cysteine and serine production by targeting two genes simultaneously. RE-CRISPR is an important addition to the genome engineering and synthetic biology toolbox for the construction of efficient C. glutamicum cell factories.
References
Becker J, Rohles CM, Wittmann C (2018) Metabolically engineered Corynebacterium glutamicum for bio-based production of chemicals, fuels, materials, and healthcare products. Metab Eng 50:122–141. https://doi.org/10.1016/j.ymben.2018.07.008
Binder S, Siedler S, Marienhagen J, Bott M, Eggeling L (2013) Recombineering in Corynebacterium glutamicum combined with optical nanosensors: a general strategy for fast producer strain generation. Nucleic Acids Res 41:6360–6369. https://doi.org/10.1093/nar/gkt312
Breinig M, Schweitzer AY, Herianto AM, Revia S, Schaefer L, Wendler L, Cobos Galvez A, Tschaharganeh DF (2019) Multiplexed orthogonal genome editing and transcriptional activation by Cas12a. Nat Methods 16:51–54. https://doi.org/10.1038/s41592-018-0262-1
Cho JS, Choi KR, Prabowo CPS, Shin JH, Yang D, Jang J, Lee SY (2017) CRISPR/Cas9-coupled recombineering for metabolic engineering of Corynebacterium glutamicum. Metab Eng 42:157–167. https://doi.org/10.1016/j.ymben.2017.06.010
Cleto S, Jensen JVK, Wendisch VF, Lu TK (2016) Corynebacterium glutamicum metabolic engineering with CRISPR interference (CRISPRi). ACS Synth Biol 5:375–385. https://doi.org/10.1021/acssynbio.5b00216
Dahlman JE, Abudayyeh OO, Joung J, Gootenberg JS, Zhang F, Konermann S (2015) Orthogonal gene knockout and activation with a catalytically active Cas9 nuclease. Nat Biotechnol 33:1159–1161. https://doi.org/10.1038/nbt.3390
DiCarlo JE, Norville JE, Mali P, Rios X, Aach J, Church GM (2013) Genome engineering in Saccharomyces cerevisiae using CRISPR-Cas systems. Nucleic Acids Res 41:4336–4343. https://doi.org/10.1093/nar/gkt135
Dong C, Fontana J, Patel A, Carothers JM, Zalatan JG (2018) Synthetic CRISPR-Cas gene activators for transcriptional reprogramming in bacteria. Nat Commun 9:2489. https://doi.org/10.1038/s41467-018-04901-6
Gaitonde MK (1967) A spectrophotometric method for the direct determination of cysteine in the presence of other naturally occurring amino acids. Biochem J 104:627–633. https://doi.org/10.1042/bj1040627
Grant SG, Jessee J, Bloom FR, Hanahan D (1990) Differential plasmid rescue from transgenic mouse DNAs into Escherichia coli methylation-restriction mutants. Proc Natl Acad Sci U S A 87:4645–4649. https://doi.org/10.1073/pnas.87.12.4645
Heider SAE, Wendisch VF (2015) Engineering microbial cell factories: metabolic engineering of Corynebacterium glutamicum with a focus on non-natural products. Biotechnol J 10:1170–1184. https://doi.org/10.1002/biot.201400590
Hermann T (2003) Industrial production of amino acids by coryneform bacteria. J Biotechnol 104:155–172. https://doi.org/10.1016/S0168-1656(03)00149-4
Ikeda M, Nakagawa S (2003) The Corynebacterium glutamicum genome: features and impacts on biotechnological processes. Appl Microbiol Biotechnol 62:99–109. https://doi.org/10.1007/s00253-003-1328-1
Jiang Y, Qian F, Yang J, Liu Y, Dong F, Xu C, Sun B, Chen B, Xu X, Li Y, Wang R, Yang S (2017) CRISPR-Cpf1 assisted genome editing of Corynebacterium glutamicum. Nat Commun 8:15179. https://doi.org/10.1038/ncomms15179
Kiani S, Chavez A, Tuttle M, Hall RN, Chari R, Ter-Ovanesyan D, Qian J, Pruitt BW, Beal J, Vora S, Buchthal J, Kowal EJK, Ebrahimkhani MR, Collins JJ, Weiss R, Church G (2015) Cas9 gRNA engineering for genome editing, activation and repression. Nat Methods 12:1051–1054. https://doi.org/10.1038/nmeth.3580
Kim SK, Kim H, Ahn W-C, Park K-H, Woo E-J, Lee D-H, Lee S-G (2017) Efficient transcriptional gene repression by type V-A CRISPR-Cpf1 from Eubacterium eligens. ACS Synth Biol 6:1273–1282. https://doi.org/10.1021/acssynbio.6b00368
Lee SS, Shin H, Jo S, Lee S-M, Um Y, Woo HM (2018) Rapid identification of unknown carboxyl esterase activity in Corynebacterium glutamicum using RNA-guided CRISPR interference. Enzym Microb Technol 114:63–68. https://doi.org/10.1016/j.enzmictec.2018.04.004
Lian J, HamediRad M, Hu S, Zhao H (2017) Combinatorial metabolic engineering using an orthogonal tri-functional CRISPR system. Nat Commun 8:1688. https://doi.org/10.1038/s41467-017-01695-x
Liu J, Wang Y, Lu Y, Zheng P, Sun J, Ma YJMCF (2017) Development of a CRISPR/Cas9 genome editing toolbox for Corynebacterium glutamicum. Microb Cell Factories 16:205. https://doi.org/10.1186/s12934-017-0815-5
Nielsen J, Keasling Jay D (2016) Engineering cellular metabolism. Cell 164:1185–1197. https://doi.org/10.1016/j.cell.2016.02.004
Park J, Shin H, Lee S-M, Um Y, Woo HM (2018) RNA-guided single/double gene repressions in Corynebacterium glutamicum using an efficient CRISPR interference and its application to industrial strain. Microb Cell Factories 17:4. https://doi.org/10.1186/s12934-017-0843-1
Park J, Yu BJ, Choi J-i, Woo HM (2019) Heterologous production of squalene from glucose in engineered Corynebacterium glutamicum using multiplex CRISPR interference and high-throughput fermentation. J Agric Food Chem 67:308–319. https://doi.org/10.1021/acs.jafc.8b05818
Peng F, Wang X, Sun Y, Dong G, Yang Y, Liu X, Bai Z (2017) Efficient gene editing in Corynebacterium glutamicum using the CRISPR/Cas9 system. Microb Cell Factories 16:201. https://doi.org/10.1186/s12934-017-0814-6
Peters-Wendisch P, Stolz M, Etterich H, Kennerknecht N, Sahm H, Eggeling L (2005) Metabolic engineering of Corynebacterium glutamicum for L-serine production. Appl Environ Microbiol 71:7139–7144. https://doi.org/10.1128/aem.71.11.7139-7144.2005
Pickar-Oliver A, Gersbach CA (2019) The next generation of CRISPR–Cas technologies and applications. Nat Rev Mol Cell Biol 20:490–507. https://doi.org/10.1038/s41580-019-0131-5
Pu Y, Dong C, Hang B, Huang L, Cai J, Xu Z (2016) Novel approach for the evolution of pyrroloquinoline quinone glucose dehydrogenase by multiplex-site in situ engineering. Process Biochem 51:2011–2016. https://doi.org/10.1016/j.procbio.2016.08.003
Rey DA, Pühler A, Kalinowski J (2003) The putative transcriptional repressor McbR, member of the TetR-family, is involved in the regulation of the metabolic network directing the synthesis of sulfur containing amino acids in Corynebacterium glutamicum. J Biotechnol 103:51–65. https://doi.org/10.1016/S0168-1656(03)00073-7
Schäfer A, Tauch A, Jäger W, Kalinowski J, Thierbach G, Pühler A (1994) Small mobilizable multi-purpose cloning vectors derived from the Escherichia coli plasmids pK18 and pK19: selection of defined deletions in the chromosome of Corynebacterium glutamicum. Gene 145:69–73. https://doi.org/10.1016/0378-1119(94)90324-7
Schweitzer J-E, Stolz M, Diesveld R, Etterich H, Eggeling L (2009) The serine hydroxymethyltransferase gene glyA in Corynebacterium glutamicum is controlled by GlyR. J Biotechnol 139:214–221. https://doi.org/10.1016/j.jbiotec.2008.12.008
Shang X, Chai X, Lu X, Li Y, Zhang Y, Wang G, Zhang C, Liu S, Zhang Y, Ma J, Wen T (2018) Native promoters of Corynebacterium glutamicum and its application in L-lysine production. Biotechnol Lett 40:383–391. https://doi.org/10.1007/s10529-017-2479-y
Simic P, Willuhn J, Sahm H, Eggeling L (2002) Identification of glyA (encoding serine hydroxymethyltransferase) and its use together with the exporter ThrE to increase L-threonine accumulation by Corynebacterium glutamicum. Appl Environ Microbiol 68:3321–3327. https://doi.org/10.1128/aem.68.7.3321-3327.2002
Singh D, Mallon J, Poddar A, Wang Y, Tippana R, Yang O, Bailey S, Ha T (2018) Real-time observation of DNA target interrogation and product release by the RNA-guided endonuclease CRISPR Cpf1 (Cas12a). Proc Natl Acad Sci 115:5444–5449. https://doi.org/10.1073/pnas.1718686115
van der Rest ME, Lange C, Molenaar D (1999) A heat shock following electroporation induces highly efficient transformation of Corynebacterium glutamicum with xenogeneic plasmid DNA. Appl Microbiol Biotechnol 52:541–545. https://doi.org/10.1007/s002530051557
Vertès AA, Inui M, Yukawa H (2005) Manipulating corynebacteria, from individual genes to chromosomes. Appl Environ Microbiol 71(12):7633–7642. https://doi.org/10.1128/AEM.71.12.7633-7642.2005
Wada M, Awano N, Haisa K, Takagi H, Nakamori S (2002) Purification, characterization and identification of cysteine desulfhydrase of Corynebacterium glutamicum, and its relationship to cysteine production. FEMS Microbiol Lett 217:103–107. https://doi.org/10.1016/s0378-1097(02)01042-x
Wang HH, Isaacs FJ, Carr PA, Sun ZZ, Xu G, Forest CR, Church GM (2009) Programming cells by multiplex genome engineering and accelerated evolution. Nature 460:894–898. https://doi.org/10.1038/nature08187
Wei L, Wang H, Xu N, Zhou W, Ju J, Liu J, Ma Y (2019) Metabolic engineering of Corynebacterium glutamicum for L-cysteine production. Appl Microbiol Biotechnol 103:1325–1338. https://doi.org/10.1007/s00253-018-9547-7
Yoon J, Woo HM (2018) CRISPR interference–mediated metabolic engineering of Corynebacterium glutamicum for homo-butyrate production. Biotechnol Bioeng 115:2067–2074. https://doi.org/10.1002/bit.26720
Zhang B, Liu Z-Q, Liu C, Zheng Y-G (2016) Application of CRISPRi in Corynebacterium glutamicum for shikimic acid production. Biotechnol Lett 38:2153–2161. https://doi.org/10.1007/s10529-016-2207-z
Zhang X, Xu G, Li H, Dou W, Xu Z (2014) Effect of cofactor folate on the growth of Corynebacterium glutamicum SYPS-062 and L-serine accumulation. Appl Biochem Biotechnol 173:1607–1617. https://doi.org/10.1007/s12010-014-0945-8
Zhao N, Qian L, Luo G, Zheng S (2018) Synthetic biology approaches to access renewable carbon source utilization in Corynebacterium glutamicum. Appl Microbiol Biotechnol 102:9517–9529. https://doi.org/10.1007/s00253-018-9358-x
Acknowledgments
This work was financially supported by National Natural Science Foundation of China (Nos. 21576232, 21606205, and 21808199), National Key Technology Research and Development Program of the Ministry of Science and Technology of China (2018YFC1603900 and 2018YFA0901800), the Natural Science Foundation of Zhejiang Province (No. LY18B060002), the Fundamental Research Funds for the Central Universities (2018QNA4039), and the Fundamental Research Funds for the Zhejiang Provincial Universities (2019XZZX003-12).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Human and animal rights
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(PDF 1870 kb)
Rights and permissions
About this article

Cite this article
Liu, W., Tang, D., Wang, H. et al. Combined genome editing and transcriptional repression for metabolic pathway engineering in Corynebacterium glutamicum using a catalytically active Cas12a. Appl Microbiol Biotechnol 103, 8911–8922 (2019). https://doi.org/10.1007/s00253-019-10118-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-019-10118-4