
Abstract
The environmental accumulation of polycyclic aromatic hydrocarbons (PAHs) is of great concern due to potential carcinogenic and mutagenic risks, as well as their resistance to remediation. While many fungi have been reported to break down PAHs in environments, the details of gene-based metabolic pathways are not yet comprehensively understood. Specifically, the genome-scale transcriptional responses of fungal PAH degradation have rarely been reported. In this study, we report the genomic and transcriptomic basis of PAH bioremediation by a potent fungal degrader, Dentipellis sp. KUC8613. The genome size of this fungus was 36.71 Mbp long encoding 14,320 putative protein-coding genes. The strain efficiently removed more than 90% of 100 mg/l concentration of PAHs within 10 days. The genomic and transcriptomic analysis of this white rot fungus highlights that the strain primarily utilized non-ligninolytic enzymes to remove various PAHs, rather than typical ligninolytic enzymes known for playing important roles in PAH degradation. PAH removal by non-ligninolytic enzymes was initiated by both different PAH-specific and common upregulation of P450s, followed by downstream PAH-transforming enzymes such as epoxide hydrolases, dehydrogenases, FAD-dependent monooxygenases, dioxygenases, and glycosyl- or glutathione transferases. Among the various PAHs, phenanthrene induced a more dynamic transcriptomic response possibly due to its greater cytotoxicity, leading to highly upregulated genes involved in the translocation of PAHs, a defense system against reactive oxygen species, and ATP synthesis. Our genomic and transcriptomic data provide a foundation of understanding regarding the mycoremediation of PAHs and the application of this strain for polluted environments.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Fungi play a major role as the decomposers of recalcitrant organic matters in nature (de Boer et al. 2005). The potent degrading abilities of fungi can be attributed to low specificity of catabolic enzymes and the formation of mycelial networks that can enhance chemical bioavailability (Harms et al. 2011). Mycoremediation, which is based on the use of fungi and mushrooms for the bioremediation of polluted areas, is a promising method to remove many hazardous chemicals of environmental and public health concern (Deshmukh et al. 2016).
Polycyclic aromatic hydrocarbons (PAHs) are a group of chemicals with two or more fused aromatic rings of carbon and hydrogen atoms (Kadri et al. 2017). Among the various toxic materials, PAHs have been one of the major targets for mycoremediation due to their possible carcinogenic and mutagenic risks (Mastrangelo et al. 1996; Schützendübel et al. 1999). The high hydrophobicity and chemical stability make them persistent in environments and cause bioaccumulation. While PAHs can be naturally formed by forest fires or volcanic eruptions, the incomplete combustion of organic materials during industrial and other human activities account for the majority of PAH formation (Johansson and van Bavel 2003). Several physical and chemical treatment methods including incineration, UV oxidation, fixation, and solvent extraction have been developed to remove PAHs, but they have many drawbacks since they are not cost-effective and environment friendly (Gan et al. 2009). The biodegradation of PAHs using fungi and other microorganisms has been regarded as an alternative method to remove PAHs without causing significant ecological damages (Abe et al. 1995; Gran-Scheuch et al. 2017).
It has been reported that many wood-degrading fungi can efficiently degrade a wide variety of PAHs (Field et al. 1992). Both ligninolytic and non-ligninolytic fungi can degrade PAHs with initial oxidation of substrates, but the metabolic pathways they use may differ (Marco-Urrea et al. 2015; Pozdnyakova 2012). Ligninolytic fungi can produce extracellular ligninolytic enzymes including manganese peroxidase (MnP), lignin peroxidase (LiP), and laccase to produce quinone intermediates. Non-ligninolytic degradation of PAHs may use intracellular cytochrome P450 monooxygenases (P450s) to produce unstable arene oxides which can be subsequently converted into phenols or trans-dihydrodiols. It was previously reported that even some ligninolytic fungi, including Phanerochaete and Pleurotus, can use P450s instead of ligninolytic enzymes for PAH degradation (Ghosal et al. 2016).
Although great efforts have been made to demonstrate PAH degradation in wood-degrading fungi through proteomic and metabolic assays, whole genome and transcriptome studies have rarely been conducted. A genome-scale survey of potential PAH-responsive genes and their regulation at transcriptional level can contribute to the comprehensive understanding of complex genetic networks in a fungal PAH degradation system and identification of novel PAH-responsive genes encoded in the genome.
Dentipellis sp. KUC8613, one of white rot fungi in the order Russulales, has previously been screened by high tolerance to various PAHs and regarded as a potential host for mycoremediation (Lee et al. 2014). In this study, we report the efficient PAH removal by the KUC8613 strain over various PAHs. In order to expand our knowledge of the genetic basis for the PAH-removal capability of this fungus, we sequenced and analyzed the genome of Dentipellis sp. KUC8613 as part of the 1000 Fungal Genomes Project (1k FGP) (Grigoriev et al. 2014) at the US Department of Energy Joint Genome Institute (JGI) (http://jgi.doe.gov/fungi). From the genome, we investigated genetic repertoires potentially involved in the metabolism of PAHs. We further performed transcriptomic analysis to observe differential expression of these genes by four different PAHs and to identify novel PAH-responsive genes. Our genomic and transcriptomic analyses to elucidate PAH removal by KUC8613 will give us a more comprehensive understanding of PAH degradation by wood-degrading fungi.
Materials and methods
Fungal strains, chemicals, and media
Dentipellis sp. KUC8613 was originally isolated from South Korea and identified by the Korea University Culture collection (KUC). Anthracene (ANT), fluoranthene (FLU), phenanthrene (PHE), pyrene (PYR), and all solvents including ethyl acetate, chloroform, and acetonitrile were purchased from Sigma-Aldrich (St. Louis, MO, USA). Malt extract (ME) medium contained 20 g malt extract for 1 l distilled water. Fifteen grams of agar was additionally added for malt extract agar (MEA) medium.
Determination of tolerance to PAHs
Four different PAHs (ANT, FLU, PHE, and PYR) were used to determine the tolerance of the fungus to PAHs. Fungal mycelia were cultured on MEA containing 100 mg/l of individual PAH. PAHs were first dissolved in acetone prior to being added into the culture medium. Acetone was evaporated from the medium by letting it stand for a few days. An actively growing fungal disk was inoculated at the center of a culture plate and incubated at 25 °C. Each sample was prepared in triplicate, and the growth rate of fungal mycelia was determined by measuring the radius of the mycelia at between 0 and 10 days.
Liquid culture condition and determination of mycelial growth
Fungal mycelium was grown on MEA at 25 °C and maintained for less than a week at 5 °C before use. Liquid ME containing 100 mg/l of individual PAH was prepared. PAHs were first dissolved in acetone and added into the ME medium. Acetone was evaporated from the medium by letting it stand for a few days before fungal inoculation. An actively growing mycelium disc (4 cm diameter) was homogenized for 10 s in a sterile blender cup, containing 50 ml of ME medium. Two milliliters of the homogenate was used to inoculate 50 ml ME medium in a 250-ml Erlenmeyer flask. Mycelium was grown at 25 °C in a rotary shaker with shaking at 180 rpm. Heat-killed mycelia (400 mg in dry weight) and cultures without fungal inoculation were also used for the determination of PAH adsorption to mycelia and natural degradation. Cultures were incubated for between 0 and 10 days.
For the determination of mycelial growth during liquid culture, mycelium was harvested after incubation by centrifugation and washed twice with 50 ml distilled water to a final volume of 10 ml. The sample was then filtered through pre-weighted dried filter paper in a vacuum filtration apparatus. The mycelium and filter paper were oven-dried at 65 °C for one day, and dry weight was measured.
HPLC analysis of PAH removal
After liquid culture, both medium and mycelium were subjected to ultrasonic disruption. The lysate was then extracted with 2 volumes of ethyl acetate. The extraction was carried out on a rotary shaker at 180 rpm for 1 h. The aqueous phase was acidified with concentrated HCl to pH 2 and extracted again in the same manner. The extracts from different pHs were combined together, dried over anhydrous sodium sulfate to 5 ml, and further dried in a rotary vacuum evaporator. The final extract was stored at − 20 °C. The dried extracts were dissolved in 1 ml chloroform. For the quantitative analysis of residual PAHs, a Waters high-pressure liquid chromatography (HPLC) system (Milford, MA, USA) equipped with a Waters 2487 UV detector was used. Data acquisition was carried out using the Waters Empower 2 software. Ten microliters of each extract was injected onto the Waters C18 column with a flow rate of 1 ml/min. The mobile phase was acetonitrile:water (80:20) and the PAHs were detected at 254 nm.
Genomic DNA extraction and sequencing
The fungus was cultured on solid MEA media at 25 °C in the dark. Genomic DNA was extracted from mycelium using a cetyl trimethyl ammonium bromide (CTAB)-based fungal DNA isolation protocol (Fulton et al. 1995). The concentration of prepared DNA was determined using a Qubit fluorometer (Invitrogen, Carlsbad, CA). For the whole genome sequencing of Dentipellis sp. KUC8613, two Illumina libraries with insert sizes 5.5 kb and 370 bp, were prepared and sequenced using 2 × 150 bp reads from HiSeq-1TB. The sequence data was filtered so as to remove low-quality reads and subsequently assembled with AllPathsLG release version R44008 (Gnerre et al. 2011). The genome was annotated using the JGI Annotation Pipeline (Grigoriev et al. 2014) and FunGAP (Min et al. 2017). From the JGI-MycoCosm repository (https://genome.jgi.doe.gov/Densp1/Densp1.home.html), we could retrieve the genome-wide annotation data such as InterPro, CAZy (Carbohydrate-Active enZYmes database), KOG (euKaryotic Orthologous Groups), and KEGG (Kyoto Encyclopedia of Genes and Genomes) for functional analysis.
RNA extraction and mRNA sequencing
For transcriptomic analysis, RNA samples from 5 days of culture were used. The grown mycelia prepared in triplicate were harvested from the culture media by centrifugation and washed twice with 50 ml distilled water. The mycelium pellet was immediately frozen under liquid nitrogen and homogenized using a mortar and a pestle. Total RNA was isolated using an RNeasy plus mini kit (Qiagen, Hilden, Germany) according to the manufacturer’s protocol. RNA concentration was determined using a Qubit fluorometer (Invitrogen, Carlsbad, CA). An RNA library was constructed using a TruSeq RNA sample preparation kit (Illumina, San Diego, CA, USA). The sequencing of the RNA library was carried out in MiSeq (2 × 200 cycles). Low-quality reads were filtered using the Trim Galore program. The filtered RNA-seq data was mapped against the assembled genome using CLC Genomics Workbench software v.12.0 (Qiagen, the Netherlands). Read alignment was performed with the following parameters: minimum length fraction and minimum similarity fraction = 0.8, strand specific = both, maximum number of hits for a read = 3. Reads per kilobase of transcript per million mapped reads (RPKM) value was generated using default settings of CLC genomics and further used for the analysis of differential expression of genes. To check if the RNA-seq data from different conditions correlate each other, a principal component analysis (PCA) was performed by an internal routine of the CLC Genomics Workbench.
Gene ontology enrichment analysis of upregulated genes
Upregulated genes by PAHs were identified according to the following criteria: fold change > 2 and p value < 0.01. To further reveal the enriched functions among upregulated genes, the gene ontology (GO) enrichment analyses of upregulated genes were performed using the R package clusterProfiler (version 3.2.1) (Yu et al. 2012). The threshold for the enrichment analysis was p value < 0.01.
Accession numbers, availability of the genome and transcriptome sequences, and the strain
This whole genome sequence of Dentipellis sp. KUC8613 has been deposited at DDBJ/EMBL/GenBank under the accession number NSJX00000000. The raw RNA-sequencing data have been deposited in NCBI Sequence Read Archive (SRA) under the BioProject ID PRJNA555151. The datasets are available under the following IDs: SRX6459337 for control samples, SRX6459338 for ANT samples, SRX6459335 for FLU samples, SRX6459336 for PHE samples, and SRX6459339 for PYR samples. The genome assembly and annotation are available at DOE JGI Genome Portal MycoCosm (Grigoriev et al. 2014) (http://genome.jgi.doe.gov/Densp1). The strain is available from Korean Collection for Type Cultures (KCTC) with the accession number KCTC46678 (http://kctc.kribb.re.kr/).
Results
PAH tolerance and removal capability by Dentipellis sp. KUC8613
We measured the PAH tolerance of the fungus according to the radial growth of mycelia in solid media (Fig. 1). ANT, FLU, PHE, and PYR were used as the representative PAHs having two to four fused aromatic rings. The average growth rate of the fungus at 25 °C was 4.7 mm/day without PAH. When four different PAHs were individually added in the growth media, KUC8613 showed high tolerance to all four PAHs with different degrees. In more detail, no detectable growth inhibition by ANT, FLU, and PYR was observed at any time point within 10 days. A moderate decrease (14%) in growth was observed only in PHE-added media. No significant change in mycelial morphology or pigmentation by any of the PAHs was recognized (data not shown).

Radial growth of the fungal mycelia in the presence of different PAHs
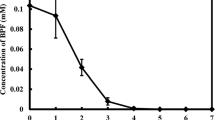
In order to verify PAH removal by this fungus, the removal of PAHs in liquid culture was examined (Fig. 2). HPLC-mediated quantification showed that 44% (ANT), 49% (FLU), 33% (PHE), and 46% (PYR) of PAHs were removed after 5 days. The removal of 90.1% (ANT), 99% (FLU), 94.3% (PHE), and 94.4% (PYR) of PAHs was observed after 10 days, while 17.4% (ANT), 25% (FLU), 24.4% (PHE), and 27.7% (PYR) were removed by autonomous chemical decomposition. We also observed that less than 10% of PAHs could be removed by adsorption to heat-killed mycelia. Fungal biomass (dry weight) in liquid culture was also measured (Supplementary Fig. S1). Only PHE induced a moderate growth retardation, which was congruent with our observation in PAH tolerance test at solid media.

PAH-removal efficiency by Dentipellis sp. KUC8613. The residual amount of PAH during the fungal culture was measured by HPLC. ANT, anthracene; FLU, fluoranthene; PHE, phenanthrene; PYR, pyrene; Control 1, 10 days of incubation without fungal inoculation; Control 2, 10 days of incubation with dead mycelia
Genome properties of Dentipellis sp. KUC8613
In order to elucidate genetic contents for PAH removal in this fungus, whole-genome sequencing was carried out under the JGI 1k FGP program. The genome sequence was 36.71 Mbp long, comprising 1184 contigs and 425 scaffolds (Table 1). The GC content of the genome was 55.4%. In total, 14,320 protein-coding genes were predicted in the genome with an average gene length of 1737 bp. Both the genome size and the number of gene models of KUC8613 were typical among the compared white rot fungi available from the JGI mycocosm (Grigoriev et al. 2014). Among 14,320 gene models, 6305 (44%) and 7538 (52.6%) were assigned to different GO terms and KOG classes, respectively.
A genomic repository of CAZymes was first surveyed in order to analyze the pattern of carbohydrate metabolism in this fungus. The genome harbored a total of 417 CAZymes including 71 auxiliary activities (AA), 49 carbohydrate-binding modules (CBM), and 180 glycoside hydrolases (GH) (Supplementary Table S1). The cellulolytic CAZyme composition in typical white rot fungi is generally represented by the presence of lytic polysaccharide monooxygenases (LMPOs) (AA9), cellobiohydrolases (GH6 and GH7), a single cellobiose dehydrogenase (AA3-1), frequent CBM1-containing proteins, and ligninolytic enzymes (Riley et al. 2014). Indeed, this strain revealed 11 copies of LMPOs that potentially carry out the oxidative cleavage of polysaccharide chains (Supplementary Table S2). It also contained four cellobiohydrolases, 22 genes encoding proteins carrying a CBM1 family module, and a single gene encoding cellobiose dehydrogenase. The CAZyme pattern suggests that Dentipellis sp. KUC8613 has the classical wood decay mode of previously well-characterized white rot fungi.
The genome of KUC8613 was annotated with 11 genes encoding ligninolytic enzymes, including seven fungal class II peroxidases (PODs; AA2) and four laccases (AA1_1). PODs were further classified into six MnPs and a single LiP by sequence comparison. When we compared the KUC8613 genome with eight other white rot fungal genomes, KUC8613 showed less than the average number of both PODs and laccase genes among the compared genomes (Fig. 3).

Comparison of relative tolerance to mixed PAHs (Lee et al. 2014) and the number of ligninolytic enzymes in white rot fungi. Fungal genome data were retrieved from the JGI-MycoCosm repository (https://genome.jgi.doe.gov/mycocosm/home). The species tree was built based on single-copy orthologs including Aspergillus nidulans as an out group. The bootstrap-based branch supports and the scale bar that represents the mean number of amino acid substitutions per site are shown. Gene numbers are shaded white or red based on the Z-scores computed for each column
While only a limited number of genes encoding ligninolytic enzymes were found in the genome of KUC8613, we could instead identify other genes encoding the non-ligninolytic type of enzymes that can potentially mediate the initial oxidation of aromatic ring structures. The presence of 154 putative P450s was revealed from the fungal genome (Supplementary Table S3). Using the KEGG database analysis, we could further sort out 77 P450s potentially involved in the metabolism of xenobiotics, specifically those from CYP1, CYP2, and CYP3 families (Lewis 2003).
Additional genes that are potentially involved in the downstream steps for PAH transformation were also investigated (Supplementary Table S4). We identified 19 putative epoxide hydrolases that might catalyze a reaction to produce trans-dihydrodiols from arene oxides. Oxidoreductase enzymes including 16 alcohol dehydrogenases, 17 aldehyde dehydrogenases, one trans-1,2-dihydrobenzene-1,2-diol dehydrogenase, and 31 FAD-dependent monooxygenases may catalyze a series of oxidation reactions to produce metabolic intermediates. Multicomponent dioxygenases comprising eight ferredoxins, four ferredoxin reductases, two hydroxyquinol 1,2-dioxygenases, and two aromatic ring-opening dioxygenases were also found in the fungal genome. Aromatic intermediates formed by the above enzymes then can act as substrates for additional ring cleavage or conjugation steps (Casillas et al. 1996; Habe and Omori 2003). Glutathione S-transferases (GSTs), sulfotransferases, or glycosyltransferases (GTFs) can catalyze the addition of glutathione, sulfate, or glycosyl donors to PAH molecules, respectively, thus making them less toxic and more water soluble. In the genome of KUC8613, ten putative GSTs, two sulfotransferases, and 13 GTFs were encoded for the potential conjugation of PAH intermediates.
Genome-wide transcriptomic responses during PAH removal
In order to further investigate which of the potential genes are actually upregulated during PAH removal in this fungus, a genome-wide transcriptomic analysis was performed. Mycelial discs grown for 5 days in liquid ME media supplemented with ANT, FLU, PHE, and PYR (100 mg/l) were subjected to RNA-seq. PCA of the gene expression data clearly separated PAH-added groups from the control group (Fig. 4). The PCA data also showed a separation within PAH samples. While the ANT, FLU, and PYR samples were clustered together, the PHE samples formed an independent group distinct from other PAH samples, suggesting that the gene expression profile of the PHE sample is unlike those of other PAHs.

Principal component analysis (PCA) of transcriptomics data. ANT, anthracene; FLU, fluoranthene; PHE, phenanthrene; PYR, pyrene
Of the 14,320 gene models predicted in the genome, we identified 1922 genes whose expression was upregulated by at least one of four different PAHs (fold change > 2 and p value < 0.01) (Supplementary Table S5). The numbers of total genes upregulated by ANT, FLU, PHE, and PYR were 915, 950, 1539, and 1095, respectively (Fig. 5a). Among the upregulated genes, the molecular function of 881 genes could not be predicted. Upregulated genes were assigned to 23 specific KOG classes and dominantly (48.13% of 1041 KOG-assigned genes) categorized into five KOG classes including posttranslational modification, signal transduction, energy production and conversion, carbohydrate transport and metabolism, and transcription (Fig. 5b; Supplementary Table S6). As illustrated by a Venn diagram in Fig. 5a, upregulated genes showed common and different PAH-specific upregulation during PAH removal. A total of 550 genes were commonly upregulated by all four PAHs. The numbers of PAH-specific genes by ANT, FLU, PHE, and PYR were 67, 72, 548, and 59, respectively. The number of PHE-induced genes were significantly higher than genes induced by other PAHs. A heatmap plotted by log2RPKM of upregulated gene expression showed a similar pattern as well (Fig. 5c). The majority of upregulated genes were commonly upregulated in all PAH samples while PHE showed a distinct gene expression pattern compared to those of the other three PAHs.

Transcriptomic analysis during removal of four different PAHs. a Venn diagram representing the number of upregulated genes by four different PAHs. b The five most common KOG classes among upregulated genes and their expression patterns. C, energy production and conversion; G, carbohydrate transport and metabolism; K, transcription; O, posttranslational modification; T, signal transduction. c Heatmap showing the transcription pattern (log2RPKM) of 1922 upregulated genes by four different PAHs. Columns represent different genes and rows represent different PAH-treated groups. ANT, anthracene; FLU, fluoranthene; PHE, phenanthrene; PYR, pyrene
We first investigated the expression profiles of ligninolytic enzymes (LiP, MnP, and laccase) and P450s for initial aromatic-ring oxidation. No ligninolytic enzymes were upregulated (fold change > 2 and p value < 0.01) during PAH removal, while one laccase gene (ProtID 934625) showed constant expression across the conditions (Supplementary Table S7). On the other hand, P450s showed both common and different PAH-specific upregulation. Among 154 putative P450 genes found in the KUC8613 genome, transcription of 15 genes was induced by one or more PAHs (Table 2). Six P450 genes were constitutively upregulated by two or three PAHs, while the remaining nine genes showed different PAH-specific upregulation. These nine genes could be further categorized into six PHE-inducible (ProtID 548471, 772453, 879478, 840982, 841965, and 873894) and one for each ANT-, FLU-, or PYR-inducible genes (ProtID 861037, 798081, and 832704 for ANT, FLU, and PYR, respectively).
We also identified a total of 27 other PAH-responsive genes potentially responsible for the downstream steps for PAH transformation. The expression patterns of these genes showed that most of them were commonly upregulated regardless of PAH type (Supplementary Fig. S2). These 27 PAH-responsive genes were three epoxide hydrolases (ProtID 581771, 840197, and 871191), three ferredoxins (ProtID 226033, 839077, and 887248), one ferredoxin reductase (ProtID 832353), five alcohol dehydrogenases (ProtID 843873, 859290, 867273, 712202, and 886660), three aldehyde dehydrogenases (ProtID 837250, 884278, and 857188), five FAD-dependent monooxygenases (ProtID 820609, 772612, 838248, 870155, and 879414), two dioxygenases (ProtID 841503 and 842583), three GSTs (ProtID 847321, 697729, and 830936), one GTF (ProtID 879011), and one sulfotransferase (ProtID 838043).
In addition to the PAH-transforming genes, the 550 commonly upregulated genes by all four PAHs included 41 genes involved in lipid transport and metabolism; these include a lipase (ProtID 835364), a sterol desaturase (ProtID 864816), a methyltransferase (ProtID 833162), and a phospholipid/glycerol acyltransferase (ProtID 39743) (Supplementary Table S8). This observation is consistent with previous studies in which the bioavailability of hydrophobic PAHs can be enhanced by the cellular production of lipid biosurfactants to emulsify PAHs and promote solubility (Cao et al. 2015).
Since our genomic and transcriptomic analysis suggests that this fungus utilizes the intracellular non-ligninolytic type of enzymes instead of extracellular ligninolytic enzymes for PAH-transformation, translocation of PAHs and metabolic intermediates across the cell membranes and organelles should be controlled by membrane-bound transporters. It is well known that major facilitator superfamily (MFS) transporters and ATP-binding cassette (ABC) transporters are two major transporter families that mediate the import and export of drugs and xenobiotics (Carmona et al. 2009; Jeong et al. 2017). We observed the common upregulation of 14 MFS and five ABC transporters by all four PAHs (Supplementary Table S9). The active transcription of MFS transporters was particularly noticeable by PHE. The more than tenfold elevated expression of four MFS transporters (ProtID 787334, 857773, 800757, and 328771) by PHE suggests an important role of these transporters for cellular response to PHE.
While a significant number of genes were commonly upregulated by all four PAHs, certain genes also showed PAH specificity within different PAH samples. Among four PAHs, PHE induced 35% more genes than the average number of upregulated genes by other PAHs. This resulted in the PHE-specific upregulation of 548 genes (Fig. 4). According to GO functional enrichment analysis, enriched GO terms among PHE-specific genes were ATP synthesis coupled proton transport and different types of cytochrome c oxidases (Fig. S3). We also identified 10 additional MFS and two ABC transporters, suggesting their restricted involvement in the translocation of PAHs other than PHE (Supplementary Table S10). PAHs are known to increase the production of reactive oxygen species (ROS), leading to oxidative stress (Alkio et al. 2005). Since only PHE showed detectable cytotoxicity during fungal growth, significant upregulation of ROS scavenging enzymes is expected. Indeed, we observed the PHE-specific upregulation of genes such as manganese and iron superoxide dismutase (ProtID 648712 and 860732) and alkyl hydroperoxide reductase (ProtID 194063), which are potentially involved in enzymatic antioxidant defense mechanisms.
Discussion
PAH transformation by wood-degrading fungi might be a ubiquitous phenomenon (Field et al. 1992; Mao and Guan 2016). In this study, we demonstrated that Dentipellis sp. KUC8613 has a high capability (> 90% of 100 mg/l PAH) of removing four different types of PAHs within a short period of time. Our genomic and transcriptomic analysis to further elucidate the PAH-transforming system encoded in the genome revealed a total of 1922 upregulated genes many of which have unknown molecular functions. To our knowledge, genome-scale transcriptomic responses by a wood-degrading fungus during PAH transformation have not been previously described.
While the ligninolytic system in white rot fungi was often described as the key for PAH transformation (Ghosal et al. 2016), our data showed that KUC8613 was capable of removing PAHs without upregulating ligninolytic genes. While one laccase gene showed constant expression across the conditions, it remains to be determined whether ligninolytic enzymes are involved in PAH transformation in this fungi. Another possibility is that they may actively take part in PAH transformation at certain growth conditions. It has been previously shown that the production of ligninolytic enzymes by white rot fungi can be dependent on carbon and nitrogen concentration in the growth media (Ben Hamman et al. 1997; D'Souza et al. 1999). It would be worthwhile to check the PAH-removal pattern by this fungus under different conditions that favor the production of ligninolytic enzymes. Taken together, these data strongly suggest that the screening of strong mycoremediation hosts primarily based on ligninolytic enzyme activities should be reconsidered.
Our transcriptomic analysis revealed that P450s might be responsible for the initial oxidation of aromatic rings in this fungus. A previous genome-scale identification of P450 genes in Phanerochaete chrysosporium showed PAH-oxidizing activity of these enzymes with varying PAH specificity (Syed et al. 2010). Similarly, upregulated P450s in this fungus showed both PAH-specific and common upregulation patterns. PHE induced the highest number of P450 genes, suggesting metabolic complexity of this compound compared to other three PAHs. In addition to P450s, we observed upregulation of many other genes encoding potential PAH-transforming activities such as epoxide hydrolase, alcohol dehydrogenase, aldehyde dehydrogenase, FAD-dependent monooxygenase, dioxygenase, GTF, GST, and sulfotransferase. The biotransformation of PAHs by dioxygenases is mainly known to occur in many PAH-degrading bacteria (Peng et al. 2008), but in some Trichoderma species, involvement of dioxygenase was also reported during phenanthrene degradation (Hadibarata et al. 2007). The shared upregulation of many potential PAH-transforming genes suggests some overlap between the metabolic pathways of different PAHs. The elucidation of the exact molecular mechanisms associated with the key enzymes discovered in this study requires further study. In addition, upregulation of many genes with unknown molecular functions also gives a clue for future research into the identification of novel genes involved in PAH transformation.
Since the solubility of PAHs in water is extremely low, the bioavailability of PAHs is an important factor for the efficient biodegradation of PAHs. Comparing to the other three PAHs used in this study, PHE is known to be more soluble (up to 20 times) in water (Yalkowsky et al. 2004). However, different water solubilities did not affect much of the removal efficiency of various PAHs by this fungus. Our transcriptomics data suggested that the bioavailability of PAHs might have been greatly increased owing to the production of lipid biosurfactants. The microbial production of biosurfactants in response to the presence of hydrocarbons can decrease the interfacial tension between aromatic compounds and the aqueous phase (Ron and Rosenberg 2001). Biosurfactants are mainly produced by bacteria, but it has been reported that filamentous fungi are also potential producers of biosurfactants (Sena et al. 2018). The application of microbial biosurfactants on the bioremediation process of polluted areas can be an alternative to synthetic counterparts (such as Tween 60) which can be toxic and poorly biodegradable in nature (Mulligan 2005).
The distinct transcriptomic response induced by PHE in this fungus was explained by a large number of PHE-specific genes involved in translocation of PAHs, defense against ROS, and ATP synthesis. The reason why PHE induced the transcription of a significantly larger number of genes is not clear, but our experimental evidences suggested that it might be associated with PHE-induced cellular cytotoxicity. A similar observation that PHE upregulated a larger number of genes than other PAHs was also previously reported in soil fungus (Gao et al. 2019).
Based on the global gene expression pattern, we constructed a tentative pathway for the transformation and detoxification of PAHs in Dentipellis sp. KUC8613 (Fig. 6). The pathway includes the initial ring-oxidation step followed by the downstream transformation step as well as transporters for the translocation of PAHs and their metabolites, lipases for the enhanced solubility of PAHs, and antioxidant enzymes for reduced oxidative stress. Our data based on whole-genomic and transcriptomic analysis will provide a strong insight into the complex gene regulation of wood-degrading fungi for PAH degradation, and may serve as a guide for the efficient screening and utilization of fungal hosts for mycoremediation.

A schematic view of PAH transformation in Dentipellis sp. KUC8613. Blue and red arrows represent the common and PAH-specific upregulation of PAH-transforming genes, respectively. The PAH-transformation pathway was divided into (1) initial ring oxidation and (2) downstream transformation steps based on key enzymatic reactions. GTF, glycosyltransferase; GST, glutathione S-transferase; SULT, sulfotransferase; SOD, superoxide dismutase; AHP, alkyl hydroperoxide reductase
References
Abe A, Inoue A, Usami R, Moriya K, Horikoshi K (1995) Degradation of polyaromatic hydrocarbons by organic solvent-tolerant bacteria from deep sea. Biosci Biotechnol Biochem 59(6):1154–1156. https://doi.org/10.1271/bbb.59.1154
Alkio M, Tabuchi TM, Wang XC, Colón-Carmona A (2005) Stress responses to polycyclic aromatic hydrocarbons in Arabidopsis include growth inhibition and hypersensitive response-like symptoms. J Exp Bot 56(421):2983–2994. https://doi.org/10.1093/jxb/eri295
Ben Hamman O, de La Rubia T, Martinez J (1997) Effect of carbon and nitrogen limitation on lignin peroxidase and manganese peroxidase production by Phanerochaete flavido-alba. J Appl Microbiol 83(6):751–757
Cao J, Lai Q, Yuan J, Shao Z (2015) Genomic and metabolic analysis of fluoranthene degradation pathway in Celeribacter indicus P73T. Sci Rep 5:7741. https://doi.org/10.1038/srep07741
Carmona M, Zamarro MT, Blázquez B, Durante-Rodríguez G, Juárez JF, Valderrama JA, Barragán MJ, García JL, Díaz E (2009) Anaerobic catabolism of aromatic compounds: a genetic and genomic view. Microbiol Mol Biol Rev 73(1):71–133. https://doi.org/10.1128/MMBR.00021-08
Casillas RP, Crow SA, Heinze TM, Deck J, Cerniglia CE (1996) Initial oxidative and subsequent conjugative metabolites produced during the metabolism of phenanthrene by fungi. J Ind Microbiol 16(4):205–215. https://doi.org/10.1007/Bf01570023
D'Souza TM, Merrit CS, Reddy CA (1999) Lignin-modifying enzymes of the white rot basidiomycete Ganoderma lucidum. Appl Environ Microbiol 65(12):5307–5313
de Boer W, Folman LB, Summerbell RC, Boddy L (2005) Living in a fungal world: impact of fungi on soil bacterial niche development. FEMS Microbiol Rev 29(4):795–811. https://doi.org/10.1016/j.femsre.2004.11.005
Deshmukh R, Khardenavis AA, Purohit HJ (2016) Diverse metabolic capacities of fungi for bioremediation. Indian J Microbiol 56(3):247–264. https://doi.org/10.1007/s12088-016-0584-6
Field JA, de Jong E, Feijoo Costa G, de Bont JA (1992) Biodegradation of polycyclic aromatic hydrocarbons by new isolates of white rot fungi. Appl Environ Microbiol 58(7):2219–2226
Fulton TM, Chunwongse J, Tanksley SD (1995) Microprep protocol for extraction of DNA from tomato and other herbaceous plants. Plant Mol Biol Report 13(3):207–209. https://doi.org/10.1007/Bf02670897
Gan S, Lau EV, Ng HK (2009) Remediation of soils contaminated with polycyclic aromatic hydrocarbons (PAHs). J Hazard Mater 172(2–3):532–549. https://doi.org/10.1016/j.jhazmat.2009.07.118
Gao R, Hao DC, Hu WL, Song S, Li SY, Ge GB (2019) Transcriptome profile of polycyclic aromatic hydrocarbon-degrading fungi isolated from Taxus rhizosphere. Curr Sci India 116(7):1218–1228. https://doi.org/10.18520/cs/v116/i7/1218-1228
Ghosal D, Ghosh S, Dutta TK, Ahn Y (2016) Current state of knowledge in microbial degradation of polycyclic aromatic hydrocarbons (PAHs): a review. Front Microbiol 7:1369. https://doi.org/10.3389/fmicb.2016.01369
Gnerre S, MacCallum I, Przybylski D, Ribeiro FJ, Burton JN, Walker BJ, Sharpe T, Hall G, Shea TP, Sykes S, Berlin AM, Aird D, Costello M, Daza R, Williams L, Nicol R, Gnirke A, Nusbaum C, Lander ES, Jaffe DB (2011) High-quality draft assemblies of mammalian genomes from massively parallel sequence data. Proc Natl Acad Sci USA 108(4):1513–1518. https://doi.org/10.1073/pnas.1017351108
Gran-Scheuch A, Fuentes E, Bravo DM, Jiménez JC, Pérez-Donoso JM (2017) Isolation and characterization of phenanthrene degrading bacteria from diesel fuel-contaminated Antarctic soils. Front Microbiol 8:1634. https://doi.org/10.3389/fmicb.2017.01634
Grigoriev IV, Nikitin R, Haridas S, Kuo A, Ohm R, Otillar R, Riley R, Salamov A, Zhao X, Korzeniewski F, Smirnova T, Nordberg H, Dubchak I, Shabalov I (2014) MycoCosm portal: gearing up for 1000 fungal genomes. Nucleic Acids Res 42(Database issue):D699–D704. https://doi.org/10.1093/nar/gkt1183
Habe H, Omori T (2003) Genetics of polycyclic aromatic hydrocarbon metabolism in diverse aerobic bacteria. Biosci Biotechnol Biochem 67(2):225–243. https://doi.org/10.1271/bbb.67.225
Hadibarata T, Tachibana S, Itoh K (2007) Biodegradation of phenanthrene by fungi screened from nature. Pak J Biol Sci 10(15):2535–2543
Harms H, Schlosser D, Wick LY (2011) Untapped potential: exploiting fungi in bioremediation of hazardous chemicals. Nat Rev Microbiol 9(3):177–192. https://doi.org/10.1038/nrmicro2519
Jeong CB, Kim DH, Kang HM, Lee YH, Kim HS, Kim IC, Lee JS (2017) Genome-wide identification of ATP-binding cassette (ABC) transporters and their roles in response to polycyclic aromatic hydrocarbons (PAHs) in the copepod Paracyclopina nana. Aquat Toxicol 183:144–155. https://doi.org/10.1016/j.aquatox.2016.12.022
Johansson I, van Bavel B (2003) Levels and patterns of polycyclic aromatic hydrocarbons in incineration ashes. Sci Total Environ 311(1–3):221–231. https://doi.org/10.1016/S0048-9697(03)00168-2
Kadri T, Rouissi T, Kaur Brar S, Cledon M, Sarma S, Verma M (2017) Biodegradation of polycyclic aromatic hydrocarbons (PAHs) by fungal enzymes: a review. J Environ Sci (China) 51:52–74. https://doi.org/10.1016/j.jes.2016.08.023
Lee H, Jang Y, Choi YS, Kim MJ, Lee J, Lee H, Hong JH, Lee YM, Kim GH, Kim JJ (2014) Biotechnological procedures to select white rot fungi for the degradation of PAHs. J Microbiol Methods 97:56–62. https://doi.org/10.1016/j.mimet.2013.12.007
Lewis DFV (2003) Human cytochromes P450 associated with the phase 1 metabolism of drugs and other xenobioties: a compilation of substrates and inhibitors of the CYP1, CYP2 and CYP3 families. Curr Med Chem 10(19):1955–1972. https://doi.org/10.2174/0929867033456855
Mao J, Guan WW (2016) Fungal degradation of polycyclic aromatic hydrocarbons (PAHs) by Scopulariopsis brevicaulis and its application in bioremediation of PAH-contaminated soil. Acta Agr Scand B-S P 66(5):399–405. https://doi.org/10.1080/09064710.2015.1137629
Marco-Urrea E, García-Romera I, Aranda E (2015) Potential of non-ligninolytic fungi in bioremediation of chlorinated and polycyclic aromatic hydrocarbons. New Biotechnol 32(6):620–628. https://doi.org/10.1016/j.nbt.2015.01.005
Mastrangelo G, Fadda E, Marzia V (1996) Polycyclic aromatic hydrocarbons and cancer in man. Environ Health Perspect 104(11):1166–1170. https://doi.org/10.2307/3432909
Min B, Grigoriev IV, Choi IG (2017) FunGAP: fungal genome annotation pipeline using evidence-based gene model evaluation. Bioinformatics 33(18):2936–2937. https://doi.org/10.1093/bioinformatics/btx353
Mulligan CN (2005) Environmental applications for biosurfactants. Environ Pollut 133(2):183–198. https://doi.org/10.1016/j.envpol.2004.06.009
Peng RH, Xiong AS, Xue Y, Fu XY, Gao F, Zhao W, Tian YS, Yao QH (2008) Microbial biodegradation of polyaromatic hydrocarbons. FEMS Microbiol Rev 32(6):927–955. https://doi.org/10.1111/j.1574-6976.2008.00127.x
Pozdnyakova NN (2012) Involvement of the ligninolytic system of white-rot and litter-decomposing fungi in the degradation of polycyclic aromatic hydrocarbons. Biotechnol Res Int 2012:243217–243220. https://doi.org/10.1155/2012/243217
Ron EZ, Rosenberg E (2001) Natural roles of biosurfactants. Environ Microbiol 3(4):229–236
Riley R, Salamov AA, Brown DW, Nagy LG, Floudas D, Held BW, Levasseur A, Lombard V, Morin E, Otillar R, Lindquist EA, Sun H, LaButti KM, Schmutz J, Jabbour D, Luo H, Baker SE, Pisabarro AG, Walton JD, Blanchette RA, Henrissat B, Martin F, Cullen D, Hibbett DS, Grigoriev IV (2014) Extensive sampling of basidiomycete genomes demonstrates inadequacy of the white-rot/brown-rot paradigm for wood decay fungi. Proc Natl Acad Sci U S A 111(27):9923–9928. https://doi.org/10.1073/pnas.1400592111
Sena HH, Sanches MA, Rocha DFS, Segundo W, de Souza ES, de Souza JVB (2018) Production of biosurfactants by soil fungi isolated from the Amazon forest. Int J Microbiol 2018:5684261–5684268. https://doi.org/10.1155/2018/5684261
Schützendübel A, Majcherczyk A, Johannes C, Hüttermann A (1999) Degradation of fluorene, anthracene, phenanthrene, fluoranthene, and pyrene lacks connection to the production of extracellular enzymes by Pleurotus ostreatus and Bjerkandera adusta. Int Biodeterior Biodegradation 43(3):93–100. https://doi.org/10.1016/S0964-8305(99)00035-9
Syed K, Doddapaneni H, Subramanian V, Lam YW, Yadav JS (2010) Genome-to-function characterization of novel fungal P450 monooxygenases oxidizing polycyclic aromatic hydrocarbons (PAHs). Biochem Bioph Res Co 399(4):492–497. https://doi.org/10.1016/j.bbrc.2010.07.094
Yalkowsky SH, He Y, Jain P (2004) Handbook of aqueous solubility data. Choice 41(5):941
Yu GC, Wang LG, Han YY, He QY (2012) clusterProfiler: an R package for comparing biological themes among gene clusters. Omics 16(5):284–287. https://doi.org/10.1089/omi.2011.0118
Acknowledgments
This work is supported by the Cooperative Research Program for Agriculture Science & Technology Development (Project No. PJ01337602) Rural Development Administration and New and Renewable Energy Core Technology Program of the Korea Institute of Energy Technology Evaluation and Planning (KETEP) grants from the Ministry of Trade, Industry and Energy (No. 20173010092460), Republic of Korea. The work conducted by the U.S. Department of Energy Joint Genome Institute, a DOE Office of Science User Facility, is supported by the Office of Science of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Park, H., Min, B., Jang, Y. et al. Comprehensive genomic and transcriptomic analysis of polycyclic aromatic hydrocarbon degradation by a mycoremediation fungus, Dentipellis sp. KUC8613. Appl Microbiol Biotechnol 103, 8145–8155 (2019). https://doi.org/10.1007/s00253-019-10089-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-019-10089-6