
Abstract
The MtrA-MtrB two-component regulatory system is highly conserved in Actinobacteria and plays crucial roles in cell cycle progression, cell morphology, antibiotic resistance, and osmoprotection. Previously, we revealed that the MtrA protein of Saccharopolyspora erythraea E3 strain (a high erythromycin-producing strain) had a two amino acid (H197 and V198) deletion in the DNA recognition helices of the C-terminal domain compared to the wild type S. erythraea strain NRRL2338. Here, we identified mepA (encoding a membrane protein related to metalloendopeptidases) as an MtrA target gene, and found that deleting the two amino acids in MtrA (MtrAdel) resulted in the loss of its DNA-binding activity for the mepA gene. The mutant MtrAdel lost its regulatory activity and affected various physiological functions consistent with mtrA deletion, including increased erythromycin biosynthesis, enhanced antibiotic resistance, deregulated osmoprotection, and improved transport of substances. The introduction of the wild type mtrA gene into the S. erythraea E3 strain with the mtrAdel gene decreased the erythromycin yield by approximately 50%, confirming that MtrA repressed erythromycin production. These findings demonstrate that MtrA is an important pleiotropic regulator of erythromycin biosynthesis, antibiotic resistance, osmoprotection, and substance transport in S. erythraea and provide new insights for improving erythromycin production. Future studies linking the molecular effects of MtrA to these phenotypes will improve our understanding of the MtrA-MtrB two-component regulatory system in Actinobacteria.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
ORCID: Bang-Ce Ye: 0000-0002-5555-5359
Introduction
Sensing and responding to the environment is essential for the survival of all living organisms (Papandreou et al. 2005). Bacteria have developed beneficial two-component regulatory systems which respond to various environmental stimuli, including nutrients, pH, temperature, and antibiotics (Stock et al. 2000). In the classical two-component system, a sensor histidine kinase is autophosphorylated by sensing a specific signal, then transfers its phosphoryl group to its corresponding response regulator which regulates the expression of relevant genes and affects physiological function (Laub and Goulian 2007). Multiple important actinobacterial two-component systems have been widely researched, including VanRS, AbsA1/2, SenRS, σE-CseABC, and MtrAB (Anderson et al. 2001; Hutchings 2007; Hutchings et al. 2006; Lucana et al. 2005; Paget et al. 1999). Among these, MtrAB has received the most attention as it is conserved in all actinobacterial genomes and thus plays a significant role in survival (Hutchings 2007).
In the past few decades, MtrAB has been extensively studied in some microorganisms, such as Mycobacterium tuberculosis (Li et al. 2010; Nguyen et al. 2010; Via et al. 1996; Zahrt and Deretic 2000), Corynebacterium glutamicum (Brocker and Bott 2006; Hoskisson and Hutchings 2006; Moker et al. 2004; Moker et al. 2007), and Streptomyces coelicolo (Zhang et al. 2017). These studies revealed that MtrAB is involved in many different cellular functions. The dnaA gene, which regulates DNA replication, was the first direct target gene of MtrA to be confirmed in M. tuberculosis (Fol et al. 2006), suggesting that MtrAB plays an essential role in cell cycle progression. The oriC and fbpB genes were also identified as MtrAB target genes, indicating a major role for MtrAB in coordinating the fecundity and pathogenicity of M. tuberculosis (Rajagopalan et al. 2010). Furthermore, it was shown that mtrA gene transcription is activated by incubating M. tuberculosis with macrophages (Via et al. 1996) and that the mtrA expression level differed for virulent and avirulent strains grown in macrophages (Zahrt and Deretic 2000), suggesting that MtrAB function affects the infectivity of M. tuberculosis. More recently, the study has provided evidence for the role of MtrAB in the multidrug resistance of Mycobacterium species, thus providing new possibilities for drug design (Li et al. 2010). Meanwhile, MtrAB studies have also revealed the relationship between the two-component system and cell wall homeostasis in Mycobacterium species (Nguyen et al. 2010). In C. glutamicum, MtrA repressed the expression of mepA and nlpC, which encode putative cell wall peptidases (Brocker and Bott 2006), revealing that MtrAB is involved in regulating cell wall turnover and suggesting that MtrAB might contribute to multidrug resistance (Moker et al. 2004). Conversely, MtrA activated the expression of betP and proP, which encode transporters for the uptake of osmoprotectants (Brocker and Bott 2006; Hoskisson and Hutchings 2006; Moker et al. 2004), indicating that MtrAB is involved in osmoprotection (Moker et al. 2007). Streptomyces is the largest Actinobacteria genus and produces over two-thirds of clinically useful antibiotics. MtrA was found to regulate the expression of genes involved in antibiotic biosynthesis in Streptomyces (Som et al. 2017b). However, MtrAB research has so far been limited to Corynebacteria, Mycobacteria, and Streptomycetes; therefore, investigating the role of MtrAB in different bacteria would improve our understanding of the overall ability of Actinobacteria to respond to their environment (Hoskisson and Hutchings 2006).
Saccharopolyspora erythraea is an excellent strain for producing erythromycin A (Staunton and Wilkinson 1997), a widely used antibiotic against gram-positive bacteria. A detailed investigation of S. erythraea would therefore be highly valuable to the antibiotic industry. Previously, we found that the MtrA protein of S. erythraea E3 (a high erythromycin-producing strain) had two amino acids missing (H197 and V198) compared to S. erythraea NRRL2338 (a low erythromycin-producing strain), and hypothesized that this might be related to high erythromycin productivity (Li et al. 2013).
In this study, we investigated the effect of deleting the mtrA gene or the two amino acids in MtrA on erythromycin level in S. erythraea, finding that mutant MtrAdel reduced the erythromycin yield by approximately 50%. MtrA repressed the expression of its target gene mepA, which encodes a membrane protein related to the metalloendopeptidases. The deletion of H197 and V198 resulted in the loss of MtrA DNA-binding activity for its target genes, its repressive activity, and affected various physiological functions consistent with mtrA deletion, including increased erythromycin biosynthesis, enhanced antibiotic resistance, deregulated osmoprotection, and improved transport of substances.
Materials and methods
Bacterial strains, plasmids, and growth conditions
The strains and plasmids used in this study are listed in Table S1. The collection number of S. erythraea NRRL23338 is DSM 40517.To prepare for seeding, S. erythraea was grown in tryptone soya broth at 30 °C and 200 rpm for 24 h. Then, 500 μL of the seed culture was inoculated into a 500-ml flask containing 50 ml of industrial fermentation medium (36 g/L soybean flour, 36 g/L corn starch, 2.4 g/L (NH4)2SO4, 7.2 g/L CaCO3, 5 g/L soybean oil, 20 g/L glucose, 6.7 g/L n-propanal, and distilled H2O) and incubated at 30 °C and 200 rpm for 4 days in order to evaluate erythromycin production. Bacillus subtilis was grown in the test medium (5 g/L peptone, 3 g/L Beef Extract Powder, 3 g/L K2HPO4, and distilled H2O) at 37 °C and 200 rpm for 14–16 h as the control organism for erythromycin production. A total of 500 μl of the seed culture was inoculated into a 500-ml flask containing 50 ml of tryptic soy broth (TSB) and incubated at 30 °C and 200 rpm for 48 h prior to RNA extraction. A total of 20 μL of the seed culture was inoculated onto MM agar plates (10 g/L glucose, 0.5 g/L K2HPO4, 1 g/L (NH4)2SO4, 0.2 g/L MgSO4, 0.01 g/L FeSO4, 20 g/L agar, and distilled H2O; pH 7.0) and incubated at 30 °C for 40 h before observing the cells using a scanning electron microscope (SEM). Escherichia coli strains were grown in Luria-Bertani (LB) medium at 37 °C. All media were sterilized by autoclaving at 121 °C for 20 min, except for MM agar which was autoclaved at 115 °C for 20 min.
Construction of the wild type mtrA complementation and deletion strain
The complementation experiments in S. erythraea E3 were performed as described previously (Liao et al. 2014). A 687 nt fragment of the mtrA gene (SACE_6447; protein ID: CAM05617.1) from S. erythraea NRRL2338 was amplified by PCR using the primer pairs mtrA-fw and mtrA-rev as shown in Table S2. The pIB-mtrA plasmid was created by cloning the PCR products into the NdeI/NotI sites of pIB139. The complementary plasmid was introduced into S. erythraea E3 by polyethyleneglycol (PEG)-mediated transformation. The desired complementary strains (S. erythraea E3::mtrA strain) were confirmed using apramycin-resistance screening and PCR.
The deletion experiments in S. erythraea NRRL2338 were performed as described previously (Liu et al. 2018). KO6447Up, tsr, and KO6447Down were amplified using PCR with the primer pairs provided in Table S2. The sgRNAII sequence was designed as shown in Table S3. The 6447sgRNAII-(KO6447Up-tsr-KO6447Down) cassette was produced and ligated into the pKECas9 plasmid using the Hieff Clone Multi One Step Cloning Kit (Yeasen, Shanghai, China) to generate pKECas9-6447sgRNA-(KO6447Up-tsr-KO6447Down). mtrA deletion was confirmed by PCR and sequencing (Fig. S1).
Erythromycin determination by high-performance liquid chromatography
High-performance liquid chromatography (HPLC) was carried out as described previously (Liu et al. 2018). The seed cultures were inoculated into the industrial fermentation medium, incubated at 30 °C and 200 rpm for 7 days, and erythromycin production was evaluated by HPLC. Freeze-dried fermentation supernatant powder was dissolved in absolute ethanol, filtered using a hydrophilic PVDF membrane (0.22 mm pore size), and analyzed. The HPLC conditions were as follows: solvent B (55% acetonitrile) against solvent A (1 L water with 8.7 g K2HPO4; pH 8.2) at a flow rate of 1.0 mL/min and 40 °C. UV spectra were measured at 215 nm. A standard curve (Y = aX + b) was obtained by analyzing a series of erythromycin standard concentrations, allowing the erythromycin levels to be calculated. Y represented the peak area and X represented the erythromycin levels.
Overexpression, purification, and phosphorylation of MtrA
The primers for PCR were provided in Table S2. The mtrA (from S. erythraea NRRL23338 genomic DNA) and mtrAdel (from S. erythraea E3 genomic DNA) genes were amplified by PCR. The PCR products were cloned into pET-28a to generate recombinant pET28a-mtrA/pET28a-mtrAdel plasmids. The cheA gene (from E. coli K-12 MG1655 genomic DNA) was amplified by PCR and cloned into pET-42a to generate recombinant pET42a-cheA plasmids. The recombinant plasmids were then confirmed by DNA sequencing and introduced into E. coli BL21 (DE3). The E. coli cells were inoculated into 100 ml LB with 50 μg/mL kanamycin, grown at 37 °C and 220 rpm to an OD600 of 0.3, induced with isopropyl-beta-D-thiogalactopyranoside (IPTG) at a final concentration of 0.4 mM, and incubated at 20 °C for 12–16 h. For protein purification, cells were harvested by centrifugation, washed twice with PBS buffer (pH 8.0), and then disrupted using an ultrasonic cell crusher. Cell debris and membrane fractions were separated from the soluble fractions by centrifugation (10 min, 10,000 rpm, 4 °C). His6-proteins were purified using Ni-NTA Superflow columns (Qiagen). Purified proteins were eluted with 250 mM imidazole (50 mM NaH2PO4, 300 mM NaCl; pH 8.0), dialyzed in protein preservation Buffer D (50 mM Tris, 0.5 mM EDTA, 50 mM NaCl, 20% glycerol, 1 mM DTT; pH 8.0) at 4 °C overnight, and then stored at − 80 °C. The quality of the purified proteins was determined by SDS-PAGE and their concentrations were determined using the Bradford reagent.
MtrA phosphorylation was carried out as described previously (Deretic et al. 1992). CheA, a typical histidine protein kinase, was expressed, purified, and used to phosphorylate MtrA. CheA was autophosphorylated by incubating with ATP at room temperature for 20 min, and then mixed with 10 μM MtrA/MtrAdel for 1 h at 37 °C in a buffer containing 100 mM Tris HCl (pH 7.5), 50 mM KCl, and 5 mM MgCl2, to allow the phosphoryl group to be transferred from CheA to MtrA/MtrAdel.
Electrophoretic mobility shift assays
The promoter regions (− 300 to + 50) of the predicted target genes were amplified using PCR with the primers listed in Table S2. The PCR products were labeled with a biotinylated universal primer (5′-biotinAGCCAGTGGCGATAAG-3′). The biotin-labeled PCR products were then analyzed by agarose gel electrophoresis and purified using a PCR purification kit (Shanghai Generay Biotech). The DNA probe concentrations were measured using a microplate reader (Biotek, USA). Electrophoretic mobility shift assays (EMSAs) were carried out as described previously (Yao et al. 2014) according to the protocol for the Chemiluminescent EMSA Kit (Beyotime Biotechnology, China). The binding reaction was carried out in 10 mM Tris HCl (pH 8.0), 25 mM MgCl2, 50 mM NaCl, 1 mM DTT, 1 mM EDTA, 0.01% Nonidet P40, 50 μg/ml poly[d(I-C)], and 10% glycerol. A biotin-unlabeled specific probe (200-fold) and a non-specific competitor DNA (200-fold, sonicated salmon sperm DNA) were used as controls. After binding, the samples were loaded and separated on 6% non-denaturing PAGE gels in ice-cold 0.5Χ Tris-borate-EDTA at 100 V, and bands were detected by BeyoECL Plus.
RNA extraction and real-time RT-PCR
Cell pellets were collected by centrifugation at 4000×g and 4 °C for 20 min. Total RNA was extracted using the RNAprep pure cell/bacterium kit (Tiangen Biotech Co., Ltd., Beijing, China). RNA quality was analyzed by 1% agarose gel electrophoresis and concentration was determined using a microplate reader (BioTek). Total RNA (1 μg) was reverse transcribed using a PrimeScript RT Reagent Kit with gDNA Eraser (Takara, Japan). Genomic DNA was extracted for 5 min at 42 °C by DNase digestion. Real-time RT-PCR was carried out using the primers listed in Table S2 with an SYBR Premix Ex Taq GC kit (Takara, Japan) and approximately 100 ng template cDNA. All procedures were performed according to the manufacturer’s instructions. PCR was performed using the CFX96 real-time system (Bio-Rad, Hercules, CA, USA) with the following conditions: 95 °C for 10 min, then 40 cycles of 95 °C for 10 s, 60 °C for 10 s, and 72 °C for 30 s.
Antibiotic resistance assays
Seed cultures of S. erythraea NRRL2338, ΔmtrA, E3, and E3::mtrA (300 μL) were grown in a 250-ml flask containing 30 ml of tryptone soya broth at 30 °C and 200 rpm for 48 h, then their OD600 values (A) were measured. The OD600 values were measured again (B) after treatment with 30 μg/mL thiostrepton and 50 μg/mL vancomycin for 24 h. The survival percentage was calculated as follows:
Osmosis assays and DiOC2(3) fluorescent probe assays
Seed cultures of S. erythraea NRRL2338 and E3 (300 μL) were grown in a 250-ml flask containing 30 ml of tryptone soya broth at 30 °C and 200 rpm for 48 h. NaCl solution was then added to increase the osmotic pressure, resulting in a final concentration of 1 M NaCl. Cells were collected after 0 min, 90 min, and 180 min and the total RNA was extracted. The relative transcript levels of the target gene mepA (SACE_5687; Protein ID: CAM04873.1) were detected according to the “RNA extraction and real-time RT-PCR” method described previously.
Seed cultures of S. erythraea NRRL2338, ΔmtrA, E3, and E3::mtrA (300 μL) were grown in a 250-ml flask containing 30 ml of tryptone soya broth at 30 °C and 200 rpm for 48 h. A total of 1 mL of the cultures was transferred to sterile 50 mL tubes, then 10 μL DiOC2(3) fluorescent probe (3 mM) was added to the tubes and incubated at 37 °C and 200 rpm for 30 min. Cells were collected by centrifugation at 4000×g, washed twice with 500 μL PBS, and flow cytometry was carried out. Data were obtained using a BD FACSCalibur Flow Cytometer with a 488 nm argon ion gas laser and an FITC channel and were analyzed using FlowJo software.
Results
Wild type MtrA decreases erythromycin levels in high-producing S. erythraea with mutant MtrA
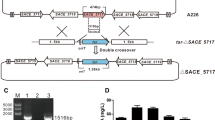
Previously, we revealed that the MtrA protein of high erythromycin-producing S. erythraea E3 had two amino acids missing (Li et al. 2013). In this study, we confirmed the deletion of 6 bp DNA (translated as H197 and V198) from MtrA (MtrAdel) in S. erythraea E3 by sequence alignment after gene sequencing (Fig. 1a, b). To investigate the effect of the MtrAdel mutant on erythromycin biosynthesis, the wild type mtrA from S. erythraea NRRL2338 was transferred into S. erythraea E3 with the mtrA mutant, producing the S. erythraea E3::mtrA strain. As a control, the empty plasmid vector PIB139 was transferred into S. erythraea E3 with the mtrA mutant. The erythromycin levels produced by S. erythraea NRRL2338, E3, and E3::mtrA were measured by HPLC. The standard curve equation was as follows: Y = 508281X − 120278 (Y was the peak area, X was the erythromycin levels). The linear correlation coefficient R2 was 0.9914. The mean erythromycin levels produced by S. erythraea NRRL2338, E3, E3::PIB139, and E3::mtrA were 7.00 mg/L, 296.50 mg/L, 282.95 mg/L, and 151.90 mg/L, respectively, when calculated using the standard curve (Fig. 1c). The erythromycin level of S. erythraea E3::mtrA was approximately 50% lower than that of S. erythraea E3, while the effect of the PIB139 plasmid on erythromycin level was shown to be negligible. The erythromycin level of S. erythraea E3::pSET152-mtrA was approximately 50% lower than that of S. erythraea E3 which was similar to that of S. erythraea E3::mtrA (Fig. S2). These results suggest that the wild type mtrA gene significantly decreased the erythromycin level of high-producing S. erythraea with the mtrA mutant.

Wild type MtrA decreases erythromycin levels in high-producing Saccharopolyspora erythraea with mutant MtrA. a Multiple DNA sequence alignment of mtrA genes from S. erythraea NRRL2338 and E3, Mycobacterium tuberculosis, Corynebacterium glutamicum, and S. venezuelae. The black rectangle indicates the 6-bp (591-596 sites) deletion in mtrA. b The amino acids H197 and V198 and the DNA-binding helix are indicated by the black rectangle. c The mean erythromycin yields of S. erythraea NRRL2338, E3, E3::PIB139, and E3::mtrA were 7.00 mg/L, 296.50 mg/L, 282.95 mg/L, and 151.90 mg/L, respectively. d Erythromycin yields of S. erythraea NRRL2338 and ΔmtrA. The mean erythromycin level of S. erythraea NRRL2338 ΔmtrA was 11.25 mg/L
The wild type mtrA of S. erythraea NRRL2338 was deleted to produce the S. erythraea NRRL2338 ΔmtrA strain. The level of erythromycin produced by S. erythraea NRRL2338 ΔmtrA was 11.25 mg/L (Fig. 1d), an increase of approximately 60% compared with S. erythraea NRRL2338 (7.00 mg/L), indicating that MtrA may be involved in erythromycin biosynthesis.
mepA is an MtrA target gene in S. erythraea
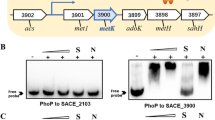
Several MtrA target genes, such as dnaA, oriC, and fbpB in M. tuberculosis (Fol et al. 2006; Rajagopalan et al. 2010) and mepA, nlpC, betP, and proP in C. glutamicum (Brocker and Bott 2006), have been reported previously. Furthermore, the 9-bp “GTCACAgcg-like” repeat has been identified as the MtrA-binding motif in M. tuberculosis (Rajagopalan et al. 2010). To identify the target genes of MtrA in S. erythraea, we used the “GTCACAgcg-like” MtrA-binding motif to screen the sequences upstream (+ 50 to − 300) of putative MtrA target genes in S. erythraea NRRL23338 (Table 1). mepA is one such predicted target gene, encoding a membrane protein related to metalloendopeptidases whose target could be cell wall peptides. The MepA protein may play a role in cell wall metabolism, cell shape phenotype, antibiotic susceptibility, and osmoprotection (Papandreou et al. 2005; Stock et al. 2000). We hypothesized that MepA might improve erythromycin transport in high erythromycin-producing strains. Two “GTCACAgcg-like” motifs (GTCACGGTTCGGTCACGAGC) were located 76-bp upstream of the translational start site (− 77 to − 96) of the mepA gene (SACE_5687) (Fig. 2a). To validate this target gene, EMSA (Fig. 2b and Fig. S3) was performed with 1.8 μM MtrA, an unlabeled specific probe (200-fold), and a non-specific competitor DNA (200-fold, sonicated salmon sperm DNA) as controls (Fig. 2b). A range of purified MtrA protein concentrations (0.6 μM, 1.8 μM, and 3 μM) were investigated, with the results indicating that MtrA can bind directly to the upstream region containing the predicted motifs and suggesting that mepA might be an MtrA target gene in S. erythraea.

MtrA directly regulates the mepA gene in Saccharopolyspora erythraea. a Genetic structure of the mepA gene in S. erythraea and its predicted MtrA-binding site. b Electrophoretic mobility shift assays of a range of pure S. erythraea MtrA protein concentrations (0.6 μM, 1.8 μM, and 3 μM) with biotin-labeled PCR products. The concentration of PCR products was 2 nM. S represents the unlabeled specific probe (200-fold) and N represents the non-specific competitor DNA (200-fold; sonicated salmon sperm DNA) used as controls, and the corresponding MtrA concentration was 1.8 μM
To further investigate the effect of MtrA on erythromycin production, we determined the transcriptional levels of SACE_0721, SACE_0723, and SACE_0724 of the ery cluster involved in erythromycin biosynthesis in S. erythraea NRRL2338 (WT), NRRL2338 ΔmtrA, E3, and E3::mtrA (Fig. S4). The results indicated that MtrA has a significant effect on erythromycin biosynthesis. To identify direct MtrA targets that might be involved in erythromycin biosynthesis, ChIP sequencing was performed using MtrA-3 x Flag. SACE_0438, SACE_3737 (ama2), SACE_4140, SACE_5429 (coaE), SACE_6295 (rho1), and SACE_6447 (mtrA) were identified as MtrA target genes, but not SACE_5687 (mepA) (Fig. S5 and Table S4). No proteins directly regulating erythromycin biosynthesis were found.
Deletion of two amino acids in MtrA (MtrAdel) results in loss of MtrA DNA-binding activity
MtrA, a response regulator of the OmpR family, contains a conserved DNA recognition helix (HTH domain) (Martinez-Hackert and Stock 1997). The deletion of two amino acids (H197 and V198) in the DNA recognition helix of the C-terminal domain altered the α-helical structure of the DNA recognition helix as predicted (Fig. 3a, b).

Deletion of two amino acids in MtrA (MtrAdel) results in loss of MtrA DNA-binding activity. a The structure of MtrA from Saccharopolyspora erythraea NRRL2338 was predicted based on the crystal structure of Rv3246c in Mycobacterium tuberculosis (PDB: 2gwr). b The deletion of His197 and Val198 disrupts the α-helix of the DNA-binding region. c Electrophoretic mobility shift assays of pure S. erythraea MtrA and MtrAdel proteins with biotin-labeled mepA. The concentration of PCR products was 2 nM, the concentration of MtrA was 3 μM, and a series of MtrAdel concentrations were used (0.6 μM, 1.8 μM, 3 μM, 4.2 μM, and 5 μM)
To confirm the effect of the deletion of these two amino acids in MtrA, EMSAs were performed on purified S. erythraea MtrA and MtrAdel proteins with biotin-labeled mepA (Fig. 3c). The concentration of the PCR products was 2 nM. A range of MtrAdel protein concentrations (0.6 μM, 1.8 μM, 3 μM, 4.2 μM, and 5 μM) were used. As shown in Fig. 3c, the MtrAdel protein could not bind the region upstream of mepA, suggesting that the deletion of the two amino acids in MtrA (MtrAdel) resulted in the loss of MtrA DNA-binding activity by altering the structure of the DNA recognition helix.
Introducing mtrA into S. erythraea with mtrAdel represses mepA transcription
To determine the regulatory effects of MtrA on mepA in S. erythraea, the transcriptional levels of mepA in S. erythraea NRRL2338, ΔmtrA, E3, E3::PIB139, and E3::mtrA were measured using real-time RT-PCR with the internal 16S rRNA gene as a control. Total RNA was extracted from the samples collected at 48 h. In S. erythraea ΔmtrA, E3, and E3::mtrA the transcription of mepA increased by approximately 3.5-fold, 2-fold, and 1.5-fold, respectively, compared with S. erythraea NRRL2338. Furthermore, deleting mtrA in the S. erythraea NRRL2338 strain promoted the transcription of mepA (Fig. 4 and Fig. S6), suggesting that MtrA is a transcriptional repressor of mepA.

mepA transcript levels in Saccharopolyspora erythraea strains mepA transcript levels in S. erythraea ΔmtrA, E3, and E3::mtrA were increased by approximately 3.5-fold, 2-fold, and 1.5-fold compared with S. erythraea NRRL2338, respectively. The fold change for mepA expression levels in S. erythraea NRRL2338 was set to 1.0. Error bars indicate the SD of three independent experiments
Effects of MtrA on antibiotic resistance
In order to understand the effects of MtrA on antibiotic resistance, we tested the susceptibility of S. erythraea NRRL2338, ΔmtrA, E3, E3::PIB139, and E3::mtrA to thiostrepton and vancomycin (Fig. 5 and Fig. S7). Vancomycin inhibits transpeptidation by inhibiting transpeptidases and binding to the terminal D-Ala-D-Ala residues of non-cross-linked peptides in the peptidoglycan network and in lipid II (Hubbard and Walsh 2003). Thiostrepton prevents protein synthesis on procaryotic ribosomes by inhibiting the GTPase activation of elongation factor. Thus, both antibiotics are related to the cell wall. When treated with thiostrepton (30 μg/mL), the survival percentage of S. erythraea NRRL2338, ΔmtrA, and E3 was approximately 63%, 26%, and 39% at 24 h, respectively. When treated with vancomycin (50 μg/mL), the survival percentage of S. erythraea NRRL2338, ΔmtrA, and E3 was approximately 83%, 47%, and 68% at 24 h, respectively. S. erythraea ΔmtrA and E3 were both more susceptible to thiostrepton and vancomycin than S. erythraea NRRL2338. S. erythraea E3::mtrA was slightly more resistant to thiostrepton and vancomycin than S. erythraea E3 (Fig. 5 and Fig. S7), and its antibiotic resistance was more similar to that of S. erythraea NRRL2338. The effects of the plasmid were shown to be negligible. These results demonstrate that MtrA has a clear effect on antibiotic resistance in S. erythraea NRRL2338.

Effects of MtrA on antibiotic resistance percentage survival of Saccharopolyspora erythraea NRRL2338, ΔmtrA, E3, and E3::mtrA after treatment with 30 μg/mL thiostrepton and 50 μg/mL vancomycin for 24 h. Error bars indicate the SD of three independent experiments
Effects of MtrA on cell osmoprotection
To investigate the effects of MtrA on osmoprotection, S. erythraea NRRL2338 was subjected to high osmotic pressure. As shown in Fig. 6a, the transcriptional level of mepA increased by approximately 2-fold at 90 min compared to 0 min and was then recovered at 180 min, suggesting that mepA transcription responds to osmotic changes in S. erythraea NRRL2338. However, the transcriptional level of mepA in S. erythraea E3 showed no obvious changes due to high osmotic pressure (Fig. 6b), indicating that S. erythraea E3 had lost the MtrA-mediated regulation of mepA in response to osmotic stress. These results were consistent with the loss of mepA DNA-binding activity in MtrAdel.

Effects of MtrA on cell osmoprotection. amepA transcription levels in Saccharopolyspora erythraea NRRL2338 at 0 min, 90 min, and 180 min after increased osmotic pressure, normalized using the internal 16S rRNA gene. The fold change in mepA expression level at 0 min was set to 1.0. Error bars indicate the SD of three independent experiments. bmepA transcription levels in S. erythraea E3 were measured at 0 min, 90 min, and 180 min after increased osmotic pressure, normalized using the internal 16S rRNA gene. c Representative measurements of the DiOC2(3) fluorescent probe absorbed by S. erythraea NRRL2338, ΔmtrA, E3, and E3::mtrA cells obtained using BD FACSCalibur. d The FITC geometric means were produced using FlowJo software. Error bars indicate the SD of three independent experiments
To a certain extent, osmosis represents the transport of solvent molecules. To determine whether MtrA was involved in diffusion and transport, DiOC2(3) fluorescent probe assays were performed. DiOC2(3) is a lipophilic fluorescent dye used to label cell membranes as its fluorescence intensity increases when bound to the cell membrane,. The fluorescence intensity of S. erythraea ΔmtrA and E3 was stronger than that of S. erythraea NRRL2338 (Fig. 6c, d), indicating that more DiOC2(3) fluorescent probes were bound to the cell membrane. The fluorescence intensity of S. erythraea E3::mtrA was weaker than of S. erythraea E3, and similar to the intensity of S. erythraea NRRL2338. The effects of the plasmid were found to be negligible (Fig. S8). MtrA may therefore affect diffusion and transport, and thereby improve the transport of erythromycin in high erythromycin-producing strains.
Discussion
MtrAB is one of the major two-component signal transduction pathways through which Actinobacteria sense and respond to their environment in order to survive. MtrAB is involved in cell cycle progression, cell morphology, antibiotic resistance, and osmoprotection (Fol et al. 2006; Li et al. 2010; Moker et al. 2007; Nguyen et al. 2010). Previously, we found that MtrA had a two amino acid (H197 and V198) deletion in erythromycin-overproducing S. erythraea E3 compared with low-producing S. erythraea NRRL2338, indicating that MtrA might be associated with erythromycin production (Li et al. 2013). In this study, we investigated the effects of mtrA deletion and the deletion of the two MtrA amino acids on erythromycin production, antibiotic susceptibility, osmoprotection, and transport of substances. The MtrAdel mutant and ΔmtrA affected various physiological functions, enhancing antibiotic resistance, deregulating osmoprotection, and improving the transport of substances by reversing the inhibition of mepA expression by MtrA. The mepA gene is predicted to encode a protein with an extracytoplasmic metallopeptidase domain belonging to the M23B family (Rawlings et al. 2002), which could target cell wall peptides (Moker et al. 2004). An obvious relationship between MtrA and the expression of the ery cluster has been observed in S. erythraea. However, the molecular mechanisms underlying the effect of MtrA on erythromycin production remains unknown. MtrA has been reported to directly regulate the production of secondary metabolites in Streptomyces species (Som et al. 2017b), and can bind to sites spanning more than 70% of the biosynthetic gene clusters in S. coelicolor and S. venezuelae to activate antibiotic production. For example, in S. venezuelae MtrA can bind to the intergenic region between the cmlF and cmlN genes, activating the expression of cmlN which encodes an efflux permease to transport chloramphenicol (Fernandez-Martinez et al. 2014). These observations demonstrate that MtrA plays an important role in the biosynthesis of secondary metabolites in Streptomyces (Som et al. 2017a). However, there is no evidence that MtrA directly regulates the expression of the erythromycin biosynthesis gene cluster in S. erythraea, and exactly how MtrAdel increases erythromycin production remains to be elucidated.
In summary, our results demonstrated that MtrA is an important pleiotropic regulator of secondary metabolism, antibiotic susceptibility, and osmoprotection in S. erythraea. Inactivation of MtrA affected various physiological functions, increasing erythromycin biosynthesis, enhancing antibiotic resistance, deregulating osmoprotection, and improving transport of substances. Future studies linking the molecular effects of MtrA to these phenotypes will improve our understanding of the MtrA-MtrB two-component regulatory system in Actinobacteria.
References
Anderson TB, Brian P, Champness WC (2001) Genetic and transcriptional analysis of absA, an antibiotic gene cluster-linked two-component system that regulates multiple antibiotics in Streptomyces coelicolor. Mol Microbiol 39(3):553–566. https://doi.org/10.1046/j.1365-2958.2001.02240.x
Brocker M, Bott M (2006) Evidence for activator and repressor functions of the response regulator MtrA from Corynebacterium glutamicum. FEMS Microbiol Lett 264(2):205–212. https://doi.org/10.1111/j.1574-6968.2006.00456.x
Deretic V, Leveau JH, Mohr CD, Hibler NS (1992) In vitro phosphorylation of AlgR, a regulator of mucoidy in Pseudomonas aeruginosa, by a histidine protein kinase and effects of small phospho-donor molecules. Mol Microbiol 6(19):2761–2767
Fernandez-Martinez LT, Borsetto C, Gomez-Escribano JP, Bibb MJ, Al-Bassam MM, Chandra G, Bibb MJ (2014) New insights into chloramphenicol biosynthesis in Streptomyces venezuelae ATCC 10712. Antimicrob Agents Chemother 58(12):7441–7450. https://doi.org/10.1128/Aac.04272-14
Fol M, Chauhan A, Nair NK, Maloney E, Moomey M, Jagannath C, Madiraju MVVS, Rajagopalan M (2006) Modulation of Mycobacterium tuberculosis proliferation by MtrA, an essential two-component response regulator. Mol Microbiol 60(3):643–657. https://doi.org/10.1111/j.1365-2958.2006.05137.x
Hoskisson PA, Hutchings MI (2006) MtrAB-LpqB: a conserved threecomponent system in actinobacteria? Trends Microbiol 14(10):444–449. https://doi.org/10.1016/j.tim.2006.08.005
Hubbard BK, Walsh CT (2003) Vancomycin assembly: nature’s way. Angew Chem Int Ed 42(7):730–765. https://doi.org/10.1002/anie.200390202
Hutchings MI (2007) Unusual two-component signal transduction pathways in the actinobacteria. Adv Appl Microbiol 61:1–26. https://doi.org/10.1016/S0065-2164(06)61001-0
Hutchings MI, Hong HJ, Buttner MJ (2006) The vancomycin resistance VanRS two-component signal transduction system of Streptomyces coelicolor. Mol Microbiol 59(3):923–935. https://doi.org/10.1111/j.1365-2958.2005.04953.x
Laub MT, Goulian M (2007) Specificity in two-component signal transduction pathways. Annu Rev Genet 41:121–145. https://doi.org/10.1146/annurev.genet.41.042007.170548
Li YQ, Zeng JM, Zhang H, He ZG (2010) The characterization of conserved binding motifs and potential target genes for M. tuberculosis MtrAB reveals a link between the two-component system and the drug resistance of M. smegmatis. Bmc Microbiology 10 doi:Artn 242 https://doi.org/10.1186/1471-2180-10-242
Li YY, Chang X, Yu WB, Li H, Ye ZQ, Yu H, Liu BH, Zhang Y, Zhang SL, Ye BC, Li YX (2013) Systems perspectives on erythromycin biosynthesis by comparative genomic and transcriptomic analyses of S-erythraea E3 and NRRL23338 strains. Bmc Genomics 14 doi:Artn 523 https://doi.org/10.1186/1471-2164-14-523, 523
Liao CH, Yao LL, Ye BC (2014) Three genes encoding citrate synthases in Saccharopolyspora erythraea are regulated by the global nutrient-sensing regulators GlnR, DasR, and CRP. Mol Microbiol 94(5):1065–1084. https://doi.org/10.1111/mmi.12818
Liu Y, Wei WP, Ye BC (2018) High GC content Cas9-mediated genome-editing and biosynthetic gene cluster activation in Saccharopolyspora erythraea. ACS Synth Biol 7(5):1338–1348. https://doi.org/10.1021/acssynbio.7b00448
Lucana DOD, Zou PJ, Nierhaus M, Schrempf H (2005) Identification of a novel two-component system SenS/SenR modulating the production of the catalase-peroxidase CpeB and the haem-binding protein hbpS in Streptomyces reticuli. Microbiol-Sgm 151:3603–3614. https://doi.org/10.1099/mic.0.28298-0
Martinez-Hackert E, Stock AM (1997) The DNA-binding domain of OmpR: crystal structures of a winged helix transcription factor. Structure 5(1):109–124
Moker N, Brocker M, Schaffer S, Kramer R, Morbach S, Bott M (2004) Deletion of the genes encoding the MtrA-MtrB two-component system of Corynebacterium glutamicum has a strong influence on cell morphology, antibiotics susceptibility and expression of genes involved in osmoprotection. Mol Microbiol 54(2):420–438. https://doi.org/10.1111/j.1365-2958.2004.04249.x
Moker N, Kramer J, Unden G, Kramer R, Morbach S (2007) In vitro analysis of the two-component system MtrB-MtrA from Corynebacterium glutamicum. J Bacteriol 189(9):3645–3649. https://doi.org/10.1128/Jb.01920-06
Nguyen HT, Wolff KA, Cartabuke RH, Ogwang S, Nguyen L (2010) A lipoprotein modulates activity of the MtrAB two-component system to provide intrinsic multidrug resistance, cytokinetic control and cell wall homeostasis in Mycobacterium. Mol Microbiol 76(2):348–364. https://doi.org/10.1111/j.1365-2958.2010.07110.x
Paget MSB, Chamberlin L, Atrih A, Foster SJ, Buttner MJ (1999) Evidence that the extracytoplasmic function sigma factor sigma(E) is required for normal cell wall structure in Streptomyces coelicolor A3(2). J Bacteriol 181(1):204–211
Papandreou I, Powell A, Lim AL, Denko N (2005) Cellular reaction to hypoxia: sensing and responding to an adverse environment. Mutat Res 569(1-2):87–100. https://doi.org/10.1016/j.mrfmmm.2004.06.054
Rajagopalan M, Dziedzic R, Al Zayer M, Stankowska D, Ouimet MC, Bastedo DP, Marczynski GT, Madiraju MV (2010) Mycobacterium tuberculosis Origin of replication and the promoter for immunodominant secreted antigen 85B are the targets of MtrA, the essential response regulator. J BiolChem 285(21):15816–15827. https://doi.org/10.1074/jbc.M109.040097
Rawlings ND, O'Brien E, Barrett AJ (2002) MEROPS: the protease database. Nucleic Acids Res 30(1):343–346. https://doi.org/10.1093/nar/30.1.343
Som NF, Heine D, Holmes N, Knowles F, Chandra G, Seipke RF, Hoskisson PA, Wilkinson B, Hutchings MI (2017a) The MtrAB two-component system controls antibiotic production in Streptomyces coelicolor A3(2). Microbiol-Sgm 163(10):1415–1419. https://doi.org/10.1099/mic.0.000524
Som NF, Heine D, Holmes NA, Munnoch JT, Chandra G, Seipke RF, Hoskisson PA, Wilkinson B, Hutchings MI (2017b) The conserved actinobacterial two-component system MtrAB coordinates chloramphenicol production with sporulation in Streptomyces venezuelae NRRL B-65442. Front Microbiol 8 doi:ARTN 1145 https://doi.org/10.3389/fmicb.2017.01145
Staunton J, Wilkinson B (1997) Biosynthesis of erythromycin and rapamycin. Chem Rev 97(7):2611–2630
Stock AM, Robinson VL, Goudreau PN (2000) Two-component signal transduction. Annu Rev Biochem 69:183–215. https://doi.org/10.1146/annurev.biochem.69.1.183
Via LE, Curcic R, Mudd MH, Dhandayuthapani S, Ulmer RJ, Deretic V (1996) Elements of signal transduction in Mycobacterium tuberculosis: in vitro phosphorylation and in vivo expression of the response regulator MtrA. J Bacteriol 178(11):3314–3321
Yao LL, Liao CH, Huang G, Zhou Y, Rigali S, Zhang BC, Ye BC (2014) GlnR-mediated regulation of nitrogen metabolism in the actinomycete Saccharopolyspora erythraea. Appl Microbiol Biotechnol 98(18):7935–7948. https://doi.org/10.1007/s00253-014-5878-1
Zahrt TC, Deretic V (2000) An essential two-component signal transduction system in Mycobacterium tuberculosis. J Bacteriol 182(13):3832–3838. https://doi.org/10.1128/Jb.182.13.3832-3838.2000
Zhang PP, Wu LL, Zhu YP, Liu M, Wang YM, Cao GX, Chen XL, Tao MF, Pang XH (2017) Deletion of MtrA inhibits cellular development of Streptomyces coelicolor and alters expression of developmental regulatory genes. Front Microbiol 8 doi:ARTN 2013 https://doi.org/10.3389/fmicb.2017.02013
Funding
This study was supported by the National Natural Science Foundation of China (No. 31730004 and 21575089).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
This article does not contain any studies with human participants or animals performed by any of the authors.
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(PDF 665 kb)
Rights and permissions
About this article

Cite this article
Pan, Q., Tong, Y., Han, YJ. et al. Two amino acids missing of MtrA resulted in increased erythromycin level and altered phenotypes in Saccharopolyspora erythraea. Appl Microbiol Biotechnol 103, 4539–4548 (2019). https://doi.org/10.1007/s00253-019-09825-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-019-09825-9