
Abstract
Expression of recombinant proteins in Escherichia coli often requires use of inducible promoters to shorten the lag phase and improve protein productivity and final protein titer. Synthetic molecules that cannot be metabolized by E. coli, such as isopropyl thiogalactopyranoside (IPTG), have been frequently used to trigger the protein expression during early exponential growth phase. This practice has many drawbacks, including high cost and toxicity of IPTG, complex operating procedure, and non-uniform protein expression pattern (some cells in the population do not express recombinant proteins). A few auto-inducible protein expression systems have been developed recently to overcome some of these limitations, but they required use of an additional plasmid or presence of large (a few kilobases) DNA part to be functional, making plasmid construction to be difficult, especially when multiple genes need to be expressed. In this study, by using RNA sequencing, we identified a short, endogenous promoter (PthrC) that can be auto-induced during early exponential growth phase, and improved its performance by use of native and mutated regulatory elements. We found that the developed mutants of PthrC drove uniform protein expression—close to 100% of cells were fluorescent when green fluorescence protein was used as target protein—and cells carrying them could achieve much higher cell density than those with T7 promoter (PT7), a commonly used inducible promoter. In terms of promoter strength (product protein quantity per cell), the developed promoter mutants can cover a range of strength, from 30 to 150% of maximal strength of PT7. One strong mutant (PthrC3_8) was found to work well at a large range of temperature (22, 30, 37 °C) and in various media, and was also confirmed to cause less stress to host cell than PT7 when they were used to express a toxic protein. We foresee that PthrC3 and its mutants will be useful genetic parts for various applications including metabolic engineering and biocatalysis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Inducible promoter has been proven to be crucial in expressing recombinant proteins in bacterium, especially when the protein product poses stresses on the host (Nocadello and Swennen 2012; Briand et al. 2016). To date, isopropyl thiogalactopyranoside (IPTG) has been the most popular inducer, and has been used in laboratory-scale fermentation to control a large variety of promoters that are based on lac regulon (de Boer et al. 1983; Amann and Brosius 1985; Hansen et al. 1998). IPTG, however, is not an ideal inducer for industrial fermentation, because of its high cost and toxicity to some commonly used bacterium hosts, such as Escherichia coli (Nocadello and Swennen 2012; Briand et al. 2016).
Although there are many IPTG-independent inducible promoters for E. coli—one of the most commonly used bacterium host—many of them still use relatively expensive chemicals as inducers, e.g., L-arabinose (Guzman et al. 1995), tetracycline analogue (Skerra 1994), and uric acid (Liang et al. 2015). In addition, most, if not all, of such systems are based on use of repressor, which requires expression of recombinant repressor protein to reduce basal expression level, and thus increases size of plasmid or even demands use of an additional plasmid.
Recently, a few auto-inducible expression systems have been developed for heterologous protein expression, which did not need addition of any inducer. For example, Nocadello and Swennen developed a quorum sensing-based system that only induced target protein expression when cell density passed a threshold (Nocadello and Swennen 2012); Briand et al. accidently identified a novel auto-induction mechanism that has yet been fully studied, and used it to build an auto-inducible protein expression system (Briand et al. 2016). These methods also required maintaining an additional plasmid and/or expressing heterologous proteins as part of the expression system, which burden the host cell and make plasmid construction to be more difficult.
Theoretically, one may isolate an E. coli endogenous promoter that is tightly controlled and would only be turned on when cells reach a specific growth phase, or when a cheap chemical is added. Such promoter would be desired, because (1) it would solely rely on endogenous transcription machinery and thus minimize the burden on the host cell, and (2) its length would be short—most E. coli promoters are less than 400 bp including core regulatory regions—reducing the expression vector size.
Materials and methods
Bacterial strains and culture conditions
E. coli BL21 (DE3) (New England Biolabs) was used for expressing green fluorescent protein (GFP). To prepare the seed culture of E. coli, a colony of the E. coli BL21 (DE3) was inoculated into 10 mL of Luria Broth (LB) medium (Biomed Diagnostics Pte Lt) containing 50 μg/mL spectinomycin (Sigma) and grown at 37 °C/200 r.p.m. overnight. Grown cell suspension with optical density at 600 nm (OD600) was around 5–6 and inoculated (1%, v/v) into 10 mL of chemical defined medium (13.3 g/L KH2PO4, 4 g/L (NH4)2HPO4, 1.7 g/L citric acid, 0.0084 g/L EDTA, 0.0025 g/L CoCl2, 0.015 g/L MnCl2, 0.0015 g/L CuCl2, 0.003 g/L H3BO3, 0.0025 g/L Na2MoO4, 0.008 g/L Zn(CH3COO)2), 0.06 g/L Fe(III) citrate, 0.0045 g/L thiamine, 1.3 g/L MgSO4, pH 7.0) containing 50 μg/mL spectinomycin, 1 g/L yeast extract (BD), and indicated carbon source. At early exponential phase (OD600 between 0.5 and 0.8), E. coli strains were induced by the addition of glucose if such induction was needed. Lysogeny Broth (10 g/L tryptone, 5 g/L yeast extract, 5 g/L NaCl) and Terrific Broth (24 g/L yeast extract, 20 g/L tryptone, 5 g/L glycerol, 2.3 g/L KH2PO4, 12.6 g/L K2HPO4) were used to study the effects of growth media on performance of the promoters.
RNA extraction and sequencing
The E. coli BL21 (DE3) was inoculated into 5 mL of LB medium and grown overnight at 37 °C/200 r.p.m. Overnight, cells were inoculated (1%, v/v) into 10 mL of chemically defined medium with 20 g/L xylose (Sigma). The first batch of samples were collected when OD600 reached around 0.5 (Time point 1), then final concentration of 40 g/L of glucose (Sigma) was added immediately and the second batch of samples were collected after 45-min cultivation (Time point 2).
Cells were harvested by centrifugation at 12,000×g for 2 min at room temperature. The cell pellets were immediately placed on ice. Total RNA was extracted by using the GeneJET RNA purification kit (Thermo Fisher Scientific) according to the manufacturer’s protocol. Contaminating DNA was removed by column and in-solution DNase digestion with an RNase-free DNase treatment (Promega) according to the manufacturer’s instructions.
The purity and concentration of the total RNA were determined by a Nano Drop 2000 spectrophotometer (Thermo Fisher Scientific). For all the samples, the A260/A280 ratios ranged 2.08–2.13 and the concentrations were 462.3–725.4 ng/μL. A total of six (Time point 1 and Time point 2 in triplicates) samples were submitted to AIT Novogene for library construction and sequencing. Raw sequencing data were assembled and analyzed by the company, which produced gene expression quantification results (Supplementary Table S1). Averaged read counts of the triplicates and the mean values were used to calculate the fold change.
Plasmid construction and E. coli transformation
In this study, constructed plasmids were summarized in Table 2 and the useful ones have been deposited into Addgene. Most plasmids were constructed by using a method that was not published when this manuscript was prepared, so the details of the plasmid construction procedure were not described in this manuscript. Since all the plasmid information have been provided and useful ones can be obtained from Addgene, we believe not including the plasmid construction procedure would not hinder reproducing this work or utilizing the promoter variants created in this work.
Each of the constructed plasmids was introduced into BL21 (DE3) chemically competent cell (New England Biolabs) via the heat-shock procedure according to the manufacturer’s instruction. All the plasmids contain spectinomycin resistance gene, so 50 μg/mL spectinomycin was used in LB agar plate to isolate the E. coli strains that carry the desired plasmids.
Quantification of fluorescence signal, cell density, and DXS cellular content
Cell growth was monitored by OD600 on a UV spectrophotometer (Novaspec III, SciMed (Asia) Pte Ltd). For quantifying fluorescence signal, 100 μL cell suspension was loaded into a well in 96-well optical plate and monitored by microplate reader, Infinite 200PRO (Tecan) with excitation wavelength at 483 nm, emission wavelength at 535 nm, and gain at 74 to reach optimal results. Specific fluorescence signal was computed by dividing fluorescence signal by OD600. Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) was used to visualize DXS expressed by the cells. Stain-Free gel (4–15% Mini-PROTEAN® TGX Stain-Free™ Protein Gel, Bio-Rad) was used and the proteins were visualized by using Gel Doc™ EZ Gel Documentation System (Bio-Rad). Samples loaded into each lane were derived from a fixed quantity of cell biomass.
Fluorescence microscopy
The samples taken from the batch culture were diluted to the concentration of 2 (OD600) using centrifugation. Zero-point, 3 μL of the concentration sample was loaded on a glass slide and covered by a slip. A fluorescence microscope (Ts2R-FL, Nikon) with × 40, phase contrast objective lens, and a GFP filter cube was used to acquire phase contrast image and fluorescence image (auto-exposure was used), which were overlaid by using software ImageJ. Brightness and contrast of the images were adjusted for all pixels if needed.
Results
RNA sequencing to identify promoter candidates
In this study, we started a search for short, auto-inducible promoters by using RNA sequencing, which could reliably evaluate transcription levels of thousands of endogenous genes at different conditions. Since E. coli cells were induced in most protein expression experiments when its OD600 was between 0.5 and 1, we collected samples of E. coli (BL21, DE3) in triplicates from a typical shake flask culture during this OD range, and sampled the same cultures again after 45 mins. RNA from the cells collected at the two conditions were sequenced, and the genes that were highly upregulated during the 45 mins were selected as candidates for the promoter search. To increase the likelihood that substantial transcription changes would be observed, we added 40 g/L of glucose to the cultures immediately after we took the first samples (20 g/L of xylose was used as carbon source upon cell inoculation). As a result, the promoters we searched for were either responsive to natural growth phase transition, or addition of glucose—a cheap chemical frequently used as sole carbon source in E. coli culture. Both induction modes would be desired. In total, we have detected transcripts from 2352 genes that had at least 500 read counts (Supplementary Table S1), and selected Top 5 of the highly upregulated genes (Table 1) as candidates for promoter characterization.
Test of top promoter candidates
For each candidate, we cloned its core promoter based on annotations that were available on ecocyc.org, and inserted each candidate promoter in front of a green fluorescence protein (GFP)-encoding gene on a high-copy number plasmid. E. coli (BL21, DE3) cells harboring these plasmids were characterized at the same condition as the RNA sequencing experiment. We measured optical density (OD, representing biomass concentration) and fluorescence signal, and used them to compute specific fluorescence signal (fluorescence signal per OD), which indicates strength of the candidate promoters. Out of five candidate promoters, only PthrC were upregulated over time (Fig. 1), so we selected PthrC for further optimization.

Specific fluorescence signal of promoter candidates. Mean and standard error are used in the bar chart (n = 3)
Using additional regulatory elements to improve PthrC
We first removed glucose induction (xylose was used as sole carbon source) to test if the transcription upregulation was triggered by glucose induction or by growth phase transition. At this condition, PthrC was upregulated to a larger extent (this phenomenon is termed as better promoter regulatability in the following text, Fig. 2), indicating that growth phase transition alone was able to upregulate the promoter. We further created five variants of PthrC containing various combinations of adjacent regulatory elements, and characterized them at the same condition.

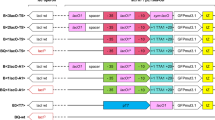
Characterization of variants of PthrC and comparison with PT7 in the xylose medium. Pthr, core promoter of thr operon; thrL, RBS and coding gene of thrL; TthrL, terminator following thrL; Down, the downstream sequence of TthrL containing DksA-ppGpp; Up, the upstream sequence of Pthr containing promoter and coding gene of yjtD. GFP, RBS and coding gene of enhanced green fluorescence protein. SFS, specific fluorescence signal; the basis is that at 0 h. Cells with T7 promoter were induced with 1 mM IPTG in early exponential growth phase. Mean and standard error are plotted in the bar charts (n = 3)
In E. coli chromosome, PthrC is followed by a short, threonine/isoleucine-rich leader peptide (encoded by thrL), a terminator (TthrL), and threonine biosynthetic genes (Gardner 1979, 1982). When TthrL was added to the tail of PthrC (new promoter named as PthrC5, Fig. 2c), expression of GFP was almost completely abolished, suggesting that this terminator was very effective. When the leader peptide and terminator were together inserted after PthrC, the new promoter (named as PthrC3) restored 20% of the maximal strength of PthrC, and, more importantly, PthrC3 had better promoter regulatability than PthrC. This observation is consistent with the early characterization of the thr operon in the 1970s and 1980s (Gardner 1979, 1982), which revealed that thrL-coding mRNA can form a stem-loop structure with TthrL to prevent formation of the terminator structure, when ribosome was paused at thrL due to low availability of threonine and isoleucine. This mechanism is employed by E. coli to dynamically regulate synthesis of threonine. We hypothesize that when cells entered exponential growth phase, intracellular availability of threonine and isoleucine dropped, and triggered the aforementioned mechanism, resulting in reduction in effectiveness of TthrL and subsequent increase in transcription of target gene (gfp).
This hypothesis was supported by inserting thrL alone after PthrC (new promoter termed as PthrC4), which did not improve the promoter regulatability as much as using both thrL and TthrL. PthrC4 had much higher maximal strength (that at 24 h) than PthrC, which might be due to improved access to ribosome when ribosome is paused at adjacent thrL region.
The other two variants created were PthrC1 and PthrC2, which compared to PthrC3 had additional upstream sequence and additional upstream and downstream sequence respectively. They did not have better regulatability, and were much longer, than PthrC3, so they were not further studied.
Comparison of PthrC3 with PT7
PthrC3 was the best one among the variants in terms of promoter regulatability, and was compared under the same growth condition with T7 promoter (PT7), one of the most frequently used IPTG-inducible promoters. PT7-carrying cells were induced by using 1 mM in early exponential phase in all experiments. PthrC3 had much lower basal expression level (expression level in this manuscript all refers to specific protein expression level, i.e., recombinant protein quantity per cell) than PT7, and had ~ 60% of regulatability of PT7 (Fig. 2), which made it a good promoter for expressing highly toxic proteins.
A drawback of our system at this point was use of xylose, which is relatively expensive, so we characterized performance of PthrC3 in the same chemically defined medium with glucose or glycerol—both of which are cheap substrates—as sole carbon source, and compared its performance with that of PT7 at the same conditions (Fig. 3a). The glycerol medium was found to be even better than the xylose one—both PthrC3 and PT7 showed better regulatability in the glycerol medium (Fig. 3b). The regulatability of PthrC and PT7 at 24 h was increased from 3.1 and 5.1 in the xylose medium to 5.2 and 8.2 in the glycerol medium respectively (regulability was defined as the ratio of specific fluorescence signal at 24 h to that at 0 h).

Comparison between PthrC3 and T7 promoter in glucose and glycerol medium, in terms of promoter strength (a) and regulatability (b). Percentage of fluorescent cells in the population in different media (c) and representative images from fluorescence microscopy (d) were also presented. Mean and standard error are plotted in the bar charts (n = 3)
We further compared PthrC and PT7 in terms of cell-to-cell variation of protein expression. At 24 h after induction, we observed fluorescence of single cells under a fluorescence microscope. Though literature reported that IPTG-induced uniform protein expression (Khlebnikov and Keasling 2002), we found that the conclusion depended on carbon source. When xylose was used as carbon source, 85% of cells that contained PT7 were fluorescent (Fig. 3c). The percentage, however, dropped sharply to 41 and 27% respectively when glucose and glycerol were used as carbon source. In contrast, close to 100% of the cells that contained PthrC3 were fluorescent across all the conditions (Fig. 3c), which should lead to smaller metabolic burden per cell as it is shared by all the cells.
Mutagenesis of PthrC3 created more promoter variants
We attempted to improve PthrC3’s regulatability and increase its maximal strength by mutating the first stem-loop region (the one with a larger loop, Fig. 4b), which once formed can repress formation of the second stem-loop that is part of TthrL (Fig. 4b). We increased the stem length of the first stem-loop by mutating one or more of the four adjacent nucleotides (…ACGGTGCGGGCTGA…). To reduce reagent cost, we used a degenerated oligo (…MMGGTGCGGGCTKW…) to create a small library (2^4 = 16), and randomly characterized 32 colonies (two times the library size). We obtained a number of promoters that were stronger than PT7 at 24 h and had regulatability similar to that of PthrC3 (Fig. 4c). We ranked the colonies based on specific fluorescence signal at 24 h and sequenced the Top 3 variants (PthrC3_8, PthrC3_7, and PthrC3_25), all of which had a mutation in the larger loop region (Fig. 4b). The importance of these mutations was expected, since they can not only improve stability of the first stem-loop structure, but also decrease that of the second one—these mutations affect stem formation of both stem-loops. As a comparison, the mutation in the smaller loop region only affected stem formation of the first stem-loop, and did not have substantial effect on behavior of the promoter based on comparison between PthrC3_8 and PthrC3_25 (Fig. 4c).

Mutagenesis of PthrC3 generated useful promoter variants. a Cells with PthrC3 or one of the Top 3 promoter variants achieved much higher cell density than that with PT7 at 24 h. b Possible secondary structures of PthrC3 and Top 3 variants. The first plot shows four mutations that might be introduced to PthrC3 by using the degenerated primers. The other three plots in b are based on sequencing results of the Top 3 variants. Each plot in b includes two mutually exclusive stem-loops. c Specific fluorescence signal of 32 strains that have been screened. d Temporal profile of cell density of strains expressing GFP by using PthrC3_8 or T7P. e Temporal profile of volumetric fluorescence signal of strains expressing GFP by using PthrC3_8 or T7P. Mean and standard error are plotted in a, d, e (n = 3). There is no replicate in the screening experiment whose results are shown in c
Although the Top 3 strains had much higher basal expression level (at 0 h) than the strain carrying PT7 (Fig. 3a and Fig. 4c), they achieved much higher cell density at 24 h (Fig. 4a), suggesting that the high basal expression level did not pose high metabolic burden on the host cells, possibly because strains carrying PthrC3 variants had uniform protein expression and well shared the burden of recombinant protein production among the whole population.
We further compared the strain harboring the strongest promoter mutant (PthrC3_8) with that carrying PT7 over 24 h, in terms of cell density and protein titer (indicated by fluorescence signal from a given volume of culture). The strain carrying PthrC3_8 started to have substantially higher cell density and protein titer after 4 h (Fig. 4d–e).
Effects of growth medium and temperature on the promoters
As recombinant proteins may be produced in different media and at various temperatures, we studied the effects of growth medium and temperature on expression level and regulatability of the promoters, to determine if they are generally applicable. We changed one parameter at a time. First, we fixed the medium as K3 medium (the chemically defined medium we used in earlier parts of the study), and tested the effects of temperature. PthrC3 behaved similarly at 30 and 37 °C, but failed to work at 22 °C (Fig. 5a). The mutant promoter (PthrC3_8) worked at all temperatures, and even had better regulatability at 30 than 37 °C (Fig. 5a). A simple theory to explain the results is that the terminator structure became more stable at lower temperature and cannot be melted by the competing structure even when ribosome paused in the region. The mutations PthrC3_8 had decreased Gibbs free energy of the structure competing with the terminator structure, enabling it to melt the terminator structure even at lower temperature. Since lowering temperature also slowed down cell growth and altered threonine synthesis, there may be other mechanisms controlling performance of these promoters.

Effects of culture temperature (a) and medium (b) on performance of the promoters. Mean and standard error are plotted (n = 3)
We next fixed the culture temperature as 37 °C and studied the effects of medium. We found the protein expression level achieved by using PthrC3 was much lower in rich medium (Lysogeny Broth or Terrific Broth) than the chemically defined medium. Because rich media contained substantial amount of threonine and isoleucine, one would expect lower frequency of ribosome pausing in the region when rich media were used, which should result in the observed lower expression level (the terminator structure would only be melted to allow RNA polymerase to continue when ribosome was paused in the region). Interestingly, the expression level achieved by using PthrC3_8 was, however, not affected by the medium, suggesting that the terminator structure can be melted even when the frequency of ribosome pausing was reduced in the rich media, possibly due to the decreased Gibbs free energy of the structure competing with the terminator structure.
Together, the PthrC3 mutant (PthrC3_8) was insensitive to temperature and medium change, and constantly had high expression level and good regulatability in all the tested conditions.
Expression of a toxic protein by using PthrC3_8
To further test if PthrC3_8 was generally applicable, we used it to express a protein that is toxic to the host cells and compared its performance with PT7’s. The model protein was deoxy-xylulose phosphate synthase (DXS), which was reported to slow down cell growth when overexpressed (Zhou et al. 2012). We constructed the plasmids that can express DXS by using PthrC3_8 or PT7, and characterized the strains harboring the plasmids as well as the parental strain (BL21 [DE3]) in the chemically defined medium at 37 °C. We cultured the strain carrying PT7 at three conditions: no induction, induction in early log phase, and induction upon inoculation. The cells carrying PthrC3_8 had the same cell density as the parental strain, which was substantially (~2× in some cases) higher at both time points studied than that of cells having PT7 and being induced (Fig. 6b). We analyzed cellular DXS content (the quantity of DXS per cell) by using SDS-PAGE, which revealed that substantial amount of DXS was produced when PthrC3_8 was used (Fig. 6a). Although cellular DXS content was lower when PthrC3_8 was used than that when PT7 was used and induced in early log phase, the titer of DXS (the quantity of DXS per culture volume) could be higher when PthrC3_8 was used because cells carrying it have much higher cell density. Considering using PthrC3_8 does not require monitoring cell density and manual induction of the cell culture, we foresee that PthrC3_8 will be a useful genetic part for recombinant protein expression and other related applications (Table 2).

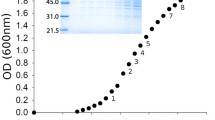
a SDS-PGAE analysis of E. coli strains expressing deoxy-xylulose phosphate synthase (DXS) by using PthrC3_8 or PT7. The samples were collected at 22 h. Lane information: 1: PthrC3_8-DXS; 2: T7P-DXS (induced in early exponential growth phase); 3: T7P-DXS (induced in early exponential growth phase); 4: T7P-DXS (no induction); 5: the parental strain; M: protein markers. The arrow indicated DXS protein. b Cell density of the strains used in this experiment. Mean and standard error are plotted (n = 3)
Discussion
In this study, we have, for the first time, identified a set of short (300 bp), auto-inducible promoters for expressing recombinant proteins in E. coli (Figs. 2 and 4). The promoters do not require expression of any repressor protein and maintaining any additional plasmid, which simplifies plasmid construction and reduces metabolic burden on the host cells. The promoters neither require any change in operating conditions during the fermentation, unlike temperature-inducible promoters (Qoronfleh et al. 1992), which are not suitable for large-scale fermentation as changing temperature of large reactors is slow and energy intensive.
The identified promoters should have broad applications in biotechnology and bioengineering. We have benchmarked our promoters with PT7, one of the most frequently used promoters in E. coli. PthrC3, a promoter variant in our collection, had the same basal expression level as PT7 and reached 50% of expression level of PT7 at the end of fermentation (24 h). This promoter can replace PT7 in metabolic engineering of E. coli, in which an intermediate expression level was often desired. PthrC3_8, a mutant of PthrC3, could reach ~1.5 fold higher expression level (Fig. 3a and Fig. 4c) and ~2 fold higher cell density than PT7 at the end of fermentation, making it to produce ~3 times as much protein as PT7 in a given volume. These data support that PthrC3_8 can replace PT7 for production of recombinant proteins for biocatalysis and other applications, in which the goal is to maximize recombinant protein quantity. Most of these promoter characterization works were done with a chemically defined medium that contained glycerol as sole carbon source. The low cost of this medium supports that our promoters might be used in industrial-scale fermentation.
A unique feature of the identified promoters is the uniform expression of proteins among the cell population, which usually cannot be achieved by using promoters induced by addition of chemicals, due to uncertainties in cellular uptake of the chemicals (Khlebnikov and Keasling 2002). We suspect that the auto-inducible promoters based on quorum sensing (Nocadello and Swennen 2012) may be subject to this problem as well, since they also involve transport of inducing-molecules across cellular membranes. Experiments are needed to confirm this hypothesis, as no such analysis was done at single cell level in the study. Such experiments can be done by using fluorescence microscopy, as in this study, or flow cytometry-based techniques (Alper et al. 2005). Having homogenous protein expression pattern is important, because it eliminates non-producers in the population and could help avoid over-burdening working cells.
Although transcription of E. coli thr operon was well characterized in biochemistry studies in the 1970s and 1980s (Gardner 1979, 1982), its promoter and regulatory elements have never been used for the purpose of expressing recombinant proteins in biotechnological applications. The promoter would never come to our attention, if we did not perform the global differential transcriptional analysis, which suggests: (a) there are many useful knowledges in classic biochemistry literature to be translated into valuable bioengineering tools, and (b) omics technologies can facilitate better utilization of these knowledges through unbiased discovery of lead candidates, especially as they become more accessible to more labs.
The way PthrC3 (and its derivatives) achieved regulatability is very different from that of currently used inducible promoters, which are mostly based on use of repressor to achieve low basal expression level. A follow-up study of our work could be further mutagenesis or de novo design of the regulatory elements of PthrC3, including the terminator and the leader peptide, to improve the promoter regulatability or change the induction timing, e.g., initiation of recombinant protein synthesis from mid-log phase or stationary phase. The regulatory elements could also be inserted after other constitutive promoters to test if they can be turned into auto-inducible ones, which would further expand the collection of the small, auto-inducible promoters for E. coli and other bacteria.
References
Alper H, Fischer C, Nevoigt E, Stephanopoulos G (2005) Tuning genetic control through promoter engineering. Proc Natl Acad Sci 102:12678–12683. https://doi.org/10.1073/pnas.0504604102
Amann E, Brosius J (1985) ATG vectors’ for regulated high-level expression of cloned genes in Escherichia coli. Gene 40:183–190. https://doi.org/10.1016/0378-1119(85)90041-1
Briand L, Marcion G, Kriznik A, Heydel JM, Artur Y, Garrido C, Seigneuric R, Neiers F (2016) A self-inducible heterologous protein expression system in Escherichia coli. Sci Rep 6:1–11. https://doi.org/10.1038/srep33037
de Boer HA, Comstock LJ, Vasser M (1983) The tac promoter: a functional hybrid derived from the trp and lac promoters. Proc Natl Acad Sci 80:21–25. https://doi.org/10.1073/pnas.80.1.21
Gardner J (1979) Regulation of the threonine operon: tandem threonine and isoleucine codons in the control region and translational control of transcription termination. Proc Natl Acad Sci 76:1706–1710. https://doi.org/10.1073/pnas.76.4.1706
Gardner JF (1982) Initiation, pausing, and termination of transcription in the threonine operon regulatory region of Escherichia coli. J Biol Chem 257:3896–3904
Guzman L-M, Belin D, Carson MJ, Beckwith J (1995) Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J Bacteriol 177:4121–4130. https://doi.org/10.1128/jb.177.14.4121-4130
Hansen LH, Knudsen S, Sørensen SJ (1998) The effect of the lacY gene on the induction of IPTG inducible promoters, studied in Escherichia Coli and Pseudomonas fluorescens. Curr Microbiol 36:341–347. https://doi.org/10.1007/s002849900320
Khlebnikov A, Keasling JD (2002) Effect of lacY expression on homogeneity of induction from the Ptac and Ptrc promoters by natural and synthetic inducers. Biotechnol Prog 18:672–674. https://doi.org/10.1021/bp010141k
Liang C, Xiong D, Zhang Y, Mu S, Tang SY (2015) Development of a novel uric-acid-responsive regulatory system in Escherichia coli. Appl Microbiol Biotechnol 99:2267–2275. https://doi.org/10.1007/s00253-014-6290-6
Nocadello S, Swennen EF (2012) The new pLAI (lux regulon based auto-inducible) expression system for recombinant protein production in Escherichia coli. Microb Cell Factories 11:1–10. https://doi.org/10.1186/1475-2859-11-3
Qoronfleh MW, Debouck C, Keller J (1992) Identification and characterization of novel low-temperature-inducible promoters of Escherichia coli. J Bacteriol 174:7902–7909. https://doi.org/10.1128/jb.174.24.7902-7909.1992
Skerra A (1994) Use of the tetracycline promoter for the tightly regulated production of a murine antibody fragment in Escherichia coli. Gene 151:131–135. https://doi.org/10.1016/0378-1119(94)90643-2
Zhou K, Zou R, Stephanopoulos G, Too HP (2012) Metabolite profiling identified methylerythritol cyclodiphosphate efflux as a limiting step in microbial isoprenoid production. PLoS One 7:e47513. https://doi.org/10.1371/journal.pone.0047513
Funding
This was funded by National University of Singapore Start-up Grant (R-279-000-452-133) and Ministry of Education Tier 1 Grants (R-279-000-478-112 and R-279-000-494-114). The study was done at the National University of Singapore.
Author information
Authors and Affiliations
Contributions
O.A. was an undergraduate student at Kingston University London and worked on this project during her research attachment at the National University of Singapore. O.A., H.L., and K.Z. conceived the project and designed the experiments. O.A., H.L., and X.Q.M. did the experiments. L.M.Y. supervised execution of some experiments and maintained many critical instruments for this project. O.A., H.L., and K.Z. analyzed the data and wrote the manuscript. All the authors have read the final version of this manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Informed consent
This article does not contain any studies with human participants or animals performed by any of the authors.
Electronic supplementary material
ESM 1
(PDF 4641 kb)
Rights and permissions
About this article

Cite this article
Anilionyte, O., Liang, H., Ma, X. et al. Short, auto-inducible promoters for well-controlled protein expression in Escherichia coli. Appl Microbiol Biotechnol 102, 7007–7015 (2018). https://doi.org/10.1007/s00253-018-9141-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-018-9141-z