
Abstract
Protein engineering has been a research hotspot to improve the catalytic efficiency of industrially important enzymes. In the present study, a novel computational strategy was developed to in silico screen mutants with enhanced binding interaction between enzyme and substrate as well as catalytic efficiency. Through homology modeling and molecular dynamics (MD) simulation, four key residues related to substrate binding were identified in the endo-polygalacturonase BiPG28A from Bispora sp. MEY-1. Further analyses of the conformation, hydrogen bond interactions, and binding free energy revealed that lysine at position 129 (subsite − 2) has the strongest affinity to substrate. Biochemical and calorimetry experiments confirmed the functional role of Lys129 in substrate binding through non-covalent interactions. The common role of Lys129 was also verified in another GH28 endo-polygalacturonase. Distinguished from other protein engineering strategies involving structure resolution and construction of certain enzymes, this computational strategy represents an insightful and efficient approach to develop a “designed” enzyme with significantly enhanced binding affinity and catalytic efficiency.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Biotechnology engineering towards enzyme performance is of great importance for enzyme commercialization. One bottleneck for industrial application of an enzyme candidate is low catalytic efficiency (Choi et al. 2015). A variety of approaches have been developed to improve the enzyme catalytic performance (Korendovych and DeGrado 2014; Daczkowski et al. 2015; Parashar and Satyanarayana 2016). Of them, rational design is based on the understanding of enzyme structure, function, catalytic mechanism, and catalytic residues, and has been used to identify important factors related to catalytic efficiency. During the catalytic process, a glycoside hydrolase (GH) generally experiences ligand binding to the active site, hydrolysis of the glycoside bond, and product release (Tu et al. 2015). Thus, protein engineering strategies involving in increased binding affinity and enhanced product release can result in higher catalytic efficiency (Tinberg et al. 2013; Comeau et al. 2016). Molecular dynamics (MD) simulation has been proposed as a useful tool to assess the enzyme structure and dynamics. In the assistance of MD, Cui et al. (2015) reconstructed a dehydrogenase with dual cofactor specificity for NADH and NADPH, and Bernardi et al. (2014) identified key contacts between the substrate and catalytic pocket of mannanase. However, this approach is less efficient to identify weak-point residues related to catalytic efficiency.
Endo-polygalacturonase (Endo-PG, EC 3.2.1.15) is the most extensively studied and well-characterized pectinase that catalyzes the cleavage of α-1,4-glycosidic bonds between the adjacent polygalacturonic acid (PGA) residues within the smooth pectin regions. It has been receiving much research interest and is often preferred for technical applications due to their capability of removing spoilage and decay of the processed foods (Tu et al. 2014). Most known polygalacturonases belong to the GH28 family (CAZy database; http://www.cazy.org/GH28.html), and approximately 200 of them have been characterized. Structure analysis of 10 polygalacturonases (http://www.rcsb.org) indicated that these enzymes share a conserved β-helix framework and some key residues (van Santen et al. 1999; Shimizu et al. 2002). For example, the conserved aspartate residues (Asp153, Asp173, and Asp174) play roles in the inverting catalytic mechanism, His223 maintains the proper ionization state of catalytic residues, and Arg256 and Lys258 are involved in substrate binding. These results provide valuable insights into the structure-function relationship of endo-PG, which might be used for the improvement of catalytic efficiency.
To acquire highly active endo-PG, mining new enzymes from natural source and structure-guided rational design are the most popular practices. By using these approaches, a polygalacturonase from Aspergillus niger was expressed in Pichia pastoris, the recombinant enzyme showed a maximum activity of 10,436 U/mL in the culture supernatant (Wang et al. 2017), and a mutant enzyme of endo-PG PG63 showed improved thermostability and catalytic efficiency (Tu et al. 2016a). In the present study, we propose a novel computational strategy combining substrate-enzyme docking and MD simulation to systematically screen mutants in silico. To validate this approach, the endo-PG BiPG28A from Bispora sp. MEY-1 was selected. After homology modeling, molecular docking, and simulated saturation mutagenesis, the residue at position 129 was found to play roles in substrate binding, in which substitution with lysine increased the affinity. This protein design was verified by bench works, and its common effect was also confirmed in another endo-PG.
Materials and methods
Cloning and expression of BiPG28A in Pichia pastoris
The genomic DNA and total RNA of Bispora sp. MEY-1 (CGMCC 2500; the China General Microbiological Culture Collection Center, Beijing, China) were extracted after 3 days growth in a medium with citrus pectin as the sole carbon source at 30 °C. Using the genomic DNA as template, the core region of Bipg28A was amplified by the degenerate primer set GH28nF and GH28nR specific for the fungal GH28 endo-PGs (Tu et al. 2013). The PCR products were purified, ligated with vector pEASY-T3, and then transformed into Escherichia coli Trans1-T1 for sequencing. The 5′ and 3′ flanking regions were then obtained using the fusion primer and nested integrated (FPNI)-PCR (Wang et al. 2011) with six nested insertion-specific primers USP 1–3 and DSP 1–3 (Table S1). The full-length gene was obtained by sequence assembly of the core and flanking regions.
The cDNA of strain MEY-1 was obtained by RT-PCR according to the protocol of Ace-α-TM kit (TOYOBO, Osaka, Japan), and the cDNA fragment coding for the mature BiPG28A without the putative signal peptide sequence was amplified with specific primers BiPG28A-F and BiPG28A-R (Table S1). The PCR products of the appropriate size were digested by EcoRI and NotI and then cloned into the vector pPIC9 in frame fusion of the α-factor signal peptide to construct the recombinant plasmid pPIC9-Bipg28A. The recombinant plasmid was linearized using SacI and transformed into P. pastoris GS115 competent cells by electroporation according to the manufacturer’s instructions (Invitrogen). The positive transformants were screened via enzymatic activity assay in shake tubes, and the transformant with the highest endo-PG activity was grown in a 1-L Erlenmeyer flask (Tu et al. 2013).
Enzyme purification
The induced culture supernatants were collected by centrifugation at 12000×g for 10 min at 4 °C, followed by concentration through a Viva flow 200 ultrafiltration membrane (cutoff 10 kDa; Vivascience, Germany) and dialysis in 10 mM citric acid-Na2HPO4 (buffer A; pH 6.5) at 4 °C overnight. The crude enzyme was loaded onto a HiTrap Q Sepharose XL 5 mL FPLC column (GE Healthcare, Sweden) equilibrated with buffer A. A linear gradient of NaCl (0–1.0 M) was used to elute the proteins. Fractions bearing endo-PG activity were pooled, dialyzed in buffer A, and concentrated by ultrafiltration at 4000×g for 40 min at 4 °C using an Amicon Ultra Centrifugal Filter Device PL-10 (Millipore). Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was carried out with the 5% stacking gel and 12% separation gel. The purified recombinant BiPG28A was deglycosylated by endo-β-N-acetylglucosaminidase H (Endo H) at 37 °C for 2 h according to the manufacturer’s instructions (New England BioLabs, Ipswich, MA). The deglycosylated enzyme was also analyzed by SDS-PAGE. Protein concentration was determined using the Bradford method with bovine serum albumin as the standard.
Biochemical characterization
The endo-PG activity was determined by using the 3,5-dinitrosalicylic acid (DNS) method with d-(+)-galacturonic acid as the standard. Reactions systems containing 900 μL of 3.3 mg/mL PGA in McIlvaine buffer (100 mM citric acid, 200 mM Na2HPO4, pH 3.5) and 100 μL of appropriately diluted enzyme solution were incubated at 70 °C for 10 min, followed by the addition of 1.5 mL DNS solution. After 5-min incubation at 100 °C in a boiling water bath, the reaction mixtures were cooled to room temperature, and the absorbance at 540 nm was measured. One unit of PG activity was defined as the amount of enzyme that released reducing sugars equivalent to 1 μmol of d-(+)-galacturonic acid per minute under standard conditions (pH 3.5, 70 °C, and 10 min). All reactions were performed in triplicate.
The pH-activity profile of purified recombinant BiPG28A was determined at 70 °C in McIlvaine buffer (pH 2.2 to 5.0) for 10 min. To estimate the pH stability, the enzyme was pre-incubated in McIlvaine buffer of various pH values (2.2–8.0) at 37 °C for 1 h, and the residual activities were measured under standard conditions as described above. The temperature-activity profile was determined at optimal pH for 10 min at 30–90 °C. For the thermostability assay, the enzyme was incubated at a temperature range of 40 to 90 °C for 30 min without a substrate, and the residual activities were then measured under standard conditions.
For kinetic assays, the BiPG28A activity was measured in McIlvaine buffer (pH 3.5) containing 0.1–5.0 mg/mL of PGA at 70 °C for 5 min. The kinetic parameters Km and Vmax were determined by fitting the Michaelis-Menten plot and using the GraphPad Prism software (La Jolla, CA).
Homology modeling and molecular docking
The homology modeling of BiPG28A was performed using the Discovery Studio 2017 (Accelrys Software) and with the X-ray structure of endo-PG from Fusarium moniliforme (1HG8, 53.1% identity; Federici et al. 2001) as the template. The structure was optimized (especially loops) for energy minimization to remove steric clashes. Enzyme-substrate docking was performed using the AutoDock Vina program (Trott and Olson 2010). The ligand coordinate (mol2 file) of tetragalacturonic acid (GalpA4) was prepared in the PRODRG server (http://davapc1.bioch.dundee.ac.uk/cgi-bin/prodrg). To encompass the entire substrate-binding pocket, the docking box was set to a size of 60 Å × 60 Å × 60 Å grid points. The box center was set exactly at the C α of Lys271, the key residue to recognize the carboxyl group of substrate (Shimizu et al. 2002). The set number generated for the docking poses was 20. The favorite docked complex was selected according to the interacting energy (ΔG, kcal/mol) by combining the geometrical matching quality and binding affinity of the AutoDock Vina software. The energy was then minimized by using Discovery Studio 2017 to resolve atomic clashes.
MD simulation
MD was performed using the AMBER 14 simulation package, and each system was repeated twice. The standard AMBER force field ff99SB and GLYCAM_06j-1 were used to generate the topologies and parameters of the enzyme and substrate, respectively (Kirschner et al. 2008; Wickstrom et al. 2009). Each system was simulated to be immersed in a dodecahedral periodic box of TIP3P explicit water and extended 10 Å outside the protein on all sides. Sodium ions or chloride ions were added to neutralize the charge. Before the MD simulations, the water molecules/ions were resolved to minimize energy via 1000 steps, followed by 20,000 steps for the side chains of the protein, and then 4000 steps for the whole system to remove potentially poor contacts between the solute and solvent. After energy minimization, the systems were gradually heated from 0 to 300 K over 100 ps, followed by 10-ns production of MD simulations with a time step of 2 fs at a temperature of 300 K and pressure of 1.0 atm that were controlled by the Langevin algorithm. Long-range electrostatic interactions were treated using the particle-mesh Ewald (PME) method (Darden et al. 1993). Bonds involving hydrogen atoms were constrained by the SHAKE algorithm (Ryckaert et al. 1977).
Based on the equilibrated dynamics trajectory (the last 2 ns), the binding free energy (ΔG0) of wild-type BiPG28A- or mutant D129K-GalpA4 complex system was calculated using the Molecular Mechanics/Poisson-Boltzmann Surface Area (MM/PBSA) module. A total of 2000 snapshots from the trajectory were extracted every 4 ps. The ΔG0 was computed according to the following equations:
where ΔG gas and ΔG solv represent the vacuum and solvation binding free energies, respectively; − TΔS is the entropic contribution, which is not considered in the relative free energy analysis; ΔG gas includes an intermolecular electrostatic term (ΔG elec ), a van der Waals term (ΔG VDW ), and an internal energy (bond, angle, and dihedral) term (ΔG int ); ΔG solv is divided into the electrostatic solvation energy (ΔG PB ) and the nonpolar solvation energy (ΔG SA ); γ is the surface tension proportionality constant; and β is the offset value. The solvent accessible surface area (SASA) was estimated by the MSMS algorithm with a probe radius of 1.4 Å.
Selection of mutation sites and site-directed mutagenesis
Through MD simulations, the position and dynamics of amino acid candidates near the carboxy group of substrate in subsites − 3, − 2 and − 1 were analyzed. The residue at position 129 was found to play a role in stabilizing the conformation of subsite − 2. Then, the number of hydrogen bonds between the residue at position 129 (saturated mutation) and the carboxy group of subsite − 2 were analyzed using the simulation trajectories. And, those with the most significant strengthening effects were selected for energy calculation and experiment verification.
Site-directed mutagenesis
Site-directed mutagenesis was conducted to construct mutant D129K and D129R using the Fast Mutagenesis System (TransGen Biotech, Beijing, China) and primers shown in Table S1. Protein expression and purification were conducted as described above. To verify the common effect of Lys129, the corresponding Asp130 of another endo-PG (TePG28b, KY474618.1) from Talaromyces leycettanus JCM 12802 (Li et al. 2017) was substituted by Lys with specific primers (Table S1). The integrity of all expression plasmids was verified by DNA sequencing. Heterologous expression and biochemical characterization of all mutant enzymes followed the same procedures as described above.
Isothermal titration calorimetry (ITC) assay
ITC 200 microcalorimeter (GE Healthcare) was used to measure the binding affinity between BiPG28A or its mutant D129K and trigalacturonic acid (GalpA3) at 25 °C. To avoid catalysis during titration, the catalytic residue Asp214 was substituted with Asn by site-directed mutagenesis with specific primers (Table S1). All solutions were degassed immediately before use. Active BiPG28A/D214N and D129K/D214N at the final concentration of 20 and 11 μM in McIlvaine buffer (pH 3.5) were placed in the protein cells, respectively. GalpA3 at the desired concentration of 1 mM in McIlvaine buffer (pH 3.5) was injected into the protein cell 20 times every 120 s with the stirring speed of 1000 rpm. The Wiseman c values were then calculated to be 1.31 and 1.09, respectively, according to the equation of Wiseman et al. (1989). The titration of the ligand with McIlvaine buffer alone served as the control. All data were corrected for the heat of dilution, which was the minima, before deconvolution of the data. The association constant, Ka, was calculated using the software MicroCal Origin 7.0. The thermodynamic parameter ΔG (the Gibbs free energy) was calculated following the equations ΔG = ΔH − TΔS and ΔG = − RTlnKa.
GalpA3 hydrolysis
To evaluate the catalytic efficiency of BiPG28A and mutant D129K towards GalpA3, each enzyme (2 μM) was incubated with 200 μM of GalpA3 at pH 3.5 and 70 °C for 3 min. Aliquots (100 μL) were collected and heated at 100 °C for 5 min, followed by the filtration through the Nanosep centrifugal 3 K device (Pall). The hydrolysis products were analyzed by the high-performance anion-exchange chromatography equipped with a pulsed amperometric detector (HPAEC-PAD) using a 250 × 3 mm CarboPac PA200 guard column. NaOH (800 mM) was used to elute the oligosaccharides at the flow rate of 0.45 mL/min. GalpA3 was used as the standard. The catalytic efficiency values were then determined following the equation described by Matsui et al. (1991).
Results
Gene cloning and heterologous expression of BiPG28A
The acidophilic fungus Bispora sp. MEY-1 has been reported to be an excellent producer of acidic GHs, including β-mannanase (Luo et al. 2009), α-galactosidase (Wang et al. 2010), glucanase (Luo et al. 2010), endo-PG (Yang et al. 2011), etc. In the present study, a novel endo-PG named BiPG28A was identified in strain MEY-1. The cDNA of Bipg28a (MG520667 in the GenBank database) consists of 1134 base pairs, which encodes a polypeptide of 377 amino acids including a putative signal peptide of 22 residues at the N-terminus. The molecular mass and pI value were estimated to be 37.4 kDa and 4.29, respectively. Further sequence analysis indicated that deduced BiPG28A has three putative N-glycosylation sites (Asn181, Asn201, and Asn282; Asn-Xaa-Thr/Ser-Zaa, where Zaa is not Pro, http://www.cbs.dtu.dk/services/NetN-Glyc/).
The gene fragment coding for mature BiPG28A without the signal peptide-coding sequence was successfully expressed in P. pastoris GS115 and secreted into the culture. Substantial endo-PG activities were detected in the shake tube cultures. After large-scale cultivation and purification, the recombinant BiPG28A became electrophoretic homogeneity on SDS-PAGE (Fig. S2). The protein band had an apparent molecular mass of approximately 50 kDa, which was higher than the theoretical value (37.4 kDa). After deglycosylation with Endo H, the enzyme showed a molecular mass of 38 kDa, which is consistent with the predicted size.
By using PGA as the substrate, the purified BiPG28A exhibited maximum activities at 70 °C (Fig. 1a) and pH 3.5 (Fig. 1b), and remained more than 60% activity over the thermophilic (60–80 °C) and acidic (pH 2.5–4.5) ranges. BiPG28A was thermostable at high temperatures, retaining more than 90% of its initial activity after incubation for 30 min at 60 °C (Fig. 1a). And, it showed stability over a broad pH range of 2.2–7.0, retaining more than 90% of the initial activity after incubation at 37 °C for 1 h (Fig. 2b). The Vmax, Km, and kcat/Km values of BiPG28A with PGA as the substrate were determined to be 17,500 ± 162 μmol/min/mg, 0.89 ± 0.01 mg/mL, and 12,200 ± 114 mL/mg/s, respectively.

Enzymatic properties of purified BiPG28A. a Temperature effects on the activity and stability of BiPG28A. The enzyme activities were determined at indicated temperatures and pH 3.5 for 10 min, and shown as relative values to the maximum. Thermostability was shown as the residual relative activity to the untreated samples after 30-min treatment at indicated temperatures and pH 3.5. b pH effects on the activity and stability of BiPG28A. The activities were determined at indicated pH and 70 °C for 30 min, and shown as relative values. The pH stability was shown as relative values to the untreated samples after 1-h treatment at indicated pHs and 37 °C

Detailed view of the catalytic pocket of BiPG28A with GalpA4 binding. The residues involved in binding at subsites − 1/+ 1 are indicated. Hydrogen bonds are depicted as yellow dashed lines
Homology modeling and molecular docking
With 1HG8 (Federici et al. 2001) as the template, modeled BiPG28A folded into a right-handed parallel β-helical architecture of 10 complete coils. After energy minimization, the root mean square deviation (RMSD) between 1HG8 and BiPG28A was 0.76 Å over the C α atoms, suggesting that the refined model is sufficient for further analysis. To determine the substrate binding mode and uncover the catalytic mechanism on the atomic level, molecular docking studies were performed. After equilibration on the substrate, the conformation of BiPG28A-GalpA4 complex was generated with reference to the active-site architecture of galacturonate units co-crystallized with Stereum purpureum endo-PG I (1KCD; Fig. S3), the only endo-PG-GalpA complex reported so far (Shimizu et al. 2002). The conserved residues Asn191, Asp193, Asp214, Asp215, His236, Gly237, Arg269, Lys271, and Tyr304 (corresponding to Asn151, Asp153, Asp173, Asp174, His195, Gly196, Arg226, Lys228, and Tyr262 of 1KCD) are involved in binding GalpA at subsites − 1/+ 1 (Fig. 2). According to the crystal structure of 1KCD, the carboxy group of GalpA at subsite + 1 has electrostatic interactions and/or forms hydrogen bonds with four residues, i.e., the interactions between O61 of the carboxy group and the NH2 of Arg269 and ND1 of His236, and O62 of the carboxy group and the NZ of Lys271 and OH of Tyr304. These interactions contribute to the substrate binding.
Selection of mutation sites
To identify the potential mutation sites related to binding affinity, 10 ns MD simulations were performed to analyze the dynamics of amino acid candidates near the carboxy group of substrate (especially in subsites − 3, − 2, and − 1). The MD results indicated that the carboxy groups of subsites − 3 and − 1 were both positioned behind the catalytic residues, while a Na+ stabilized the carboxy group of subsite − 2 by coordinating to the Asp129 (Fig. 3). This double dentate mode appears to play an important role in stabilizing the conformation of subsite − 2.

A refined model of BiPG28A-GalpA4 complex by MD simulation. Hydrogen bonds are shown as broken lines
The hydrogen bond numbers between each residue at position 129 (saturated mutation) and the carboxy group of subsite − 2 were calculated after 10 ns MD simulations. In comparison to the wild type, five mutants showed more hydrogen bonds, which were in the order of wild type < D129Q < D129S < D129Y < D129K < D129R (Fig. 4). Notably, the introduction of positively charged amino acids Lys and Arg at position 129 showed the most significant strengthening effects. As shown in Fig. 5a, the most stable hydrogen bond was formed by the NE atom of Arg129 and O61 of the carboxy group of substrate − 2, not the positively charged atoms (NH1 or NH2). Oppositely, the NZ atom of Lys129 formed strong hydrogen bond and electrostatic interactions with the carboxy group of subsite − 2 (Fig. 5b). The longer side chain of Arg probably occupied a larger space. Taken together, the MD simulation results strongly supported that mutation of D129K might be greater to enhance the conformation stability and binding efficiency of BiPG28A.

Number of the hydrogen bonds between the mutant sites and the carboxyl group of subsite − 2 during the 10 ns MD simulation

The interactions of the residue at position 129 in mutants D129R (a) and D129K (b) with the carboxyl group of subsite − 2 during the 10 ns MD simulations. Hydrogen bonds are shown as broken lines
The binding free energy (MM/PBSA) of the wild-type BiPG28A or its mutant D129K with GalpA4 as the substrate was also calculated. Compared to the wild-type BiPG28A, the mutant D129K exhibited a lower ΔG0 value (− 30.11 vs. − 10.61 kcal/mol), corresponding to the stronger interaction between the mutant D129K and substrate. Through the computational simulation design and energy analysis, we speculated that the mutation D129K might contribute to higher binding affinity with substrate.
Experiment verification
To verify the computational simulation results, we performed site-directed mutagenesis at position 129, produced and characterized the mutant enzymes as described above, and compared their catalytic activities with the wild type. Mutants D129K and D129R showed quite similar temperature- and pH-activity profiles to the wild-type BiPG28A, with optimal activities at pH 3.5 and 70 °C (Fig. S4). However, great differences were detected in their substrate binding and catalysis (Table 1 and Fig. S5). The mutants D129K and D129R showed increases in the substrate affinity (decreased Km values by 43 and 10%, respectively) and catalytic efficiencies (increased kcat/Km values by ~ 4.7 and 1.5-fold, respectively). It corresponded to the computational simulation results and confirmed the contribution of the positively charged side chain at position 129 to the binding affinity. Moreover, residue Lys rather than Arg at position 129 was more favorable for catalysis.
Another widely used endo-PG in the grape juice production, i.e., the TePG28b from T. leycettanus JCM 12802, was selected for mutagenesis at the corresponding position (Asp130). The mutant TePG28b-D130K was constructed and expressed in P. pastoris GS115. In comparison to the wild-type TePG28b, mutant TePG28b-D130K showed significant increases in the substrate affinity (Km, 0.78 ± 0.01 vs. 1.29 ± 0.02 mg/mL) and catalytic efficiency (kcat/Km, 105,600 ± 919 vs. 31000 ± 288 mL/mg/s). The results are consistent with that of BiPG28A and mutant D129K, and further prove the functional role of Lys at position 129 (BiPG28A numbering) in the substrate binding of GH28 PGs.
ITC experimental verification
ITC was used to undermine the thermodynamics of enzyme-substrate system. Inactive enzymes with substitution of Asp214 with Asn, BiPG28A/D214N and D129K/D214N, were incubated with GalpA3 at 25 °C, and their raw binding curves were recorded. As shown in Fig. 6, both binding reactions were spontaneous and exothermic with ΔG under zero. The Ka value of mutant D129K to GalpA3 was 9.97 × 104/M, 50% higher than that for wild-type BiPG28A (6.55 × 104/M) (Table 2). The results indicated that mutant D129K had stronger binding affinity to GalpA3 than BiPG28A, which was consistent with the decreased Km value.

ITC analysis of the binding isotherms (lower panels) and thermograms (upper panels) of inactive BiPG28A (a) and mutant D129K (b) with GalpA3 as the substrate. Binding experiments were performed at 25 °C in 25 mM McIlvaine buffer (pH 3.5) with triplicate
According to the formula ΔG = − RTlnKa = ΔH − TΔS, enthalpy (ΔH) and entropy (ΔS) are two determinants of the binding affinity. The substrate binding of BiPG28A/D214N and D129K/D214N is both enthalpy-driven, with the negative ΔH values of − 6.16 ± 0.95 and − 13.31 ± 0.94 kcal/mol. This binding enthalpy primarily reflects the strength of interactions between the substrate and enzyme (non-covalent interactions, e.g., van der Waals, hydrogen bonds, and electrostatics) relative to those existing in the solvent (Velázquez-Campoy et al. 2004). The negative ΔH and positive ΔS values of inactive BiPG28A/D214N resulted in a negative ΔG value of − 6.57 kcal/mol. In contrast, the favorable high ΔH value offsets the negative ΔS and correspondingly the propitious ΔG value (− 6.81 kcal/mol) of inactive D129K/D214N. Thus, the highly favorable ΔH value of the mutant D129K/D214N made it more significant in substrate binding through non-covalent interactions.
The catalytic efficiencies of wild-type BiPG28A and mutant D129K with GalpA3 as the substrate were also determined. Mutant D129K showed a greater catalytic efficiency against GalpA3 than BiPG28A (280.03 ± 2.59 vs. 239.17 ± 1.63/mM/min). In combination with the ITC binding results, it suggested that mutant D129K had a stronger affinity and hydrolytic capacity against GalpA3.
Discussion
Thermophilic/thermostable enzymes with high catalytic efficiency are more favorable than mesophilic counterparts due to the high-temperature pelleting process, higher mass transfer rate, and lower contamination risk (Berka et al. 2011). To our best knowledge, only a few fungal endo-PGs have activities at ≥ 60 °C (Table 3). Compared with other thermophilic/thermostable endo-PGs, BiPG28A has a lower acidic pH optimum (3.5) and better stability (> 90% activity at 60 °C for 30 min). These properties make BiPG28A favorable for applications in various acidic and high-temperature industries. Therefore, to further improve its catalytic activity is of importance for industrial purposes.
Structural analysis of the only endo-PG-galacturonate complex (1KCD; Shimizu et al. 2002) known so far revealed the important binding interaction between the carboxyl group of GalpA at subsite + 1 and the substrate binding sites of endo-PGs. In the present study, we focused on the binding interaction related to the carboxyl group of GalpA. To select optimal mutation sites and favorable amino acids, we took the spatial orientation of amino acids and their side chains for serious consideration. Based on the 10 ns MD simulation of BiPG28A-GalpA4 complex, amino acids having side chains towards the carboxyl group of GalpA were chosen. Within the homology model, a Na+ was found to stabilize the carboxyl group of subsite − 2 by coordinating to Asp129. MD simulation analysis showed that Arg, Lys, Tyr, Ser, and Gln at position 129, especially Arg and Lys with long and positively charged side chains, can form a number of hydrogen bonds with the carboxyl group of subsite − 2. However, great steric clash was detected between the longer side chain of Arg129 and the carboxyl group of subsite − 2; thus, Lys forming the second most hydrogen bonds with substrate was chosen as the substitution candidate.
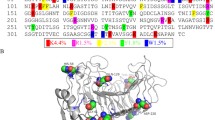
Residue 129 is located on the T3 loop, which forms a catalytic cleft with T1 loop (Bonivento et al. 2008). As shown in Fig. 7, the T3 loop is a flexible region containing 16 residues (Trp116–Phe131 according to the BiPG28A numbering) and connects the 310-helix and β-strand in the third coil. This region displays a hairpin topology with an α-ladder internal hydrogen-bonding pattern. Some residues of the T3 loop, such as Trp116, Asp117, Gly120, Asn122, Gly124, Lys127, and Pro128, are highly conserved. Previous studies have shown that Asn122 can narrow the active site cleft and plays a critical role in positioning the substrate close to the catalytic residues (Tu et al. 2015), while residue at position 118 determines the flexibility of T3 loop and influences the enzyme-substrate interaction (Tu et al. 2016b). The present study indicated that the residue at position 129 is another key substrate binding site. Most endo-PGs have positively charged Lys at this position, with the exception of BiPG28A and TePG28b (Fig. 7). Analysis of the binding free energy of wild-type BiPG18A- and mutant D129K-GalpA4 complex systems by MM/PBSA module revealed that the affinity of GalpA4 and D129K is stronger since Lys129 has more interactions with the carboxyl group of subsite − 2. In combination with the decreased Km and increased K a values, Lys129 was supported theoretically and experimentally to be a crucial residue in substrate binding.

Multiple sequence alignment of the T3 loop residues of endo-PGs (numbering according to BiPG28A) from Bispora sp. MEY-1, Talaromyces leycettanus JCM 12802 (TePG28b), S. purpureum (1K5C_A), A. aculeatus (1IA5_A), A. niger (1NHC_A), C. lupine (2IQ7_A), T. arenaria (AIZ95162.1), F. oxysporum (AAS15639.1), C. parasitica (AAB36616.1), and T. cellulolyticus (GAM33350.1)
In summary, we proposed a novel computational approach to design an endo-PG mutant with high binding affinity and catalytic efficiency. Without laborious construction of a large number of mutants, this modeled structure-based method is rapid, efficient, and reliable to identify favorable mutants. It could be extended to engineer other enzymes for improved binding affinity and catalytic efficiency.
References
Berka RM, Grigoriev IV, Otillar R, Salamov A, Grimwood J, Reid I, Ishmael N, John T, Darmond C, Moisan MC, Henrissat B, Coutinho PM, Lombard V, Natvig DO, Lindquist E, Schmutz J, Lucas S, Harris P, Powlowski J, Bellemare A, Taylor D, Butler G, de Vries RP, Allijn IE, van den Brink J, Ushinsky S, Storms R, Powell AJ, Paulsen IT, Elbourne LDH, Baker SE, Magnuson J, LaBoissiere S, Clutterbuck AJ, Martinez D, Wogulis M, de Leon AL, Rey MW, Tsang A (2011) Comparative genomic analysis of the thermophilic biomass-degrading fungi Myceliophthora thermophila and Thielavia terrestris. Nat Biotechnol 29:922–927
Bernardi RC, Cann I, Schulten K (2014) Molecular dynamics study of enhanced Man5B enzymatic activity. Biotechnol Biofuels 7:83
Bonivento D, Pontiggia D, Matteo AD, Fernandez-Recio J, Salvi G, Tsernoglou D, Cervone F, Lorenzo GD, Federici L (2008) Crystal structure of the endopolygalacturonase from the phytopathogenic fungus Colletotrichum lupini and its interaction with polygalacturonase-inhibiting proteins. Proteins 70:294–299
Cheng Z, Chen D, Lu B, Wei Y, Xian L, Li Y, Luo Z, Huang R (2016) A novel acid-stable endo-polygalacturonase from Penicillium oxalicum CZ1028: purification, characterization, and application in the beverage industry. J Microbiol Biotechnol 26:989–998
Choi JM, Han SS, Kim HS (2015) Industrial applications of enzyme biocatalysis: current status and future aspects. Biotechnol Adv 33:1443–1454
Comeau MA, Lafontaine DA, Abou Elela S (2016) The catalytic efficiency of yeast ribonuclease III depends on substrate specific product release rate. Nucleic Acids Res 44:7911–7921
Cui D, Zhang L, Jiang S, Yao Z, Gao B, Lin J, Yuan YA, Wei D (2015) A computational strategy for altering an enzyme in its cofactor preference to NAD(H) and/or NADP(H). FEBS J 282:2339–2351
Daczkowski CM, Pegan SD, Harvey SP (2015) Engineering the organophosphorus acid anhydrolase enzyme for increased catalytic efficiency and broadened stereospecificity on Russian VX. Biochemistry 54:6423–6433
Darden T, York D, Pedersen L (1993) Particle mesh Ewald: an N-log (N) method for Ewald sums in large systems. J Chem Phys 98:10089–10092
Federici L, Caprari C, Mattei B, Savino C, Di Matteo A, De Lorenzo G, Cervone F, Tsernoglou D (2001) Structural requirements of endopolygalacturonase for the interaction with PGIP (polygalacturonase-inhibiting protein). Proc Natl Acad Sci U S A 98:13425–13430
Hirose N, Kishida M, Kawasaki H, Sakai T (1999) Purification and characterization of an endo-polygalacturonase from a mutant of Saccharomyces cerevisiae. Biosci Biotechnol Biochem 63:1100–1103
Kirschner KN, Yongye AB, Tschampel SM, González-Outeiriño J, Daniels CR, Foley BL, Woods RJ (2008) GLYCAM06: a generalizable biomolecular force field. Carbohydrates. J Comput Chem 29:622–655
Korendovych IV, DeGrado WF (2014) Catalytic efficiency of designed catalytic proteins. Curr Opin Struct Biol 27:113–121
Li K, Meng K, Pan X, Ma R, Yang P, Huang H, Yao B, Su X (2015) Two thermophilic fungal pectinases from Neosartorya fischeri P1: gene cloning, expression, and biochemical characterization. J Mol Catal B-Enzym 118:70–78
Li Y, Wang Y, Tu T, Zhang D, Ma R, You S, Wang X, Yao B, Luo H, Xu B (2017) Two acidic, thermophilic GH28 polygalacturonases from Talaromyces leycettanus JCM 12802 with application potentials for grape juice clarification. Food Chem 237:997–1003
Luo H, Wang Y, Wang H, Yang J, Yang Y, Huang H, Yang P, Bai Y, Shi P, Fan Y, Yao B (2009) A novel highly acidic β-mannanase from the acidophilic fungus Bispora sp. MEY-1: gene cloning and overexpression in Pichia pastoris. Appl Microbiol Biotechnol 82:453–461
Luo H, Yang J, Yang P, Li J, Huang H, Shi P, Bai Y, Wang Y, Fan Y, Yao B (2010) Gene cloning and expression of a new acidic family 7 endo-β-1,3-1,4-glucanase from the acidophilic fungus Bispora sp. MEY-1. Appl Microbiol Biotechnol 85:1015–1023
Martins ES, Silva D, Leite RS, Gomes E (2007) Purification and characterization of polygalacturonase produced by thermophilic Thermoascus aurantiacus CBMAI-756 in submerged fermentation. Antonie Van Leeuwenhoek 91:291–299
Mathew A, Eldo AN, Molly A (2008) Optimization of culture conditions for the production of thermostable polygalacturonase by Penicillium SPC-F 20. J Ind Microbiol Biotechnol 35:1001–1005
Matsui I, Ishikawa K, Matsui E, Miyairi S, Fukui S, Honda K (1991) Subsite structure of Saccharomycopsis α-amylase secreted from Saccharomyces cerevisiae. J Biochem 109:566–569
Miyairi K, Matsue T, Kagawa O, Kutsuzawa T, Okuno T (1994) Purification and characterization of an endopolygalacturonase from Physalospora piricola. Biosci Biotechnol Biochem 58:1909–1910
Parashar D, Satyanarayana T (2016) A chimeric α-amylase engineered from Bacillus acidicola and Geobacillus thermoleovorans with improved thermostability and catalytic efficiency. J Ind Microbiol Biotechnol 43:473–484
Ryckaert JP, Ciccotti G, Berendsen HJC (1977) Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J Comput Phys 23:327–341
Schnitzhofer W, Weber HJ, Vršanská M, Biely P, Cavaco-Paulo A, Guebitz G (2007) Purification and mechanistic characterisation of two polygalacturonases from Sclerotium rolfsii. Enzyme Microb Tech 40:1739–1747
Shimizu T, Nakatsu T, Miyairi K, Okuno T, Kato H (2002) Active-site architecture of endopolygalacturonase I from Stereum purpureum revealed by crystal structures in native and ligand-bound forms at atomic resolution. Biochemistry 41:6651–6659
Tinberg CE, Khare SD, Dou J, Doyle L, Nelson JW, Schena A, Jankowski W, Kalodimos CG, Johnsson K, Stoddard BL, Baker D (2013) Computational design of ligand-binding proteins with high affinity and selectivity. Nature 501:212–216
Tounsia H, Sassia AH, Romdhanea ZB, Lajnefa M, Dupuyb JW, Lapaillerieb D, Lomenechb AM, Bonneub M, Gargouria A, Hadj-Taieb N (2016) Catalytic properties of a highly thermoactive polygalacturonase from the mesophilic fungus Penicillium occitanis and use in juice clarification. J Mol Catal B-Enzym 127:56–66
Trott O, Olson AJ (2010) AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 31:455–461
Tu T, Meng K, Bai Y, Shi P, Luo H, Wang Y, Yang P, Zhang Y, Zhang W, Yao B (2013) High-yield production of a low-temperature-active polygalacturonase for papaya juice clarification. Food Chem 141:2974–2981
Tu T, Meng K, Huang H, Luo H, Bai Y, Ma R, Su X, Shi P, Yang P, Wang Y, Yao B (2014) Molecular characterization of a thermophilic endo-polygalacturonase from Thielavia arenaria XZ7 with high catalytic efficiency and application potential in the food and feed industries. J Agric Food Chem 62:12686–12694
Tu T, Meng K, Luo H, Turunen O, Zhang L, Cheng Y, Su X, Ma R, Shi P, Wang Y, Yang P, Yao B (2015) New insights into the role of T3 loop in determining catalytic efficiency of GH28 endo-polygalacturonases. PLoS One 10:e0135413
Tu T, Li Y, Su X, Meng K, Ma R, Wang Y, Yao B, Lin Z, Luo H (2016a) Probing the role of cation-π interaction in the thermotolerance and catalytic performance of endo-polygalacturonases. Sci Rep 6:38413
Tu T, Pan X, Meng K, Luo H, Ma R, Wang Y, Yao B (2016b) Substitution of a non-active-site residue located on the T3 loop increased the catalytic efficiency of endo-polygalacturonases. Process Biochem 51:1230–1238
van Santen Y, Benen JA, Schröter KH, Kalk KH, Armand S, Visser J, Dijkstra BW (1999) 1.68-Å crystal structure of endopolygalacturonase II from Aspergillus niger and identification of active site residues by site-directed mutagenesis. J Biol Chem 274:30474–30480
Velázquez-Campoy A, Ohtaka H, Nezami A, Muzammil S, Freire E (2004) Isothermal titration calorimetry. Curr Protoc Cell Biol 17(8):1–17.8.24
Wang H, Luo H, Li J, Bai Y, Huang H, Shi P, Fan Y, Yao B (2010) An α-galactosidase from an acidophilic Bispora sp. MEY-1 strain acts synergistically with β-mannanase. Bioresour Technol 101:8376–8382
Wang Z, Ye S, Li J, Zheng B, Bao M, Ning G (2011) Fusion primer and nested integrated PCR (FPNI-PCR): a new high-efficiency strategy for rapid chromosome walking or flanking sequence cloning. BMC Biotechnol 11:109
Wang J, Zhang Y, Qin X, Gao L, Han B, Zhang D, Li J, Huang H, Zhang W (2017) Efficient expression of an acidic endo-polygalacturonase from Aspergillus niger and its application in juice production. J Agric Food Chem 65:2730–2736
Wickstrom L, Okur A, Simmerling C (2009) Evaluating the performance of the ff99SB force field based on NMR scalar coupling data. Biophys J 97:853–856
Wiseman T, Williston S, Brandts JF, Lin LN (1989) Rapid measurement of binding constants and heats of binding using a new titration calorimeter. Anal Biochem 179:131–137
Yang J, Luo H, Li J, Wang K, Cheng H, Bai Y, Yuan T, Fan Y, Yao B (2011) Cloning, expression and characterization of an acidic endo-polygalacturonase from Bispora sp. MEY-1 and its potential application in juice clarification. Process Biochem 46:272–277
Acknowledgments
This research was supported by the National Natural Science Foundation of China (31571777), the China Modern Agriculture Research System (CARS-41), and the National Key Research and Development Program of China (2016YFD0501409-02).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical statement
This article does not contain any studies with human participants or animals performed by any of the authors.
Electronic supplementary material
ESM 1
(PDF 371 kb)
Rights and permissions
About this article

Cite this article
Tu, T., Li, Y., Luo, Y. et al. A key residue for the substrate affinity enhancement of a thermophilic endo-polygalacturonase revealed by computational design. Appl Microbiol Biotechnol 102, 4457–4466 (2018). https://doi.org/10.1007/s00253-018-8948-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-018-8948-y