
Abstract
In the present study, 35 Leuconostoc mesenteroides strains isolated from vegetables and food products from South Korea were studied by multilocus sequence typing (MLST) of seven housekeeping genes (atpA, groEL, gyrB, pheS, pyrG, rpoA, and uvrC). The fragment sizes of the seven amplified housekeeping genes ranged in length from 366 to 1414 bp. Sequence analysis indicated 27 different sequence types (STs) with 25 of them being represented by a single strain indicating high genetic diversity, whereas the remaining 2 were characterized by five strains each. In total, 220 polymorphic nucleotide sites were detected among seven housekeeping genes. The phylogenetic analysis based on the STs of the seven loci indicated that the 35 strains belonged to two major groups, A (28 strains) and B (7 strains). Split decomposition analysis showed that intraspecies recombination played a role in generating diversity among strains. The minimum spanning tree showed that the evolution of the STs was not correlated with food source. This study signifies that the multilocus sequence typing is a valuable tool to access the genetic diversity among L. mesenteroides strains from South Korea and can be used further to monitor the evolutionary changes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The genus Leuconostoc consists of heterofermentative, Gram-positive, non-motile, non-sporulating, and facultative anaerobic lactic acid bacteria (LAB) that produce lactic acid as one of the key fermentation products from carbohydrate metabolism (Axelsson 2004; Hayek and Ibrahim 2013). At present, nearly 400 species of LAB have been recognized (Zhang et al. 2011) and the genus Leuconostoc is comprised of 24 different species (Kot et al. 2014). The genus Leuconostoc includes Leuconostoc mesenteroides (with four subspecies: mesenteroides, dextranicum, cremoris, and suionicum) and 23 other species, i.e., L. amelibiosum, L. argentinum, L. cremoris, L. citreum, L. carnosum, L. dextranicum, L. durionis, L. fallax, L. ficulneum, L. holzapfelii, L. pseudoficulneum, L. fructosum, L. gasicomitatum, L. gelidum, L. inhae, L. kimchii, L. lactis, L. pseudomesenteroides, L. miyukkimchii, L. oeni, L. palmae, L. paramesenteroides, and the recently reported L. rapi (www.bacterio.net). Leuconostocs can be found in various natural niches such as green vegetation and plant roots (Hemme and Foucaud-Scheunemann 2004). In addition, the genus Leuconostoc has also been found to be associated with beverages, meat, vegetables, and dairy and bakery products (Wassie and Wassie 2016). The amazing fermentative capacity of these bacteria helps to improve organoleptic attributes and safety of the food products (Gemechu 2015). It has also been reported that Leuconostoc helps in augmentation of various nutrients in the food products, including ample health benefits for humans (Steele et al. 2013). Furthermore, leuconostocs are utilized as starter cultures for various industrial processes and also as a flavoring and texturizing agents (Dan et al. 2014).
Several typing methods have been used for the identification, characterization, and utilization of LAB (Sabat et al. 2013; Tanigawa and Watanabe 2011). Generally, typing of bacteria is related to phenotypic and genotypic characteristics. Phenotyping includes traditional methods such as serotypes, biotypes, phage-types, and antibiograms (Dan et al. 2014). In contrast, genotypic methods are found to be more advantageous as they are rapid, generally applicable powerful tools to differentiate isolates on the basis of genetic variations (Unemo and Dillon 2011). Therefore, molecular approaches have been used for the proper identification and characterization of bacterial species (Botina et al. 2006). Currently, several molecular typing methods, such as random amplified polymorphic DNA PCR (RAPD-PCR), amplified fragment length polymorphism (AFLP), pulsed-field gel electrophoresis (PFGE), restriction fragment length polymorphism (RFLP), repetitive element palindromic PCR (Rep-PCR), protein fingerprinting, and multilocus sequence typing (MLST) have been utilized to characterize Leuconostoc spp. (Alegría et al. 2013; Björkroth et al. 2000; Cibik et al. 2000; Kunene et al. 2000; Nieto-Arribas et al. 2010; Pérez et al. 2002; Sánchez et al. 2005; Vihavainen and Björkroth 2009; Zhang et al. 2015). Multilocus sequence typing method was built on the procedure of multilocus enzyme electrophoresis (MLEE) by targeting the variation present at multiple housekeeping loci (Sabat et al. 2013). However, in MLST, identification of this variation was achieved by nucleotide sequencing of the housekeeping loci to differentiate between isolates of the same microbial species (Bain et al. 2007). In the last few decades, the MLST technique has been recognized to study the phylogenetic structure of many bacterial species by considering six to eight housekeeping genes (Feil and Enright 2004; Maiden et al. 1998; Oh et al. 2010). According to Konstantinidis et al. (2006), the minimum number of genes needed for multilocus differentiation between species is three because of potential events of horizontal gene transfer or recombination. Originally, the technique was used to identify different virulent lineages of 107 isolates of Neisseria meningitidis (Maiden et al. 1998). Soon after that, the approach was utilized to analyze other bacterial species, including LAB (Dingle et al. 2001; Farfán et al. 2002; Helgason et al. 2004). LAB strains that have been typed by MLST technique include Oenococcus oeni (Bilhere et al. 2009), Lactobacillus casei (Cai et al. 2007), Pediococcus (Calmin et al. 2008), Lactobacillus plantarum (de Las Rivas et al. 2006; Tanganurat et al. 2009), Lactococcus lactis (Passerini et al. 2010), Lactobacillus sanfranciscensis (Picozzi et al. 2010), Lactobacillus delbrueckii (Tanigawa and Watanabe 2011), Lactobacillus sakei (Chaillou et al. 2013), L. lactis (Dan et al. 2014), Lactobacillus fermentum (Dan et al. 2015), Carnobacterium maltaromaticum (Rahman et al. 2014), L. mesenteroides (Zhang et al. 2015), and recently Lactobacillus delbrueckii subsp. bulgaricus (Song et al. 2016).
The aim of the present study was to develop an MLST scheme for the characterization of 35 L. mesenteroides strains from South Korea. The relatedness of the selected strains was displayed as dendrogram and minimum spanning tree by means of various software using differences between their allelic profiles.
Materials and methods
Bacterial strains
In this study, a total of 35 strains, deposited in the Korean Culture Collection of Probiotics (KCCP, Seongnam, South Korea), were identified as L. mesenteroides based on sequence analysis of the 16S rRNA gene (Kaur et al. 2017). These strains were isolated from various food sources, including Kimchi (11 isolate), radish (10), Chinese cabbage kimchi (4), radish kimchi (2), young radish (4), salted clam (2), salted fish (1), and salted small octopus (1) from South Korea as described in previous study (Kaur et al. 2017). The strains were grown in MRS broth (Difco Laboratories, Detroit, MI, USA) and incubated overnight at 37 °C. Stock cultures were stored in 20% glycerol (Duksan, South Korea) at − 80 °C.
DNA extraction
Total genomic DNA was extracted and purified from 3-mL overnight cultures using the AccuPrep Genomic DNA Extraction Kit (Bioneer). The bacterial cells grown in MRS broth (Difco) at 37 °C were collected by centrifugation (16,000×g, 1 min), and DNA was extracted from the cell pellets of L. mesenteroides strains as described previously (Kaur et al. 2017; Lee and Lee 2008).
PCR amplification and sequencing of amplicons
To study the population structure of L. mesenteroides, seven housekeeping genes were selected: atpA (ATP synthase subunit alpha), gyrB (gyrase subunit B), groEL (molecular chaperonin), pheS (Phenylalanyl-tRNA synthetase), pyrG (CTP synthase), rpoA (RNA polymerase), and uvrC (Excinuclease ABC, subunit C). Primers were designed using a primer blast tool from the NCBI website (www.ncbi.nlm.nih.gov) and synthesized by Macrogen (Macrogen Inc., Seoul, South Korea). For polymerase chain reaction (PCR) amplification, 1 μL purified genomic DNA of each strain, 2 μL (10 pmoles/μL) of each primer (forward and reverse), and 17 μL of distilled water were added to the premix kit (Bioneer, Daejeon, South Korea) to make a final volume of 20 μL. The PCR procedure for the amplification of the seven housekeeping genes was done in an automatic thermocycler (Bio-Rad Gel Doc XR+, Bio-Rad, CA, USA) under the following conditions: 94 °C for 2 min, 35 cycles of denaturation at 95 °C for 20 s each, annealing for 30 s at a different temperature for each gene (atpA, 51 °C; groEL, 60 °C; gyrB, 49.2 °C; pheS, 46.3 °C; pyrG, 53.7 °C; rpoA, 50.5 °C; and uvrC, 58.3 °C), extension at 72 °C for 30 s, followed by a final elongation at 72 °C for 7 min. The PCR products were stored at 4 °C then 5 μL of each was assessed by electrophoresis on a 1% (w/v) agarose gel (Seakem, Lonza, USA), and the images were recorded by using a gel documentation system (Bio-Rad Gel Doc XR+). A 100-bp ladder (Bioneer, Daejeon, South Korea) and a 1-kb ladder (Takara, Japan) were used as size markers. The confirmed PCR products were purified with the Wizard SV Gel and PCR Clean-Up system kit (Promega, USA), and sequencing was performed by Humanizing Genomics MACROGEN (Macrogen Inc., Seoul, South Korea) and Bioneer (Bioneer, Daejeon, South Korea).
Data analysis
For MLST data analysis, the forward and reverse sequences of each gene were trimmed and analyzed with Bioedit Sequence Alignment Editor v 7.2.5 (Hall 1999), and further aligned with the ClustalX v 1.83 software. The guanine-cytosine (GC) content and the polymorphism data were calculated by DnaSP v 5.10 (Librado and Rozas 2009). The ratios of the synonymous substitutions (dS) to non-synonymous substitutions (dN) were studied by using an online software, SNAP (www.hiv.lanl.gov) (Korber 2000). The allelic profiles and the sequence types (STs) were deduced by using Bionumerics v 7.6 (Applied-Maths, Sint Martens-Latem, Belgium) software. The evolution of the population was studied with the help of minimum spanning tree (MST) using the Bionumerics software. Moreover, the relatedness of the STs were evaluated by constructing a dendrogram by the unweighted pair group method with arithmetic averages (UPGMA) using the START 2 v 0.9.0 beta software (Jolley et al. 2001). To assess the degree of tree-like structure, split decomposition analysis was performed with SPLITSTREE v 4.14 (Huson and Bryant 2006).
Nucleotide sequence accession numbers
The partial sequences of the seven MLST loci for 35 L. mesenteroides strains used in this study have been deposited in GenBank/EMBL databases under accession numbers KY224760-KY224969 and KY111323-KY111357.
Results
Sequence diversity and assignment of sequence types
The sequences of the seven chosen loci (atpA, groEL, gyrB, pheS, pyrG, rpoA, and uvrC) were determined for the 35 strains of L. mesenteroides. However, only a few genes have given desired clean sequences, while the others generated noisy sequence signals. Therefore, the latter PCR products were repurified and sequenced again. The number of alleles, polymorphic sites, guanine-cytosine content, and ratios of dN/dS were calculated. The fragment sizes of the seven housekeeping loci ranged in length from 366 to 1414 bp (Table 1). The MLST scheme defined alleles between 316 bp (uvrC) and 769 bp (gyrB) in length. The allelic combinations of the selected gene loci were determined, and a unique sequence type number was assigned for each isolate. Strains could be divided into 27 STs (ST-1 to ST-27) using combined data from the seven loci, giving a clear indication of variability among 35 strains (Table 2). Among these, ST-3 and ST-5 were the most observed STs, each was presented by five different strains, and the remaining 25 STs were represented by a single strain only. All the STs differed at various loci, except for ST-5, ST-8, ST-3; ST-13, ST-23; ST-26, ST-27; and ST-11, ST-24, differed only in the atpA locus; ST-3, ST-6 which showed differences at the pyrG locus; and ST-5, ST-23; ST-16, ST-17 with variation at the pheS locus whereas ST-3 and ST-22 showed variation at rpoA locus (Table 2). The allelic profiles of the seven loci for each strain are described in Table 2. The number of alleles per gene fragment varied from 7 for each of groEL and gyrB to 12 for rpoA. All the seven loci were found to be polymorphic (each is represented by more than five polymorphic sites), and the number of polymorphic sites ranged from 9 for groEL to 48 for pyrG (Table 3), giving a total of 220 SNPs.
Variation and recombination at MLST loci
The GC content of the partial sequence of the seven gene fragments was ranged from 37.2% (pyrG) to 42.9% (rpoA) with a mean value of 40.05% (Table 3). The dN/dS ratio for the seven gene fragments varied between 0.0245 and 1.6417 (Table 3). Except for gyrB, pyrG, and uvrC, the dN/dS ratio was found to be < 1. The allele frequencies showed that for most of the loci, allele 3 (atpA, pheS, pyrG, rpoA, and uvrC) and allele 1 (atpA, groEL, gyrB) (Fig. 1 and Table 2) were dominant in the population. In addition, the frequency of allele 2 was found to be more mainly for groEL locus. For the measurement of intragenic recombination, a split decomposition analysis was performed on each locus (atpA, gyrB, groEL, pheS, pyrG, rpoA, and uvrC) separately. As shown in split graphs (Fig. 2), the 35 L. mesenteroides strains revealed different structures in the split graphs for all the seven gene segments. In the split graphs, a parallelogram-shaped structure indicative of intergenic recombination in the evolutionary history of the gene was detected in the pheS, pyrG, rpoA, and uvrC loci. However, split graphs for atpA, gyrB, and groEL loci were appeared as tree-like structures, suggesting an absence of recombination (Fig. 2).

Polymorphic nucleotide sites in the seven Leuconostoc mesenteroides MLST genes. Variable sites are shown. The number of strains possessing the allele is indicated in parentheses

Split decomposition analysis of alleles obtained from 35 Leuconostoc mesenteroides strains for seven loci (atpA, gyrB, groEL, pheS, pyrG, rpoA, and uvrC). Numbering in the figure refers to allele numbers
UPGMA tree and clustering analysis based on MLST data
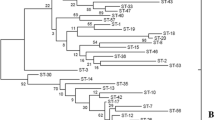
An UPGMA (Unweighted Pair Group Method with Arithmetic Mean) tree showing the genetic relatedness among the L. mesenteroides strains was displayed in Fig. 3. As deduced from the tree, 35 strains were clustered within two major groups, A and B. Group A was composed of 28 strains, included three subgroups (A1, A2, and A3). Subgroups A1, A2, and A3 were consisted of 6, 3, and 19 (11 STs) strains respectively. Group B was investigated into 7 strains that included four subgroups (B1, B2, B3, and B4). Subgroups B1 and B3 consisted of 3 and 2 strains respectively, whereas groups B2 and B4 were comprised of 1 strain individually. ST-3, ST-5, and ST-8 were closely related and clustered together in group A (subgroup A3). Among these, ST-3 and ST-5 were consisted of five different strains, whereas ST-8 was represented by a single strain only. Similarly, ST-13 and ST-23 were clustered in subgroup A3 and were found to be closely related. Phylogenetic analysis of 35 different L. mesenteroides strains based on the allelic profile data was performed using minimum spanning tree algorithm (Fig. 4). In this representation, each colored circle of the MST corresponds to an ST and the size of the circle was proportional to the number of strains with the same allelic profile. Therefore, two large circles (red and green) out of 27, depicting two STs, i.e., ST-3 (red) and ST-5 (green), were consisted of 10 strains out of 35. ST-3 was represented by 11204, 11251, 11357, 11359, 11396 strains and ST-5 was consisted of 11386, 11360, 11364, 11362, 11289 strains. In addition, the remaining 25 colored circles of similar size were represented by a single strain only, thereby completing the 35 strains in total, used in the present study (Table 2). Different circles were connected to each other by different lines representing the degree of relationship among L. mesenteroides strains. If we compare MST and UPGMA tree, the results obtained were supporting each other. However, in the present study, we could not find an appropriate association between ST and the sources from which the strains were isolated (Table 2).

UPGMA dendrogram showing the genetic relationship between 27 STs found in Leuconostoc mesenteroides via MLST typing. The phylogenetic tree was produced using START 2.0 software. Two major groups were designated as A and B

Minimum spanning tree analysis of the 35 Leuconostoc mesenteroides strains from South Korea. Each circle corresponds to a sequence type (ST), and the circle size denotes the number of strains sharing the same ST. The type of line between isolates indicates the strength of the genetic relationship between these isolates (bold—strong relationship, solid—intermediate relationship, and dotted line—weak relationship). The lines (solid, bold, and dashed) are related to genetic similarity, i.e., the number of common alleles between profiles
Discussion
In this study, MLST scheme based on an analysis of seven gene fragments from L. mesenteroides strains was described. The results of the study showed that 27 different genotypes were reported from 35 strains of South Korea. In population genetic studies, PFGE and MLST were considered as the methods of choice (Glaeser and Kämpfer 2014; Weng et al. 2013). MLST is the best method for typing the DNA sequences of selected housekeeping genes of bacterial isolates (Urwin and Maiden 2003).
The represented MLST schemes showed high discriminatory power as it was able to differentiate between highly similar strains. This was represented by strains clustered in ST-3 and ST-5, and other STs which were differed only in the one locus, out of seven used in the study. Only 10 strains were clustered in two different STs, and the rest of the 25 strains were represented by single ST. Similar results were observed in an MLST study of Carnobacterium maltaromaticum (LAB) where 5 STs were characterized by at least two strains while approximately 80% of the STs were represented by one strain only (Rahman et al. 2014). The number of alleles of the seven housekeeping loci ranged from 7 to 12 (Table 2). Our results validated the genetic heterogeneity of this species in a previous study, where the number of alleles varied in between 6 and 11 among the nine studied genes of L. mesenteroides (Zhang et al. 2015). The observed results for GC content corroborated the previously described results of the mean GC content of L. mesenteroides (Zhang et al. 2015). In support, the published GC content for the genus Leuconostoc can vary from 38 to 44% (de Bruyne et al. 2007). Analysis of synonymous and non-synonymous changes in the allele sequences of the loci can be used to determine whether they are subject to positive selection; therefore, a dN/dS ratio > 1 implies selection for the amino acid changes (Tanigawa and Watanabe 2011). By comparison, in an earlier study, the dN/dS ratio obtained from L. mesenteroides for nine loci ranged from 0.0219 to 2.4379 (Zhang et al. 2015). In contrast to earlier studies (Zhang et al. 2015), the ratio for pyrG and uvrC was found to be more than 1 in the present study. Nevertheless, the groEL, pyrG, and uvrC loci were used in the molecular typing of L. mesenteroides (Zhang et al. 2015). Although in most of the studies related to MLST, dN/dS ratio was used to detect the selection pressure. But an assumed gene evolution model was studied from MLST data by computing the dN/dS ratio (Kryazhimskiy and Plotkin 2008). However, it has been verified that the dN/dS ratio is not suitable for deducing selection pressures from single bacterial populations, in which most differences between sequences signify segregating polymorphisms rather than fixed substitutions (Kryazhimskiy and Plotkin 2008). Recombination in the sequences of the housekeeping genes can be evaluated by split decomposition analysis. This provides evidence of intraspecies recombination events which may generate diversity among isolates. Parallelogram-shaped structures have been observed in the split graphs of four (rpoA, pheS, pyrG, and uvrC) housekeeping loci, illustrating recombination had occurred in these loci. Similar parallelogram-shaped structures were also reported in murC, pheS, pyrG, and uvrC split graphs of L. lactis isolates (Dan et al. 2014), and also in the dnaA and murC loci of L. mesenteroides isolates (Zhang et al. 2015). Previously, it has been reported that the recombination could occur in Leuconostoc species because of the presence of mobile elements such as bacteriophages, genomic islands, and transposable elements in their genome sequences (Meslier et al. 2012). Furthermore, plasmids from L. mesenteroides have also been identified (Jeong et al. 2007). From the UPGMA tree analysis based on the MLST data (Fig. 3), we could not find a strong relationship between the ST obtained and the isolation source of the bacteria. Similar results have been reported for L. lactis, Lactococcus lactis, and Lactobacillus sanfranciscensis, where no significant associations between STs and the various sources of the isolates could be found (Dan et al. 2014; Passerini et al. 2010; Picozzi et al. 2010). The absence of an association between STs and the isolation source may be because of the genetic diversity of individual L. mesenteroides strains. The applicability of this technique lies in the evolutionary and population analyses for the estimation of recombination and mutation rates among bacterial population. In addition, the technique can also be utilized to investigate the evolutionary relationships among bacteria belonging to the same genus.
In conclusion, the MLST scheme presented here will be a useful tool for the precise characterization of L. mesenteroides strains, and appears to have sufficient discriminatory power for molecular typing. Phylogenetic analysis divided the 35 strains into two major groups, but it has also been concluded that the evolution of the 27 STs is not related to their origin from food sources. In the future, work will be focused on having more samples of L. mesenteroides to obtain better insight into the evolution of this bacterial species in South Korea.
References
Alegría Á, Delgado S, Flórez AB, Mayo B (2013) Identification, typing, and functional characterization of Leuconostoc spp. strains from traditional, starter-free cheeses. Dairy Sci Technol 93:657–673
Axelsson L (2004) Lactic acid bacteria: classification and physiology. In: Salminen S, Wright AV, Ouwehand A (eds) Lactic acid bacteria: microbiological and functional aspects. Marcel Dekker, New York, pp 1–66
Bain JM, Tavanti A, Davidson AD, Jacobsen MD, Shaw D, Gow NAR, Odds FC (2007) Multilocus sequence typing of the pathogenic fungus Aspergillus fumigatus. J Clinic Microbiol 45(5):1469–1477
Bilhere E, Lucas PM, Claisse O, Lonvaud-Funel A (2009) Multilocus sequence typing of Oenococcus oeni: detection of two subpopulations shaped by intergenic recombination. Appl Environ Microbiol 75:1291–1300
Björkroth KJ, Geisen R, Schillinger U, Weiss N, De Vos P, Holzapfel WH, Korkeala HJ, Vandamme P (2000) Characterization of Leuconostoc gasicomitatum sp. nov., associated with spoiled raw tomato-marinated broiler meat strips packaged under modified-atmosphere conditions. Appl Environ Microbiol 66(9):3764–3772
Botina SG, Tsygankov YD, Sukhodolets VV (2006) Identification of industrial strains of lactic acid bacteria by methods of molecular genetic typing. Russ J Genet 42(12):1367–1379
de Bruyne K, Schillinger U, Caroline L, Boehringer B, Cleenwerck I, Vancanneyt M, Vuyst LD, Franz MAPC, Vandamme P (2007) Leuconostoc holzapfelii sp. nov., isolated from Ethiopian coffee fermentation and assessment of sequence analysis of housekeeping genes for delineation of Leuconostoc species. Int J Syst Evol Microbiol 57:2952–2959
Cai H, Rodriguez BT, Zhang W, Broadbent JR, Steele JL (2007) Genotypic and phenotypic characterization of Lactobacillus casei strains isolated from different ecological niches suggests frequent recombination and niche specificity. Microbiology 153:2655–2665
Calmin G, Lefort F, Belbahri L (2008) Multi-loci sequence typing for two lacto-acid bacteria (LAB) species: Pediococcus paryulus and P. damnosus. Mol Biotechnol 40:170–179
Chaillou S, Lucquin I, Najjari A, Zagorec M, Champomier-Vergès MC (2013) Population genetics of Lactobacillus sakei reveals three lineages with distinct evolutionary histories. PLoS One 8(9):e73253
Cibik R, Lepage E, Talliez P (2000) Molecular diversity of Leuconostoc mesenteroides and Leuconostoc citreum isolated from traditional French cheeses as revealed by RAPD fingerprinting, 16S rDNA sequencing and 16S rDNA fragment amplification. Syst Appl Microbiol 23(2):267–278
Dan T, Liu W, Sun Z, Lv Q, Xu H, Song Y, Zhang H (2014) A novel multi-locus sequence typing (MLST) protocol for Leuconostoc lactis isolates from traditional dairy products in China and Mongolia. BMC Microbiol 14:150
Dan T, Liu W, Song Y, Xu H, Menghe B, Zhang H, Sun Z (2015) The evolution and population structure of Lactobacillus fermentum from different naturally fermented products as determined by multilocus sequence typing (MLST). BMC Microbiol 15:107
Dingle KE, Colles FM, Wareing DRA, Ure R, Fox AJ, Bolton FE, Bootsma HJ, Willems RJL, Urwin R, Maiden MCJ (2001) Multilocus sequence typing system for Campylobacter jejuni. J Clinic Microbiol 39:14–23
Farfán M, Miñana-Galbis D, Fusté MC, Lorén JG (2002) Allelic diversity and population structure in Vibrio cholera O139 Bengal based on nucleotide sequence analysis. J Bacteriol 184:1304–1313
Feil EJ, Enright MC (2004) Analyses of clonality and the evolution of bacterial pathogens. Curr Opin Microbiol 7(3):308–313
Gemechu T (2015) Review on lactic acid bacteria function in milk fermentation and preservation. Afr J Food Sci 9(4):170–175
Glaeser P, Kämpfer P (2014) Multilocus sequence analysis (MLSA) in prokaryotic taxonomy. Syst Appl Microbiol 38:237–245
Hall TA (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis program for windows 95/98NT. Nucleic Acids Symp Ser 41:95–98
Hayek SA, Ibrahim SA (2013) Current limitations and challenges with lactic acid bacteria: a review. Food Nutr Sci 4:73–87
Helgason E, Tourasse NJ, Meisal R, Caugant DA, Kolsto AB (2004) Multilocus sequence typing scheme for bacteria of the Bacillus cereus group. Appl Environ Microbiol 70:191–201
Hemme D, Foucaud-Scheunemann C (2004) Leuconostoc, characteristics, use in dairy technology and prospects in functional foods. Int Dairy J 14:467–494
Huson DH, Bryant D (2006) Application of phylogenetic networks in evolutionary studies. Mol Bio Evol 23(2): 254–267
Jeong SJ, Park JY, Lee HJ, Kim JH (2007) Characterization of pFMBL1, a small cryptic plasmid isolated from Leuconostoc mesenteroides SY2. Plasmid 57(3):314–323
Jolley KA, Feil EJ, Chan MS, Maiden MC (2001) Sequence type analysis and recombinational tests (START). Bioinformatics 17(12):1230–1231
Kaur J, Lee S, Park Y-S, Sharma A (2017) RAPD analysis of Leuconostoc mesenteroides strains associated with vegetables and food products from Korea. LWT - Food Sci Technol 77:383–388
Konstantinidis KT, Ramette A, Tiedje JM (2006) Towards a more robust assessment of intra-species diversity using fewer genetic markers. Appl Environ Microbiol 72:7286–7293
Korber B (2000) HIV signature and sequence variation analysis. In: Rodrigo AG, Learn GH (eds) Computational analysis of HIV molecular sequences. Kluwer Academic Publishers, Dordrecht, pp 55–72
Kot W, Neve H, Heller KJ, Vogensen FK (2014) Bacteriophages of Leuconostoc, Oenococcus, and Weissella. Front Microbiol 5:186
Kryazhimskiy S, Plotkin JB (2008) The population genetics of d N /d S . PLoS Genet 4:e1000304
Kunene NF, Geornaras I, Von Holy A, Hastings JW (2000) Characterization and determination of origin of lactic acid bacteria from a sorghum-based fermented weaning food by analysis of soluble proteins and amplified fragment length polymorphism fingerprinting. Appl Environ Microbiol 66(3):1084–1092
de Las Rivas B, Marcobal A, Munoz R (2006) Development of a multilocus sequence typing method for analysis of Lactobacillus plantarum strains. Microbiology 152:85–93
Lee HM, Lee Y (2008) A differential medium for lactic acid-producing bacteria in a mixed culture. Lett Appl Microbiol 46(6):676–681
Librado P, Rozas J (2009) DnaSPv5: a software for comprehensive analysis of DNA polymorphisms data. Bioinformatics 25:1451–1452
Maiden MC, Bygraves JA, Feil E, Morelli G, Russell JE, Urwin R, Zhang Q, Zhou J, Zurth K, Caugant DA, Feavers IM, Achtman M, Spratt BG (1998) Multilocus sequence typing: a portable approach to the identification of clones within populations of pathogenic microorganisms. Proc Natl Acad Sci U S A 95(6):3140–3145
Meslier V, Loux V, Renault P (2012) Genome sequence of Leuconostoc pseudomesenteroides strain 4882, isolated from a diary starter culture. J Bacteriol 194:696–712
Nieto-Arribas P, Sesena S, Poveda JM, Palop L, Cabezas L (2010) Genotypic and technological characterization of Leuconostoc isolates to be used as adjunct starters in Manchego cheese manufacture. Food Microbiol 27:85–93
Oh PL, Benson AK, Peterson DA, Patil PB, Moriyama EN, Roos S, Walter J (2010) Diversification of the gut symbiont Lactobacillus reuteri as a result of host-driven evolution. ISME J 4(3):377–387
Passerini D, Beltramo C, Coddeville M, Quentin Y, Ritzenthaler P, Daveran-Mingot M-L, Bourgeois PL (2010) Genes but not genomes reveal bacterial domestication of Lactococcus Lactis. PLoS One 5(12):e15306
Pérez G, Cardell E, Zárate V (2002) Random amplified polymorphic DNA analysis for differentiation of Leuconostoc mesenteroides subspecies isolated from Tenerife cheese. Lett Appl Microbiol 34:82–85
Picozzi C, Bonacina G, Vigentini I, Foschino R (2010) Genetic diversity in Italian Lactobacillus sanfranciscensis strains assessed by multilocus sequence typing and pulsed-field gel electrophoresis analyses. Microbiology 156:2035–2045
Rahman A, Cailliez-Grimal C, Bontemps C, Payot S, Chaillou S, Revol-Junelles A-M, Borgesa F (2014) High genetic diversity among strains of the unindustrialized lactic acid bacterium Carnobacterium maltaromaticum in dairy products as revealed by multilocus sequence typing. Appl Environ Microbiol 80(13):3920–3929
Sabat AG, Budimir AJ, Nashev D, Sá-Leão R, van Dijl J, Laurent F, Grundmann H, Friedrich AW (2013) Overview of molecular typing methods for outbreak detection and epidemiological surveillance. Euro Surveill 18(4):20380
Sánchez JI, Martínez B, Rodríguez A (2005) Rational selection of Leuconostoc strains for mixed starters based on the physiological biodiversity found in raw milk fermentations. Int J Food Microbiol 105:377–387
Song Y, Sun Z, Guo C, Wu Y, Liu W, Yu J, Menghe B, Yang R, Zhang H (2016) Genetic diversity and population structure of Lactobacillus delbrueckii subspecies bulgaricus isolated from naturally fermented dairy foods. Sci Reports 6:22704
Steele J, Broadbent J, Kok J (2013) Perspective on the contribution of lactic acid bacteria to cheese flavor development. Curr Opin Biotechnol 24(2):135–141
Tanganurat W, Quinquis B, Leelawatcharamas V, Bolotin A (2009) Genotypic and phenotypic characterization of Lactobacillus plantarum strains isolated from Thai fermented fruits and vegetables. J Basic Microbiol 49:377–385
Tanigawa K, Watanabe K (2011) Multilocus sequence typing reveals a novel sub speciation of Lactobacillus delbrueckii. Microbiology 157:727–738
Unemo M, Dillon J-AR (2011) Review and international recommendation of methods for typing Neisseria gonorrhoeae isolates and their implications for improved knowledge of gonococcal epidemiology, treatment and biology. Clinic Microbiol Rev 24(3):447–458
Urwin R, Maiden MC (2003) Multi-locus sequence typing: a tool for global epidemiology. Trends Microbiol 11(10):479–487
Vihavainen EJ, Björkroth KJ (2009) Diversity of Leuconostoc gasicomitatum associated with meat spoilage. Int J Food Microbiol 136(1):32–36
Wassie M, Wassie T (2016) Isolation and identification of lactic acid bacteria from raw cow milk. Int J Adv Res Biol Sci 3(8):44–49
Weng PL, Ramli R, Shamsudin MN, Cheah YK, Hamat RA (2013) High genetic diversity of Enterococcus faecium and Enterococcus faecalis clinical isolates by pulsed-field gel electrophoresis and multilocus sequence typing from a hospital in Malaysia. Biomed Res Int 2013:1–6
Zhang ZG, Ye ZQ, Yu L, Shi P (2011) Phylogenomic reconstruction of lactic acid bacteria: an update. BMC Evol Biol 11(1):1–12
Zhang W, Liu W, Song Y, Xu H, Menghe B, Zhang H, Sun Z (2015) Multilocus sequence typing of a dairy-associated Leuconostoc mesenteroides population reveals clonal structure with intragenic homologous recombination. J Dairy Sci 98:1–10
Funding
This work was supported by the Korea Institute of Planning and Evaluation for Technology in Food, Agriculture, Forestry and Fisheries (IPET) through the High Value-added Food Technology Development Program, funded by the Ministry of Agriculture, Food and Rural Affairs (MAFRA) (grant number: 314073-03-2-HD040).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Ethical statement
This article does not contain any studies with human participants or animals performed by any of the authors.
Rights and permissions
About this article

Cite this article
Sharma, A., Kaur, J., Lee, S. et al. Genetic diversity analysis of Leuconostoc mesenteroides from Korean vegetables and food products by multilocus sequence typing. Appl Microbiol Biotechnol 102, 4853–4861 (2018). https://doi.org/10.1007/s00253-018-8942-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-018-8942-4