
Abstract
Aurovertins are the structurally diverse polyketides that distribute widely in different fungal species. They feature a 2,6-dioxabicyclo[3.2.1]-octane ring in structure and exhibit the potential antitumor activity against breast cancer as F1-ATPase β subunit inhibitor. In this study, we constructed the biosynthetic pathway of aurovertin in an Aspergillus nidulans host and obtained seven aurovertin-type compounds. Surprisingly, three new aurovertin geometric isomers were characterized. By introducing an inducible promoter xylP(p) in the pathway gene acyltransferase aurG, we can control the product ratios among different aurovertin compounds by adding glucose and/or inducer xylose. The yields of aurovertins could be increased up to about 20 times by adding a constitutive promoter gpdA(p) to transcription factor AurF, which indicates AurF’s positive role in the biosynthesis of aurovertin. Taken together, our results provided not only an efficient way to generate bioactive fungal natural products but also realized the rational controlling their yields with designed promoters.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The aurovertins are highly reduced polyketide products with the basic structure of 2,6-dioxabicyclo[3.2.1]-octane ring system (DBO). Totally, near 30 aurovertin-type compounds including aurovertins A-S (Baldwin et al. 1964; Wang et al. 2005; Azumi et al. 2008; Zhou et al. 2009; Niu et al. 2010; Guo et al. 2013), asteltoxins (Adachi et al. 2015), avertoxins (Wang et al. 2015b), and citreoviridin (Franck and Gehrken 1980; Lin et al. 2016) were isolated from multiple fungal genus such as Calcarisporium, Metarhizium, Aspergillus, Penicillium, Pochonia, and mushroom Albatrellus. Due to the unusual polyketide-derived structure, the members of aurovertin-type compounds exhibit good bioactivities. Aurovertin B from Calcarisporium arbuscular (Baldwin et al. 1964) inhibits mitochondrial oxidative phosphorylation by binding to the β subunit of F1-ATPase (van Raaij et al. 1996), therefore uncompetitively inhibits ATP hydrolysis and has been implicated as potential therapeutics against cancer (Huang et al. 2008). With the similar mechanism, aurovertin D from Pochonia chlamydosporia showed strong toxicity toward the root-knot nematode Meloidogyne incognita and was found to mediate interactions between the fungus and its host (Wang et al. 2015a).
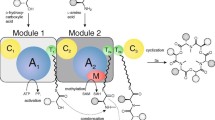
Due to the potent biological activities, the total synthesis and biosynthesis of aurovertins have been investigated since 1980s. Using D-glucose as starting material, Nishiyama et al. (1986) synthesized aurovertin B and determined its absolute configuration. The biosynthetic origin of aurovertin B was investigated by the labeling experiment to derive from a polyketide pathway (Steyn et al. 1981). Recently, the biosynthetic gene cluster of aurovertin was characterized by the genome mining of C. arbuscular and genetic disruption as well as in vitro biochemical experiments (Mao et al. 2015a, b). The cluster includes seven genes (Fig. 1a). The encoded enzymes contains a high reducing polyketide synthase (HR-PKS) AurA, a methyltransferase (MT) AurB, a flavin-dependent mono-oxygenase (FMO) AurC, a hydrolase AurD, a cyclase AurE, a transcription factor (TF) AurF, and an acyltransferase AurG. Four enzymes were reported to be required to form aurovertin E with DBO system. FMO AurC and hydrolase AurD are identified to generate the complex natural product scaffolds by iterative oxidizing and the regioselective hydrolyzing reactions (Mao et al. 2015). More recently, Wang and co-workers reported the heterologous expression in Aspergillus nidulans for the biosynthetic gene cluster of citreoviridin from A. terreus var. aureus (Lin et al. 2016). Here, our effort is aimed to establish an efficient way to produce the aurovertins, the bioactive fungal natural products, in the heterologous host A. nidulans. Then, we can determine the production of acetylated compound, e.g., aurovertin B by the controlling aurG expression using an inducible promoter.

Heterologous expression of aurovertin (aur) biosynthetic gene cluster in Aspergillus nidulans. a Aurovertin biosynthetic gene cluster from C. arbuscular. b Design for plasmid construction of different cluster gene combinations. (i) Five cluster genes aurA−F; (ii) aurA−F with overexpression transcription factor gpdA(p)::aurF; (iii) acyltransferase gene aurG with the inducible promoter xylP(p) and gpdA(p)::aurF in aurA−F (C) HPLC analysis of full aur cluster gene expression strains: TYZM14.2 (aurA−G) and A. nidulans RJMP1.59 (CK), seven new peaks were detected in the mutant at 363 nm
Materials and methods
Strain and culture conditions
The fungal strains used in this study are indicated in Table S1. Calcarisporium arbuscula NRRL 3705 was grown on potato dextrose agar medium (PDA) at 25 °C. A. nidulans strains were grown on glucose minimum medium (GMM) with or without appropriate supplementary (0.56 g uracil/L, 1.26 g uridine/L, and 0.5 μM pyridoxine HCl) at 37 °C for 72 h for sporulation. For aurovertin production, A. nidulans mutants were cultured in liquid GMM (supplemented with 5 g yeast extract/L, LMM) for 4 days at 25 °C, or were cultured in rice medium (rice 30 g, water 45 mL and supplemented with appropriate supplementary in 500-mL Erlenmeyer flasks) for 10 days at 25 °C for large fermentation. Escherichia coli DH5α strain was used routinely for DNA transformation and plasmids amplification, and LB media were used for E. coli culture. Saccharomyces cerevisiae strain BJ5464-NpgA (MATαura3-52 his3-Δ200 leu2-Δ1 trp1 pep4::HIS3 prb1Δ1.6R can1 GAL) was used for biosynthesis pathway reconstitution of plasmid construction with Yeast assembly method. For yeast culture, SD-drop out media were used for selection of plasmids transformed into S. cerevisiae at 30 °C.
Gene cloning and plasmids construction
The plasmids utilized in this work are listed in Table S1. The oligonucleotide sequences for PCR primers are given in Table S2. PCR reactions were performed with Phusion high-fidelity DNA polymerase (NEW ENGLAND Biolabs, NEB). PCR screening for transformants were carried out with 2 × TSINGKE® Master Mix (TSINGKE). The basic strategy for assembling of large PCR fragments is splicing by overlapping extension (SOE)-PCR and yeast homologous recombination (Wang et al. 2005). To clone aurovertin (aur) biosynthetic gene cluster, aur biosynthetic genes from aurA to aurF were divided into five fragments. Overlapping regions between adjacent flanking segments ranged from 230 to 250 bp and overlapping regions between the segments and the vector pYH-wA-pyrG were 40 bp. Each fragment (around 4 kb) was amplified with C. arbuscula genomic DNA by using designated primers (Table S2) respectively and then was assembled by homologous recombination to generate pWYaur in yeast (Wang et al. 2005). Positive yeast colonies were selected by screening PCR. Yeast plasmids were isolated by using Zymoprep™ (D2001) Kit (ZYMO RESEARCH) and transformed into E. coli DH5α. Bacterial plasmids were isolated by using a plasmid kit (OMEGA). Using Quick-change mutagenesis method, transcription factor gene aurF was inserted behind gpdA(p) in pYW25.16 to construct pYZM2.1 (Guo et al. 2013). Then, the inducible promoter xylP(p) and aurG gene were introduced to pYZM2.1 to get pYZM4.1 via an intermediate vector pYZM3.1. All plasmids were confirmed by restriction enzyme digestion (NEW ENGLAND Biolabs, NEB) and sequencing.
Transformation manipulation
A. nidulans RJMP1.59 was used as the heterologous host (Table S1). The protocols for protoplasts preparation and transformation were described previously (Zhou et al. 2009). pWYaur was transformed into A. nidulans RJMP1.59 to generate TYZM1. Then pYZM2.1 or pYZM4.1 was transformed into TYZM1.5 to obtain TYZM13 and TYZM14, respectively. The transformants were verified by using diagnostic PCR with appropriate primers (Table S2).
RNA preparation and semi-quantitative RT-PCR
For the analysis of function of aurF, 4 × 106 spores from TYZM1.5 and TYZM13.7 were inoculated into 20 mL LMM and cultivated at 25 °C for 4 days under dark conditions. To control acetylated aurovertin production with the expression of aurG, TYZM14.2 was inoculated into LMM with different carbon sources glucose or xylose. Three replicates were performed for each experiment. Then, mycelia were harvested and total RNA was extracted using TRIzol® Reagent (Life Technologies). Five micrograms of total RNA were reverse transcribed into first-strand cDNA using FastQuant RT-Kit (with gDNase) (TIANGEN Biotech, China). For transcription assessment, the coding regions of targeted genes were amplified using designated primer pairs, and the expression of ANactin was used as the internal control (Table S2).
Chemical analysis
A. nidulans strains were cultivated in LMM (supplemented with 5 g yeast extract/L) at 25 °C for 4 days in the dark. 2.0 × 106 spores were added to 10 mL medium per 60 mm plate. The cultures were extracted with methanol-ethyl acetate (10:90, v/v), and then evaporated under reduced pressure. The residue was dissolved in 1 mL methanol, and 20 μL of the solution was directly injected for HPLC analysis. Analytical HPLC was conducted with a Waters HPLC system (Waters e2695, Waters 2998, Photodiode Array Detector) using an ODS column (C18, 250 × 4.61 mm, COSMOSIL, 5 μm) with a flow rate of 1 mL/min. Crude extract was analyzed on a gradient of 60 to 80% methanol (0.1% formic acid) in 20 min followed by 100% methanol for 5 min. The fold differences of these compounds between control and mutants were calculated according to the following formula: [Area (Sample)-Area (Blank)]/[Area (WT)-Area (Blank)]. All MS and LC-MS analyses were performed using a Kromasil 100–5 C18 column (4.6 × 250 mm; 25 °C) on an Agilent 6520 Accurate-Mass QTOF LC/MS system (Agilent Technologies) equipped with an electrospray ionization (ESI) source. NMR spectra were recorded at room temperature with a Bruker Avance-500 spectrometer.
Purification of compounds 1–7
TYZM1.5 was cultured at 25 °C in 500-mL Erlenmeyer flasks containing rice media (rice 30 g, water 45 mL, and 90 μL 0.1% pyridoxine HCl). Totally, 1.2 kg rice was fermented. After 10 days of cultivation, the fermentation from 40 flasks was extracted three times with methanol: ethyl acetate (10:90). To discard the residual starch, the residual was extracted with EtOAc/H2O (1:1) for three times. The organic phase was evaporated to dryness under reduced pressure to afford the residue (5.44 g). The extracts were separated by a Sephadex LH-20 column with methanol. Further purification was carried out by a reverse-phase ODS column (25:75 → 100:0 MeOH/H2O) to give eight fractions. Fractions 4 and 5 were applied to another Sephadex LH-20 column with methanol. The compound 1 (7.0 mg) and 3 (17.6 mg) were obtained by semi-preparative HPLC (30:70 CH3CN/H2O, 2 mL/min) with a reverse-phase column (YMC-Pack ODS-A 250 × 10 mml S-5 μm 12 nm).
TYZM13.7 was cultivated as described above for the purification of compounds 2 and 4. 1.5 kg rice in 50 flasks was fermented in total. The crude extracts (10.9 g) were subjected to a ODS column, eluting with CH3CN-H2O (10:90–100:0 gradient system) to afford nine fractions. Fraction 3 was further fractionated into Frs. 3.1–3.8. Frs. 3.1 and 3.2 were combined and separated over a Sephadex LH-20 column using methanol as the mobile phase, and compound 2 (2.2 mg) was then isolated by a semi-HPLC eluting with methanol-water (55:45, v/v) with a flow rate of 2.5 mL/min. Compound 4 (3.5 mg) was purified from a combination of Frs. 3.3–3.6 by applying the same separation procedure.
TYZM14.2 was cultivated as described above for the purification of compounds 5, 6, and 7. Seven liters LMM media were used in total. The crude extracts (3.5 g) were subjected to a ODS column, eluting with CH3CN-H2O (10:90–100:0 gradient system) to afford six fractions. Fraction 4 was further fractionated into Frs. 4.1–4.8. Frs. 4.2 and 4.4 (24.5 mg) were combined and isolated by a semi-HPLC eluting with methanol-water (55:45, v/v) with a flow rate of 2.5 mL/min. And then compounds 5 (5.4 mg) and 6 (1.3 mg) were purified. Compound 7 (3.7 mg) was purified from Fr. 4.6 by applying the same separation procedure.
Analytical methods and equipment overview
HPLC analyses were performed on Waters 2695 Separations Module 241 with a RP C18 column (38020-41 COSMOSIL 5C18-MS-II Packed 242 Column; 4.6 × 250 mm) at a flow rate of 1 mL/min. Water and methanol were used as solvents. For analysis of the extracts, a linear gradient of methanol in water (60–80%, v/v) in 20 min was used. The column was then washed with 100% (v/v) methanol for 5 min and equilibrated with 30% (v/v) methanol for 5 min. Detection was carried out with a photodiode array detector and illustrated at 363 nm in this paper. For isolation of compound 1, 2, and 3, Shimadzu LC-6AD with a YMC-Pack ODS-A column (250 × 10 mm, 5 μm) was used. Water and methanol/acetonitrile were used as solvents. For isolation of 2 and 3, 40% acetonitrile was used with a flow rate at 3.0 mL/min. The column was then washed with 100% (v/v) acetonitrile for 10 min and equilibrated with 40% (v/v) acetonitrile for 10 min. All LC-MS analyses were performed on an Agilent 6520 Accurate-Mass QTOF LC/MS system (Agilent Technologies) equipped with a RP C18 column (Kromasil 100-5 C18 column; 4.6 × 250 mm; 25 °C) and an electrospray ionization (ESI) source. NMR spectra were recorded at room temperature with a Bruker Avance-500 spectrometer.
Statistical analysis
For statistical analyses, data were analyzed using the GraphPad Instate software package, version 5.01 (GraphPad software Inc) according to the Tukey-Kramer multiple comparison test at P ≤ 0.05. Mean values with asterisks are significant.
Results
Construction and transformantion of aurovertin biosynthetic gene cluster
The genomic DNA containing the biosynthetic cluster genes of aurovertins (Fig. 1a) described by Mao et al. (Mao et al. 2015a) was extracted from C. arbuscular NRRL 3705. We amplified the cluster gene fragment in three combinations to reach different expectations with the oligonucleotide primers (Fig. 1b): (1) clone aurA to aurF to get the simplest compound 3 (aurovertin E); (2) the use of the constitutive promoter A. nidulans gpdA(p) is to identify the function of TF AurF in the basis of (1) and control the production of aurovertins; (3) introduction of an inducible promoter xylP(p) derived from the P. chrysogenum endoxylanase (xylP) gene to the acyltransferase aurG is to control the yields of acetylated aurovertin compounds by using inducer xylose or xylan (Haas et al. 1993; Zadra et al. 2000). Then, three plasmids were constructed by using yeast recombination strategy (Colot et al. 2006) (Fig. 1b). To obtain the transformants of aurovertin cluster gene, A. nidulans RJMP1.59 protoplasts were transformed with plasmids pWYaur, which contained aurA to aurF in aurovertin cluster with auxotroph marker pyrG. Transformants were selected by auxotroph 0.5 μM pyridoxine HCl, then gDNA of the transformants were isolated and verified by PCR with the screening primers (Table S2). The PCR product showed an amplicon of 527 bp in the positive transformants named TYZM1. Then, TYZM1 protoplasts were transformed with plasmids pYZM2.1 which contained pyroA::gpdA(p)::aurF cassette and pYZM4.1 which contained pyroA::gpdA(p)::aurF and xylP(p)::aurG cassette, respectively. The gDNA of the transformants were isolated and verified by PCR with the screening primers (Table S2). The PCR product showed an amplicon of 1210 or 989 bp in the transformants, which named TYZM13 and TYZM14, respectively (Fig. S1 and Table S1).
Analysis and isolation of aurovertins in the heterologous host A. nidulans
The strains TYZM1.5 (aurA−aurF) and TYZM14.2 (aurA−aurG) were cultivated in LMM media, respectively. And the metabolites were extracted and analyzed by HPLC and LC-MS (Figs. 1c and S4). Compared to the isogenic control, four or seven new peaks were observed with UV absorptions identical to those of aurovertin-type compounds, respectively. The expression of aurG gene led to the production of the peaks 5, 6, and 7. Surprisingly, some peaks appeared in pairs, e.g., peaks 1, 2; 3, 4, and 5, 6 which suggested the production of aurovertin isomers (Fig. 1c).
To identify these new peaks, large-scale fermentation of TYZM1.5 and TYZM14.2 strains was conducted. After the extraction and isolation, peaks 1, 3, 5, and 7 were characterized as known compounds: aurovertins I (1), E (3), C (5), and B (7), respectively (Fig. 2), according to their MS and NMR data in comparison to the known compounds (Zhou et al. 2009; Niu et al. 2010; Mao et al. 2015b) (Tables S3, S5, S7, S9 and Supplementary Figures of NMR spectra). Following the above isolation, the compounds 2, 4, and 6 were purified carefully due to their instabilities and then immediately analyzed by MS and NMR. Mass analysis showed that they have the identical molecular weights at m/z = 404, 419 and 447 [M + H]+ with the related compounds 1, 3, and 5, respectively (Fig. S4), which suggest that they are paired isomers.

Structures of aurovertins identified in the heterologous host A. nidulans. Seven compounds are characterized as aurovertin I (1), iso-aurovertin I (2), aurovertin E (3), iso-aurovertin E (4), aurovertin C (5), iso-aurovertin C (6), and aurovertin B (7)
Stability assessment and structural determination of compounds 2, 4, and 6
From the previous purifications for compound 2, 4, and 6, we found that they are not stable and quickly converted into their stable forms. To assess their stabilities, we took 2 as an example for the analysis. HPLC analysis showed that compound 2 was a major peak when injection immediately after purification, then compound 2 was transformed to compound 1 near 1:1 ratio after 4 days. After 30 days, over 90% of compound 2 was transformed to compound 1 which showed compound 2’s instability (Fig. 3a, b). By comparisons of the 1H NMR spectra, almost all proton signals between compound 1 and compound 2 are consistent except for the H-13 (δ H 7.09, J = 11.3, 15.0 Hz, for 1; δ H 7.42, J = 12.0, 11.3 Hz, for 2) and H-14 (δ H 6.60, J = 15.0 Hz, for 1; δ H 6.16, J = 12.0 Hz, for 2) indicating that compound 2 was the specific 13-Z-isomer of compound 1 (Table S4 and Supplementary NMR spectra). Additionally, the Z-isomer (2) was found to isomerize readily to its E-isomeric form (1) under laboratory conditions, which could be explained by the light-driven cis to trans isomerization (Zaki et al. 2013). Compounds 4 and 6 showed a similar 1H NMR pattern at C13/C14 double bond as compound 2, which demonstrated the Z-configuration of compound 4 and compound 6 (Tables S6 and S8). Hereafter, compounds 2, 4, and 6 are named as iso-aurovertin I, iso-aurovertin E, and iso-aurovertin C, respectively.

Compound 2 is not stable and converted into 1 automatically. a 2’s stability assessment by HPLC analysis in different days after isolation. b 1H NMR spectra of the 1 and 2. 2 is mixed with 1 (i). Proton signals of 2- H13/H14 are clearly different with 1 (ii)
AurF is a positive transcription factor in the biosynthesis of aurovertin
We examined the transcription factor aurF’s function by the quantitative analysis of auroverin’s yields in the aurF overexpression (OE) strain. Both of aurA−F (TYZM1.5) and gpdA(p)::aurF, aurA−F (TYZM13.7) strains were cultivated in LMM media for 4 days at 25 °C. HPLC analysis of crude extracts showed that the yields of compound 1 and compound 3 in OE::aurF strain are much higher than the isogenic control (Fig. 4a, b). Quantitative analysis indicated that 20 times of compound 1 and compound 3 were produced in the OE::aurF strain in comparison to the control (Fig. 4c). To assess the role of aurF in the regulation of the aur cluster genes, RT-PCR was carried out and showed the transcription levels of aurB-F were greatly increased (Fig. 4d).

AurF is a positive transcription factor in the biosynthesis of aurovertin. a HPLC analysis of crude extracts from aurA−F strain and b aurF overexpressed strain; c Quantitative analyses of aurovertin I and E production in both strains. d RT-PCR analysis of aur cluster gene expression. 1: cDNAs from aurA−F strain; 2: cDNAs from aurF overexpressed strain; Anactin was used as internal standard
Promoter controlling aurovertin-type compound production
To control the production of different compounds, we constructed gpdA(p)::aurF with xylP(p)::aurG in the same vector and transformed it into the aurA−F strain. Thus, the yields of aurovertin-type compounds can be controlled by mediating of the expression of aurG with different carbon sources such as glucose and inducer xylose. By the analysis of transformants, seven peaks were found in the mutant as expected (Fig. 1c). To meidiate the production of different aurovertin-type compounds, we added glucose and xylose to the culture media at different ratios. When using glucose as sole carbon source, four major compounds 1, 3, 5, and 7 were produced whether 2 or 1% glucose in the culture media. While adding 1% xylose with 1% glucose to the media, the proportions and of 5 and 7 were greatly increased in comparison to the media only with glucose (Fig. 5a–c). When adding xylose as sole carbon source to the media, 5 and 7 are the major compounds and their yield are much higher in 2% xylose media than 1% xylose media suggesting the higher expression of aurG. Indeed, RT-PCR analysis of aurG in TYZM14.2 showed the higher expression with the induction of xylose (Fig. S3).

Promoters controlling aurovertin-type compound production in A. nidulans hosts. The strains were cultivated in the minimal media with inducer xylose. Glu: glucose, xyl: xylose. a HPLC analysis of crude extracts from aurA−G strain with different concentrations of glucose or xylose. b Quantitative analysis of the proportions of different aurovertin-type compounds. c Quantitative analysis of the relative yields of different aurovertin-type compounds
Discussion
Calcarisporium arbuscular was reported to produce the aurovertin-type compounds including aurovertins A, B, C (Mulheirn et al. 1974; Silber et al. 2013), D (Baldwin et al. 1964), E (Wang et al. 2005), J, M, T, and U (Zhao et al. 2016). However, the natural host C. arbuscular grows very slowly. The average growth rate is 1.75 mm per day in the defined media (Watson Pauline 1965). In this study, we aim to reconstruct the biosynthetic pathway of aurovertin quickly and express the cluster in an A. nidulans host.
For fungal heterologous expression, we chose A. nidulans RJMP1.59 as host (Palmer et al. 2010; Yin et al. 2012) (Table S1) which has been used for the heterologous production and activations of silent gene clusters such as immunosuppressant neosartoricins from dermatophytes and cryptic fungal pigment melanin from an endophytic fungus Pestalotiopsis fici previously (Yin et al. 2013; Zhang et al. 2017; Liu et al. 2017). The resultant plasmids containing the designated genes were transformed into A. nidulans. Transformants were obtained by PCR verification of the integration of aur cluster genes.
As expected, the transformant strain TYZM1.5 (aurA−aurF) can produce compound 3, precursor to other aurovertin derivatives, and TYZM14.2 (aurA−aurG) can produce compound 3, 5, and 7. Interestingly, compound 1 (aurovertin I) is not reported in C. arbuscular but was only found in P. chlamydosporia (Niu et al. 2010) and Albatrellus confluens (Guo et al. 2013). Heterologous expression of aur cluster indicated that the genes from C. arbuscular can synthesize compound 1 too. It may because that compound 1 turns into compound 5 almost completely so that it could not be detected in C. arbuscular. Following the above isolation, the compounds 2, 4, and 6 were purified carefully due to their instabilities and then immediately analyzed by MS and NMR, which were suggested that they are paired isomers of compounds 1, 3, and 5, respectively.
AurF was speculated to be mostly the transcriptional activator of the aur cluster (Mao et al. 2015b). In this study, improving about 20 times of compound 1 and compound 3 produced in the OE::aurF strain confirmed the function of aurF, which is consistent with the production of aurovertin-type compounds which demonstrated the positive role in the biosynthesis of aurovertins. Compounds 1 and 3 can be produced without the acyltransferase gene aurG. The acetylated aurovertin compounds such as compounds 7 often exhibited good activities (van Raaij et al. 1996). So, to acquire more product of compounds 7, we designed to control acyltransferase gene aurG by introducing of inducible promoter xylP(p). As shown in the results, we can not only improve the total products of aurovertins including compounds 3 only with glucose in the media, but also greatly increase the compound 7 which has potential therapeutics against breast cancer with adding xylose as sole carbon source to the media. Therefore, acetylated compounds of aurovertins could be controlled by the inducible expression of acyltransferase gene aurG in the heterologous A. nidulans host strain.
In summary, our studies demonstrate an efficient way to construct the biosynthetic pathway and produce the bioactive fungal natural products. Using this heterologous system, we expressed successfully seven aurovertin-type compounds, specifically; we characterized three new aurovertin isomers. Introduction of constitutive and inducible promoters to the expression system, we identified the TF AurF’s positive role in the biosynthesis of aurovertins and realized the control of acetylated aurovertins compounds production.
References
Adachi H, Doi H, Kasahara Y, Sawa R, Nakajima K, Kubota Y, Hosokawa N, Tateishi K, Nomoto A (2015) Asteltoxins from the entomopathogenic fungus Pochonia bulbillosa 8-H-28. J Nat Prod 78:1730–1734
Azumi M, Ishidoh K, Kinoshita H, Nihira T, Ihara F, Fujita T, Igarashi Y (2008) Aurovertins F-H from the entomopathogenic fungus Metarhizium anisopliae. J Nat Prod 71:278–280
Baldwin CL, Nash HA, Osborne CE, Brooker RM, Weaver LC, Jacobsen TN (1964) Biological and chemical properties of aurovertin, a metabolic product of Calcarisporium abuscula. Lloyd 27:88–95
Colot HV, Park G, Turner GE, Ringelberg C, Crew CM, Litvinkova L, Weiss RL, Borkovich KA, Dunlap JC (2006) A high-throughput gene knockout procedure for Neurospora reveals functions for multiple transcription factors. Proc Natl Acad Sci U S A 103:10352–10357
Franck B, Gehrken HP (1980) Citreoviridins from Aspergillus terreus. Angew Chem Int Ed Engl 19:461–462
Guo H, Feng T, Li Z-H, Liu JK (2013) Ten new aurovertins from cultures of the basidiomycete Albatrellus confluens. Nat Prod Bioprospect 3:8–13
Haas H, Friedlin E, Stoffler G, Redl B (1993) Cloning and structural organization of a xylanase-encoding gene from Penicillium chrysogenum. Gene 126:237–242
Huang TC, Chang HY, Hsu CH, Kuo WH, Chang KJ, Juan HF (2008) Targeting therapy for breast carcinoma by ATP synthase inhibitor aurovertin B. J Proteome Res 7:1433–1444
Lin TS, Chiang YM, Wang CC (2016) Biosynthetic pathway of the reduced polyketide product citreoviridin in Aspergillus terreus var. aureus revealed by heterologous expression in Aspergillus nidulans. Org Lett 18:1366–1369
Liu N, Hung YS, Gao SS, Hang L, Zou Y, Chooi YH, Tang Y (2017) Identification and heterologous production of a benzoyl-primed tricarboxylic acid polyketide intermediate from the zaragozic acid a biosynthetic pathway. Org Lett 19:3560–3563
Mao XM, Xu W, Li D, Yin WB, Chooi YH, Li YQ, Tang Y, Hu Y (2015) Epigenetic genome mining of an endophytic fungus leads to the pleiotropic biosynthesis of natural products. Angew Chem Int Ed Engl 54:7592–7596
Mao XM, Zhan ZJ, Grayson MN, Tang MC, Xu W, Li YQ, Yin WB, Lin HC, Chooi YH, Houk KN, Tang Y (2015) Efficient biosynthesis of fungal polyketides containing the dioxabicyclo-octane ring system. J Am Chem Soc 137:11904–11907
Mulheirn LJ, Beechey RB, Leworthy DP, Osselton MD (1974) Aurovertin B, a metabolite of Calcarisporium arbuscula. J Chem Soc Chem Comm 21:874–876
Nishiyama S, Toshima H, Kanai H, Yamamura S (1986) Total synthesis and the absolute-configuration of aurovertin B. Tetrahedron Lett 27:3643–3646
Niu XM, Wang YL, Chu YS, Xue HX, Li N, Wei LX, Mo MH, Zhang KQ (2010) Nematodetoxic aurovertin-type metabolites from a root-knot nematode parasitic fungus Pochonia chlamydosporia. J Agric Food Chem 58:828–834
Palmer JM, Mallaredy S, Perry DW, Sanchez JF, Theisen JM, Szewczyk E, Oakley BR, Wang CC, Keller NP, Mirabito PM (2010) Telomere position effect is regulated by heterochromatin-associated proteins and NkuA in Aspergillus nidulans. Microbiology 156(Pt 12):3522–3531
Pauline W (1965) Further observations on Calcarisporium arbuscula. Trans Brit mycol Soc 48(I):9–17
van Raaij MJ, Abrahams JP, Leslie AG, Walker JE (1996) The structure of bovine F1-ATPase complexed with the antibiotic inhibitor aurovertin B. Proc Natl Acad Sci U S A 93:6913–6917
Silber J, Ohlendorf B, Labes A, Nather C, Imhoff JF (2013) Calcaripeptides A-C, cyclodepsipeptides from a Calcarisporium strain. J Nat Prod 76:1461–1467
Steyn PS, Vleggaar R, Wessels PL (1981) Biosynthesis of the aurovertins B and D. The role of methionine and propionate in the simultaneous operation of two independent biosynthetic pathways. J Chem Soc Perkin Trans 1:1298–1308
Wang YL, Li LF, Li DX, Wang BL, Zhang KQ, Niu XM (2015) Yellow pigment aurovertins mediate interactions between the pathogenic fungus Pochonia chlamydosporia and its nematode host. J Agric Food Chem 63:6577–6587
Wang F, Luo DQ, Liu JK (2005) Aurovertin E, a new polyene pyrone from the basidiomycete Albatrellus confluens. J Antibiot 58:412–415
Wang M, Sun M, Hao H, Lu C (2015) Avertoxins A−D, prenyl asteltoxin derivatives from Aspergillus versicolor Y10, an endophytic fungus of Huperzia serrata. J Nat Prod 78:3067–3070
Yin WB, Amaike S, Wohlbach DJ, Gasch AP, Chiang YM, Wang CCC, Bok JW, Rohlfs M, Keller NP (2012) An Aspergillus nidulans bZIP response pathway hardwired for defensive secondary metabolism operates through aflR. Mol Microbiol 83:1024–1034
Yin WB, Chooi YH, Smith AR, Cacho RA, Hu YC, White TC, Tang Y (2013) Discovery of cryptic polyketide metabolites from dermatophytes using heterologous expression in Aspergillus nidulans. ACS Synth Biol 2:629–634
Zadra I, Abt B, Parson W, Haas H (2000) xylP promoter-based expression system and its use for antisense downregulation of the Penicillium chrysogenum nitrogen regulator NRE. Appl Environ Microbiol 66:4810–4816
Zaki MA, Balachandran P, Khan S, Wang M, Mohammed R, Hetta MH, Pasco DS, Muhammad I (2013) Cytotoxicity and modulation of cancer-related signaling by (Z)- and (E)-3,4,3′,5′-tetramethoxystilbene isolated from Eugenia rigida. J Nat Prod 76:679–684
Zhang P, Wang X, Fan A, Zheng Y, Liu X, Wang S, Zou H, Oakley BR, Keller NP, Yin WB (2017) A cryptic pigment biosynthetic pathway uncovered by heterologous expression is essential for conidial development in Pestalotiopsis fici. Mol Microbiol 105:469–483
Zhao H, Wu R, Ma LF, Wo LK, Hu YY, Chen C, Zhan ZJ (2016) Aurovertin-type polyketides from Calcarisporium arbuscula with potent cytotoxic activities against triple-negative breast cancer. Helv Chim Acta 99:543–546
Zhou ZY, Liu R, Jiang MY, Zhang L, Niu Y, Zhu YC, Dong ZJ, Liu JK (2009) Two new cleistanthane diterpenes and a new isocoumarine from cultures of the basidiomycete Albatrellus confluens. Chem Pharm Bull 57:975–978
Acknowledgements
We thank Dr. Yi Tang (University of California, in Los Angeles) for providing Calcarisporium arbuscular strain. We thank Drs. Jinwei Ren and Wenzhao Wang (Institute of Microbiology, CAS) for NMR and MS data collection. We thank Dr. Aili Fan for the helpful discussion for NMR spectra. W.B.Y. is a scholar of “the 100 Talents Project” of CAS.
Funding
This work is supported by the National Natural Science Foundation of China (31470178) and CAS/SAFEA International Partnership Program for Creative Research Teams.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interests
The authors declare that they have no competing interests.
Ethical statement
This article does not contain any studies with human participants or animals performed by any of the authors.
Electronic supplementary material
ESM 1
(PDF 2182 kb).
Rights and permissions
About this article

Cite this article
Ma, Z., Li, W., Zhang, P. et al. Rational design for heterologous production of aurovertin-type compounds in Aspergillus nidulans . Appl Microbiol Biotechnol 102, 297–304 (2018). https://doi.org/10.1007/s00253-017-8606-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-017-8606-9