
Abstract
Lutein is a commercial carotenoid with potential health benefits. Microalgae are alternative sources for the lutein production in comparison to conventional approaches using marigold flowers. In this study, a process analysis of a single-step simultaneous extraction, saponification, and primary purification process for free lutein production from wet microalgae biomass was carried out. The feasibility of binary solvent mixtures for wet biomass extraction was successfully demonstrated, and the extraction kinetics of lutein from chloroplast in microalgae were first evaluated. The effects of types of organic solvent, solvent polarity, cell disruption method, and alkali and solvent usage on lutein yields were examined. A mathematical model based on Fick’s second law of diffusion was applied to model the experimental data. The mass transfer coefficients were used to estimate the extraction rates. The extraction rate was found more significantly related with alkali ratio to solvent than to biomass. The best conditions for extraction efficiency were found to be pre-treatment with ultrasonication at 0.5 s working cycle per second, react 0.5 h in 0.27 L/g solvent to biomass ratio, and 1:3 ether/ethanol (v/v) with 1.25 g KOH/L. The entire process can be controlled within 1 h and yield over 8 mg/g lutein, which is more economical for scale-up.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Lutein is a commercially available, high-value product. It belongs to the group of the carotenoids and is responsible for their natural yellow to orange color (Taylor et al. 2012). Lutein is an important food colorant and is clinically proven to improve human eye health, especially for reducing the risk of cataract and age-related macular degeneration (Manayi et al. 2015). Traditionally, marigold flowers are used as natural lutein sources; however, microalgae have attracted increasing attention as an alternative source for lutein production due to faster growth rate, higher lutein content, less labor requirements, and reduced land requirement (Kim et al. 2013; Gong and Bassi 2016). The major challenge for commercial production of lutein from microalgae lies in downstream extraction and separation, which account for 50–80% of the total production cost (Amaro et al. 2015). This is because of the existence of a rigid cell wall in many algal species which limits the yield and rate of pigment extraction (Chan et al. 2013; McMillan et al. 2013). Thus, extraction procedures that are fast, simple, selective, and cost-effective need to be developed.
Conventionally, the natural lutein esters are first extracted from the dried source biomass, then saponification is used to produce lutein in free form (Wang et al. 2016), followed by further extraction for improved purification. In contrast to the conventional extraction methods (Lu et al. 2005; Deenu et al. 2013), where the extraction, saponification, and purification were each a separate step, we propose that operation units may be eliminated and the operational time could be greatly reduced using a single-step extraction method. By doing this, solvent usage can be reduced and potentially higher yield may be achieved. In addition, drying is extremely energy consuming; by eliminating this step, savings in cost and energy could also be expected.
Solvent extraction is widely employed to separate and purify lutein on an industrial scale. In general, during the extraction, the internal diffusion of the solute occurs from inside the algal mass to the surface, then the solute transfers to the stagnant solvent film around the particle, finally to the bulk solvent (Hojnik et al. 2008). The diffusion step in the particle is most commonly assumed as the rate-limiting step in this mass transfer mechanism, and can be predicted by the simplified unsteady-state second-order Fick’s equation (Shi et al. 2003; Hojnik et al. 2008).
The development of a new single-step method that skips drying and combines extraction, saponification, and purification approach may save both time and solvent. Previously, Wang et al. (2016) have developed a procedure for a combined procedure of lutein extraction from marigold flowers, but similar studies for more microalgal lutein extraction and purification are rare. It is of great interest to reduce the operating units and to investigate the kinetics of this process, in order to minimize the time and cost of free lutein recovery from microalgae. In this study, (i) development and process analysis of a single-step extraction, saponification, and purification method was conducted for extraction of lutein from wet microalgae biomass; (ii) the extraction kinetics of microalgal lutein extraction were first monitored under different conditions for a better understanding and optimization of the process; (iii) the experimental data was fitted using mathematical modeling; and (iv) the diffusion coefficients were determined and analyzed for different conditions to determine the extraction rate.
Methods
Microalgae type and cultivation
Chlorella vulgaris UTEX 265 (University of Texas, Austin, TX) was maintained in Bold’s Basal Medium (BBM) at 23 °C, containing (per L): 0.2 g NaNO3, 0.025 g CaCl2·2H2O, 0.075 g MgSO4·7H2O, 0.075 g K2HPO4, 0.175 g KH2PO4, 0.025 g NaCl, 0.005 g EDTA (anhydrous), 3.1 mg KOH, 0.05 mg FeSO4·7H2O, 0.11 mg H3BO3, 0.088 mg ZnSO4·7H2O, 0.014 mg MnCl2·4H2O, 0.007 mg, 0.016 mg CuSO4·5H2O, and 0.005 mg Co(NO3)2·6H2O. All the chemicals were dissolved in deionized water. The initial pH of the culture was adjusted to 6.8 ± 0.2 before sterilization at 121 °C for 20 min. Air was filter sterilized and used as the only autotrophic carbon source. The light intensity was 150 μmol photons/m2/s1, and a 16 h/8 h light/dark cycle was maintained during algal cultivation. Microalgae were harvested at the end of the growth period by centrifugation at 3500 rpm for 15 min at 4 °C in a Sorvall R40 centrifuge (ThermoScientific, USA). The wet microalgae pellet was washed three times with deionized water and frozen at −20 °C for further analysis.
Extraction method
Simultaneous extraction and saponification experiments were conducted in a batch extraction mode for 5 h in a 500-mL flat-bottom glass bottle with screw cap at 23 °C. For each experiment, 0.60 g dry weight equivalent frozen microalgae pellets were re-suspended in 10 mL ethanol, then subjected to ultrasonic treatment using a 50-W Ultra-sonication probe (Hielscher, Germany) at 0.5 s/s for a specified time (0 to 30 min). The biomass suspension was then added to the specified known volume of extraction solvent. In addition, ethanolic potassium hydroxide was added and this suspension was then well mixed using a magnetic stirrer. The binary solvents used were as follows (v/v): 1/3 ethanol/ether (C2H5OH/C2H5OC2H5), 1/1 ethanol/ether, 3/1 ethanol/ether, 1/1 ethanol/hexane (C2H5OH/C6H14), and 3/1 ethanol/hexane. The final solvent volume was 80, 160, or 320 mL, corresponding to the solvent to biomass (S/B) ratio of 0.13, 0.27, and 0.53 L/g dry cell, respectively. Various ratios were investigated, i.e., 10% (w/v) alkali in ethanol added to the ratio of dried algae biomass (R a) 1.67, 3.33, 6.67, and 13.3 L/kg; consequently, the concentration of alkali in extraction solvent (C a) was 0.313, 0.625, 1.25, or 2.5 g KOH/L solvent. The ranges were determined by preliminary experimental results (data not shown) and literature (Shi et al. 2003; Hojnik et al. 2008).
The samples were collected (1% total initial volume) at specified time intervals and centrifuged at 3500 rpm for 5 min, then the supernatants were combined. Water was added to achieve phase separation of lutein from other contents for purification purposes. The ether or hexane phase was collected and filtered through a 0.22-μm PTFE filter for UV/vis and HPLC analysis.
Solvent polarity measurements
The Nile Red dye (Sigma-Aldrich, Oakville) was used to determine the polarity of the liquids. A known quantity (10 μg/L) of dye was dissolved in the solvent for maximum absorbance wavelength of the solvent (λ max) scan from 800 to 200 nm. The λ max is then related to the polarity index \( {\mathrm{E}}_N^{\mathrm{T}} \).
Pigment quantification
The pigment analysis was carried out by HPLC (method adapted from Maxwell et al. (1994) with minor changes). An Acclaim C30 column (Thermo Scientific, USA) was used in an Agilent LC 1100 series system (Agilent Technologies, Canada). The sample was eluted at 1 mL/min (acetonitrile/methanol (9/1, v/v)) for 5 min then in a 4-min gradient change to 100% ethyl acetate. Re-equilibration between sample injections was carried out for 6 min. The total analytical run time was 14 min. Lutein, Chl a, Chl b, and β-carotene standards were purchased from Sigma-Aldrich (NJ, USA). Standard curves were obtained by injecting serial dilution samples ranging from 5 to 100 mg/mL. Each point was triplicated and the R 2 was above 0.990. Then the analytic sample retention time and peak area were used to determine the pigment contents in analytical samples. A Cary Bio 50 UV/Visible Spectrophotometer (Varian, USA) was used for simplified lutein measurement. The method was similar to that described by Hojnik et al. (2008). Absorbance at 445 nm was measured and calibrated with HPLC to obtain the calibration curve. Each solution was measured five times and the average was taken. Once the concentration of lutein was known, it was multiplied by the total volume of solvent to get the mass of lutein, then divided by the mass of microalgae dry weight to get the lutein yield (mg lutein/g cell dry weight).
Statistics
All the experiments were at least triplicated unless otherwise stated. The mean of the samples was reported and the STDEV was shown as error. A 2-factor-2-level full factorial design was used for the interaction study of solvent to biomass ratio and alkali concentration. The ranges were determined from previous studied results (Hojnik et al. 2008). ANOVA test using a confidence level of 95% was used to establish the statistical significance. The performance of the model was evaluated by the average absolute relative deviation (AARD).
Theory
The method used by Hojnik et al. (2008) was used to model the free lutein recovery process. The solvent first extracts lutein in the ester form, then saponification happens and free lutein is released. We speculate that both ester and free forms of lutein are undergoing diffusion in the entire process. But only free lutein was measured and reported as yield. The entire process that happened during the free lutein extraction can be described as (1) the solvent forms a thin layer around the solid matrix and dissolution or desorption happens, (2) the diffusion of solute/solvent mixture from the inside to the surface of the solid particle happens, (3) the solute moves across the stagnant film to the bulk solvent (Crank 1975), and in our process, an additional step needs to be considered, which is (4) the hydrolysis of the extracted lutein esters into free lutein. Since, usually, the second step is the rate-limiting step (Chan et al. 2014), the dynamic behavior of the extraction and saponification of lutein can be modeled (Hojnik et al. 2008). In this study, the assumptions of symmetrical and porous solid sphere were made for the mathematical model of lutein release from the microalgae into well-stirred bulk liquid. The external mass transfer resistance was considered negligible as we applied very low biomass to solvent ratios.
Fick’s second law of diffusion was widely employed for the modeling of the extraction process. Assuming uniform concentration of lutein in the microalgae particle, homogeneous solid-liquid mixing, and no interaction between the diffusion of solute and other compounds, the extraction of solute in solid particles depends on time, t, and radius, r. The equation can be written as
Here, the term on the left-hand side of Eq. (1) represents the extraction rate. The respective initial and boundary conditions can be written as
where C 0 is the initial lutein concentration in microalgae and C i is the lutein concentration at the solid-liquid interface. Assuming negligible mass transfer resistance of lutein in the very diluted microalgae extracts, the general solution of Eq. (1) for spherical samples can be written as
the mass of solute, M, here calculated as free lutein, transferred from the sample particle sphere at any time t, can be calculated by solving Eq. (5):
As only the first term of Eq. (6) remains significant after a short time, the equation can be expressed in the simplified form:
where c is the lutein concentration in the solution at time t and c ∞ is the concentration in the solution after infinite time. Here, the solid-liquid extraction can be divided into two stages. The first stage is the fast stage, which represents the period of fast extraction at a constant extraction rate limited by the film resistance. The internal diffusion is limiting the extraction rate in the second stage, where the extraction rate is much slower and keeps decreasing. By plotting Eq. (7) against time using experimental data, two intersecting straight lines can be drawn based on the points, representing the fast stage and the slow stage of the extraction, and the intersection of the two lines is the transition point of the two stages. As suggested by Osburn and Katz (1944), the two parallel diffusion processes happen simultaneously, and should both be considered to achieve better modeling results; Eq. (7) can be rewritten as
where f 1 and f 2 are the fractions of the solute extracted in fast and slow stages, corresponding to the diffusion coefficients (or more precisely, apparent mass transfer coefficients) D fast and D slow.
Results
Process analysis for lutein recovery
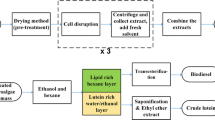
The lutein extraction process is illustrated in Fig. 1. The extraction, saponification, and primary purification steps for lutein recovery from the microalgal biomass were combined into a single-step operation. To simplify the process, a binary solvent system was used with alkali addition. As alkali was added to the reaction chamber, the saponification of lutein esters into free lutein happened simultaneously with the solid-liquid extraction. The separation and purification was achieved in the separator by using the polar solvent (ethanol) and the non-polar solvent (diethyl ether or hexane) and subsequent addition of water. As water was added to the binary solvent system, the more polar components, i.e., KOH, chlorophylls, proteins, sugars, and saponified lipids, remained in the bottom water-ethanol layer, while the non-polar solvent was in the upper layer and contained lutein and other carotenoids, mainly β-carotene. Compared to the conventional extraction methods (Lu et al. 2005; Deenu et al. 2013), two unit operations were eliminated and the operational time was reduced by up to 3 h using the single-step extraction method. The entire process does not involve temperature control; hence, it has low energy consumption compared with the more advanced super-critical CO2 extraction. Based on this method, the effects of solvent type, polarity, solvent to biomass (S/B) ratio, usage of alkali, and the presence of cell disruption pre-treatment on free lutein extraction kinetics were studied.

Schematic diagram of single-step lutein extraction, saponification and primary purification, and subsequent separation process to acquire free lutein
Kinetics modeling
The overall lutein mass transfer extraction rate was a combination of saponification and the free lutein extraction rate. The results were fitted into the model based on two term Fick’s second law of diffusion to determine the diffusivities during the free lutein extraction as described in the “Theory” section. The determination of a “fast-stage” and a “slow-stage” extraction is shown graphically in Fig. 2 based on Eq. (8). The slope and the intercept of a first-order fit were used to determine the diffusion coefficients as per Eq. (8). Since the slow extraction stage is the only process that is significant at later stages, the parameters of slow-stage fraction f 2 and diffusion coefficient D slow can be first determined from the slope and intercept of the second stage by a plot of \( \ln \left(\frac{c_{\infty }}{c_{\infty }-c}\right) \) vs. time. Then fast-stage fraction f 1 and diffusion coefficient D fast of the early fast-stage extraction can be determined as the second term in Eq. (8) is close to unity. The kinetics parameters and model statistics were calculated (Tables 1, 2, 3, 4, and 5). The diffusion coefficients D fast and D slow are good indicators of the rate of free lutein recovery. The average absolute relative deviation (AARD) was employed to estimate the model performance.

Representative first-order plot for the fast stage (solid line) and slow stage (dashed line) of lutein extraction. The points represent a typical extraction result in solvent to biomass ratio of 0.27 L/g, with 1.25 g KOH/L solvent. 23 °C, ultrasound pre-treatment of 10 min at 0.5 s/s working cycle
Effect of solvent type
The choice of solvent is known to affect the lutein extraction yield (Chan et al. 2013). Since in addition to the different solubility of lutein of various solvents, the polarity is also a significant factor for the lutein extraction yield; first, the binary solvent mixtures of identical polarities were compared for the best lutein extraction yield and extraction rate. This was achieved by adjusting the polarities of binary solvent systems to the same value using Nile Red dye as a polarity indicator. Based on the maximum absorbance wavelength of the solvent (λ max), 1:1 hexane/ethanol (v/v) has similar polarity with 3:2 ether/ethanol (v/v), with a polarity index \( {\mathrm{E}}_N^{\mathrm{T}} \) of 0.452. Therefore, these two solvent mixtures were compared (Fig. 3a).

Lutein extraction kinetics for a two different solvent mixture types: 1:1 hexane/ethanol (v/v) and 3:2 ether/ethanol (v/v) in solvent to biomass ratio of 0.27 L/g, with 0.625 g KOH/L solvent. b Lutein extraction kinetics for three different solvent mixture types in solvent to biomass ratio of 0.27 L/g: P1:1/3, P0:1/1, P-1: 3/1 ether/ethanol (v/v) with 1.25 g KOH/L solvent. c Lutein extraction kinetics for three different solvent to biomass ratios: V1 0.53 L/g, V0 0.27 L/g, V-1 0.13 L/g dry cell, in 1:3 ether/ethanol (v/v) with 1.25 g KOH/L solvent. d Lutein extraction kinetics for four alkali concentrations in extraction solvent (C a) levels: A-1, 0.313 g KOH/L solvent; A0, 0.625 g/L; A1, 1.25 g/L, and A2, 2.5 g/L in 1:3 ether/ethanol (v/v), solvent to biomass ratio of 0.53 L/g. The extraction was pre-treated with 10 min ultrasonication at 0.5 s working cycle per second. The lutein concentration in the bulk solvent is presented by a calculated lutein extraction yield ± STDEV, n = 3
In general, the extraction curves are similar to the conventional solid-liquid extraction curves of bioactive compounds (Tao et al. 2014). A fast stage occurred with a steep slope, followed by the exponential shape (Fig. 3a). The extracted lutein increased rapidly during the first 15 min, then incremented slowly after the initial extraction stage passed. However, the ether/ethanol binary solvent had a much higher lutein extraction yield than the hexane system, despite the two solvent systems having similar polarity. The final yield in the ether system was 6.5 mg/g, which was about 2.5 mg/g higher than that in hexane system after the 5-h extraction. The extraction kinetics curves were fitted into Eq. (8) to obtain D fast and D slow and the fractions of fast stage f 1 and slow stage f 2. Both D fast and D slow were higher in the ether solvent system than the hexane system (Table 1), indicating more efficient mass transfer in the ether system, so the ethanol/ether system was chosen for the subsequent experiments.
Effect of solvent polarity
Beside the solvent type, the polarity of the solvent as well plays a significant role in the lutein extraction. The polarity was adjusted by changing the ethanol to ether ratio at three levels: 1:3, 1:1, or 3:1 ether/ethanol (v/v). The higher polarity improved the extraction yield (Fig. 3b) but had a negative effect on the extraction rate (Table 2). The trend agreed with that of Ryckebosch et al. (2014). The rapid initial mass transfer of solute toward the bulk solvent is represented by the high values of D fast of the less polar solvent mixtures, 3.61–4.74 × 10−11 cm2/s. These mixtures extract the surface content faster due to their stronger solvent power, but did not extract as much lutein in the slower stage, corresponding to poor diffusion efficiencies, which is explicitly described by the decreased trend of D slow from 0.042 to 0.023 × 10−11 cm2/s as the ethanol ratio decreased.
Effect of solvent to biomass ratio
The three extraction curves (Fig. 3c) showed a similar final lutein yield after 5 h at different solvent to biomass (S/B) ratios. However, slight variance can be observed in the slope of the fast stage, indicating the extraction rates vary. This is better represented by the diffusion coefficients. As shown in the latter half of Table 2, the larger the amount of the solvent, the faster the diffusion in the fast stage. The D fast of the smallest S/B ratio, 1.08 × 10−11 cm2/s, was approximately half that of the 0.53 L/g trial. The final lutein concentration in solvent was similar; the extraction yield of free lutein was around 7.6–8.0 mg/g.
Effect of alkali usage
Since simultaneous saponification and extraction of lutein was conducted in a single step for a simpler process, it was important to also study the effect of alkali usage on the final yield in addition to the traditional mass transfer operators. The alkali used in this experiment was KOH dissolved in ethanol in a volume percentage of 10% (w/v), and added to 320 mL of solvent mixture containing 0.60 dry weight equivalent wet algae biomass. Different levels of the 10% alkali solution were added, specifically 1, 2, 4, and 8 mL, which has a concentration of alkali in extraction solvent (C a) of 0.313, 0.625, 1.25, and 2.5 g KOH/L, or alkali to dried algae biomass ratio (R a) of 1.67, 3.33, 6.67, and 13.3 L/kg, respectively. The smallest amount of alkali used, denoted as A-1(Fig. 3d), had a very low lutein yield. It was about half of the value of the other trials due to the insufficient OH− to cleave lutein esters into free form. No significant difference was observed for the C a = 0.625 or 1.25 g/L trials; both yielded around 8.0 mg/g lutein during the experiment, typical of most of the other experiments in this study. The C a = 2.5 g/L trial had a similar trend with the C a = 0.625 or 1.25 g/L ones, but degradation was observed after the initial phase; therefore, the final lutein yield was not as high. The lutein yield topped at 30 min, 7.1 mg/g, but decreased to 5.9 mg/g at the end. The rates of diffusion can be better monitored from Table 3. The highest concentration of alkali had the fastest extraction rate, with a diffusion coefficient of 4.30 × 10−11 cm2/s. The D fast of the 2- or 4-mL experiments were 1.77–2.05 × 10−11 cm2/s. The values of D slow showed an opposite trend as D fast, decreased as the alkali concentration went higher. The lowest concentration of alkali performed poor in every aspect.
Interaction of alkali and solvent to biomass ratio
It is also important to understand whether the alkali concentration or the absolute alkali amount in the solvent is more significant for extraction and hydrolysis rate. This knowledge would offer a more precise guideline for the usage of solvent and alkali amount for optimal lutein extraction yield and efficiency. A full factorial designed study was used to reveal the interaction study solvent to biomass ratio and alkali usage (Table 4). The ANOVA showed that the lutein extraction rate, as indicated by D fast, was more significantly related with the concentration of KOH in solvent than with the ratio of KOH to microalgae dry mass (see Table S1). Thus, it can be concluded that the lutein yield was limited by the amount of alkali added, though the extraction and hydrolysis rate is co-related to the concentration. The diffusion coefficients were fitted into the second-order polynomial model, and the fitted model for D fast and D slow is listed below:
where A is the coded alkali concentration and V is the coded solvent S/B ratio.
Since the factor parameters were codified, it can be easily seen that the alkali concentration has a more significant effect on the lutein extraction rate than the solvent to biomass ratio. The interaction term has a weight of 0.552, so it could not be ignored. Almost all the term coefficients for D fast are positive, indicating that the increase of both factors and their interaction within the tested range would benefit the extraction yield. The highest D fast was obtained at 0.53 L/g S/B ratio and 2.5 g KOH/L (Fig. 4a) in the experimental range. Contrary to the D fast, the slow-stage extraction rate was negatively related to the alkali concentration and solvent to biomass ratio, and the values were much smaller (Fig. 4b).

Lutein extraction kinetics fitted models for D fast (a), D slow (b), and a fraction of the fast stage (c)
In addition to the diffusion coefficients, the fraction of the fast stage within the entire extraction process was studied. Maximizing this portion would greatly enhance the extraction efficiency. The model is given below:
The optimal point was at 320 mL solvent and 2 mL KOH (Fig. 4c). The terms beside the S/B ratio are all negative, showing that the parameters beside the S/B ratio have negative effects on the fast-stage fraction.
Effect of biomass pre-treatment
The results to determine the effect of pre-treatment for lutein extraction from wet microalgae biomass are presented in Fig. 5. The initial mass transfer rate together with the lutein ester hydrolysis (saponification) rate was fast, obtained from the steep slopes of the fast stage in ultrasound-treated 30 min, 10 min, and no treatment, corresponding to the D fast values of 1.58, 1.36, and 1.03 × 10−11 cm2/s, respectively (Table S2). The increased trend of D fast as the ultrasonication time increased indicated that the treatment of cell disruption benefited the washing effect. However, the fast-stage fraction was highest at the 10-min rather than the 30-min level, showing the degradation effect existed during the ultrasonication.

Lutein extraction kinetics for three different pre-treatment methods: US1, ultrasonication for 10 min at 0.5 s working cycle per second; US3, ultrasonication for 30 min; US0, no pre-treatment. The extraction was 1:3 ether/ethanol (v/v), solvent to biomass ratio of 0.27 L/g, and 1.25 g KOH/L. The lutein concentration in the bulk solvent is presented by calculated lutein extraction yield ± STDEV, n = 3
Discussion
The use of binary solvent in this study enabled easier lutein extraction from wet biomass, because the polar solvent can easily penetrate into the wet biomass containing water, and the non-polar solvent has stronger affinity for lutein extraction (Ryckebosch et al. 2014). The non-polar solvent served two roles in the process: extracting solvent of leaching as well as the liquid-liquid extraction solvent. This single-step approach potentially can save both overall extracting time and lead to reduced solvent usage. A comparison of the operation method and the yield of different lutein extraction studies is exhibited in Table 5. The choice of the inter-miscible polar and non-polar solvent systems can also reduce the mass transfer resistance by avoiding additional liquid-liquid extraction steps. In terms of solvent selection, ethanol was reported to be the best solvent for lutein extraction from wet microalgae, and hexane is the most conventional solvent applied for lutein extraction from marigold flowers on the industrial scale (Soares et al. 2016). In addition, diethyl ether (ether) is reported as a good solvent due to the high solubility of lutein and its low boiling point (Chan et al. 2013). Therefore, ethanol/hexane and ethanol/ether solvent systems were chosen for further investigation in this study.
In the fast stage of the extraction, the extractable components located on the exposed particle surfaces are washed into the bulk solvent rapidly; while in the slow stage, the diffusion of dissolved solute inside the solid particles controls the extraction rate (Tao et al. 2014). The model derived from Fick’s law fitted the extraction curves quite well, judging from the small deviation. The fraction of lutein released was much higher in the fast stage than in the slower stage (Fig. 3, 4, and 5); however, the calculated values of f 1 (Tables 1, 2, 3, and 4, S2) were low, below 0.65, which is because a uniform c 0 8.5 mg/g was assumed to better compare the diffusion coefficients D fast and D slow. The value 8.5 mg/g was determined experimentally based on the highest lutein yield. Although f 1 does not represent the true fraction of the fast stage, the general trend still can be inferred from the values. The rate constants of diffusion (D fast of 0.5–4.3 × 10−11 cm2/s) were much smaller than those of previous reported studies with extraction from marigold flowers (Hojnik et al. 2008). It is speculated that the smaller microalgae cell size may be more difficult to extract inner contents as the solvent moves into the cell and the lutein diffuses out.
The lutein content yielded from the single-step extraction was higher than those of other studies because the improved operation procedure reduced the operation units and minimized the lutein loss during drying and transferring (Dineshkumar et al. 2015; Chiu et al. 2016). The solvatochromic dye Nile Red was used as a polarity indicator to adjust different types of binary solvent systems, so as to eliminate the effect of polarity from solvent solubility in the kinetics study. Therefore, the reason for the variation of the extraction yield and rate contributed to the difference in lutein solubility, which is 20 mg/L for hexane, and 2000 mg/L for ether (Craft and Soares 1992).
Higher polarities resulted in a higher lutein yield yet slower extraction; the potential reason is that ethanol could form hydrogen bonds and can weaken the van der Waals force between lutein-lipid associations and the cell membrane. The larger ratio of polar solvent ethanol facilitated the dissolution of the lutein bond to the chloroplast membrane and enabled solvent diffusion into the chloroplast inside the center of the cell matrix. The fractions of fast stage and slow stage of the three ratios of solvent mixture did not make a significant difference.
Generally, the concentration of active compounds in the solvent phase increases until the equilibrium is reached (Hojnik et al. 2008). The trend of changing D fast as the S/B ratio changed is in good agreement with the theory of the driving force of diffusion being the concentration gradient. As the final lutein concentration in the solvent was much lower than saturation, the amount of solvent used was more than the minimum solvent needed to fully extract lutein. This minimized the diffusion resistance from the intact film to bulk solvent, and enabled the use of Eq. (8). In general, the values of D slow showed an opposite trend as D fast, decreased as the alkali concentration went higher, probably due to the reduced concentration difference, as more lutein was extracted to the bulk solvent in the fast stage.
As the molecular size of lutein esters is larger than the cell membrane pore size, in the traditional process, it was necessary to lyse the intact cell for bioactive compound extraction (Azencott et al. 2007). However, OH– and free lutein is small enough to travel through the cell wall pores, which may also benefit the extraction rate. In the study of alkali usage, the alkali amount cannot go too low for full release of free lutein, nor too high to avoid degradation. To precisely understand the usage of solvent and alkali amount for optimal lutein extraction yield and efficiency, the interaction of alkali usage and S/B ratio was first investigated in this study. From the results, it was concluded that the lutein yield was limited by the amount of alkali added, while the extraction rate was more directly related to the concentration. To more clearly indicate the significance of experimental factors and to simplify the calculation, the factor values were codified (Table 4); the alkali concentration has a more significant effect on the lutein extraction rate (as represented by the fast-stage diffusion coefficient) than the S/B ratio. The change in D slow does not make a significant difference in practicality due to the low efficiency of the slow stage. The reason for the lower values of f 1 should be attributed to the following three reasons: (1) Lutein is located inside the chloroplast in the center of the cell matrix, so the diffusion of lutein to the particle surface takes time. (2) Saponification was conducted in parallel with extraction, which should take a long time than one-step extraction. (3) To acquire better comparisons of D fast and D slow, a uniform c 0 value of 8.5 mg/g was used instead of the infinite lutein concentration in each extraction in the diffusion model fitting.
The presence of biomass pre-treatment is widely recognized as an effective method to enhance lutein yield (Guedes et al. 2011). However, its effect on lutein extraction kinetics was seldom studied. Therefore, lutein extraction yield in the presence of cell disruption was compared with non-treated biomass. Considering the scale of operation and cell disruption efficiency, ultrasonication was chosen, since it can be adapted to larger-scale continuous extraction and was reported to maximum carotenoid yield from freeze-dried thraustochytrids biomass (Singh et al., 2015). From the results (Fig. 5 and Table 5), the fast-stage fraction was highest at the 10-min level rather than the 30-min level. This is probably due to the degradation of heat and oxygen-sensitive lutein during the ultrasonication process. The degradation was also observed in the final lutein yield: the 10-min level had a value of 8.3 mg/g while that of the 30-min level was only 6.2 mg/g. The non-treated cells had much more difficulty in releasing lutein, and the yield was less than half of those with cell disruption treatments. Additional cell disruption pre-treatment breaks up the cell wall, increases the contact surface area of biomass to solvent, and facilitates the mass transfer of intracellular solute. Therefore, the increased yield and extraction efficiency of the pre-treatment of algal biomass make it worthwhile to include an additional operation step. Meanwhile, the temperature dissipated during the cell disruption should be closely monitored to avoid lutein degradation. Moreover, although the temperature rising can accelerate the mass transfer rate, its negative effects on lutein stability, temperature-control cost, and solvent losses should be considered especially when ether was used, which has a boiling point of 34.6 °C (Tao et al. 2014). Thus, the experiments were conducted at 23 °C and the effect of temperature on extraction was not studied.
In conclusion, this is the first study on lutein extraction kinetics from microalgae. The feasibility of binary solvent for wet biomass extraction was investigated. Mixing a polar and a non-polar solvent together is energy and time saving as polar solvent better extracts lutein from the wet cell matrix while the non-polar solvent has higher solubility of lutein and could be easily separated away by subsequent water addition. Therefore, a single-step simultaneous extraction, saponification, and primary purification process for free lutein production from wet microalgae biomass, was developed and optimized. Two parallel diffusion processes, fast and slow extraction processes, were considered to model the kinetics of lutein extraction and hydrolysis. Based on the kinetics study, the free lutein recovery process can be limited within 1 h in a simple extraction plus separation manner. Higher lutein yield (over 8.0 mg/g) was achieved in the single-step extraction. The best conditions for extraction efficiency were with 10 min’ pre-treatment of ultrasonication at 0.5 s working cycle per second, react 0.5 h in 0.27 L/g S/B ratio, 1:3 ether/ethanol (v/v) with 1.25 g KOH/L. The use of alkali should correspond to the solvent volume, since the lutein extraction rate was more significantly related with the KOH concentration in solvent, rather than the ratio to biomass. Although a higher solvent to biomass ratio can increase the efficiency, considering the solvent cost, additional alkali needed, and similar lutein yield, less solvent options are recommended, especially for large-scale operations. Overall, the results obtained in this study can lead to new and improved techniques for the design and optimization of the lutein extraction process, and scale-up.
References
Amaro HM, Fernandes F, Valentão P, Andrade PB, Sousa-Pinto I, Malcata FX, Guedes AC (2015) Effect of solvent system on extractability of lipidic components of Scenedesmus obliquus (M2-1) and Gloeothece sp. on antioxidant scavenging capacity thereof. Mar Drugs 13:6453–6471
Araya B, Gouveia L, Nobre B, Reis A, Chamy R, Poirrier P (2014) Evaluation of the simultaneous production of lutein and lipids using a vertical alveolar panel bioreactor for three Chlorella species. Algal Res:218–222
Azencott HR, Peter GF, Prausnitz MR (2007) Influence of the cell wall on intracellular delivery to algal cells by electroporation and sonication. Ultrasound Med Biol 33:1805–1817
Chan CH, Yusoff R, Ngoh GC (2014) Modeling and kinetics study of conventional and assisted batch solvent extraction. Chem Eng Res Des 92:1169–1186
Chan MM-C, Ho SS-H, Lee DD-J, Chen CC-Y, Huang C-C, Chang J-S (2013) Characterization, extraction and purification of lutein produced by an indigenous microalga Scenedesmus obliquus CNW-N. Biochem Eng J 78:24–31
Chiu PH, Soong K, Chen CNN (2016) Cultivation of two thermotolerant microalgae under tropical conditions: influences of carbon sources and light duration on biomass and lutein productivity in four seasons. Bioresour Technol 212:190–198
Craft NE, Soares JH (1992) Relative solubility, stability, and absorptivity of lutein and β -carotene in organic solvents in organic solvents. J Agric Food Chem:431–434
Crank J (1975) The mathematics of diffusion. Oxford Univ Press 414
Deenu A, Naruenartwongsakul S, Kim SM (2013) Optimization and economic evaluation of ultrasound extraction of lutein from Chlorella vulgaris. Biotechnol Bioprocess Eng 18:1151–1162
Dineshkumar R, Dash SK, Sen R (2015) Process integration for microalgal lutein and biodiesel production with concomitant flue gas CO2 sequestration: a biorefinery model for healthcare, energy and environment. RSC Adv 5:73381–73394
Gong M, Bassi A (2016) Carotenoids from microalgae: a review of recent developments. Biotechnol Adv 34:1396–1412
Guedes AC, Amaro HM, Malcata FX (2011) Microalgae as sources of carotenoids. Mar Drugs 9:625–644
Ho SH, Xie Y, Chan MC, Liu CC, Chen CY, Lee DJ, Huang CC, Chang JS (2015) Effects of nitrogen source availability and bioreactor operating strategies on lutein production with Scenedesmus obliquus FSP-3. Bioresour Technol 184:131–138
Hojnik M, Škerget M, Knez Ž (2008) Extraction of lutein from marigold flower petals—experimental kinetics and modelling. LWT - Food Sci Technol 41:2008–2016
Kim J, Yoo G, Lee H, Lim J, Kim K, Kim CW, Park MS, Yang JW (2013) Methods of downstream processing for the production of biodiesel from microalgae. Biotechnol Adv 31:862–876
Lu YB, Yao H, Gong Y, Sheng Q (2005) Effects of enzymatic treatment of corn gluten meal on lutein and zeaxanthin extraction. Int J Food Prop 8:161–169
Manayi A, Abdollahi M, Raman T, Nabavi SF, Habtemariam S, Daglia M, Nabavi SM (2015) Lutein and cataract: from bench to bedside. Crit Rev Biotechnol 8551:1–11
Maxwell DP, Falk S, Trick CG, Huner NP (1994) Growth at low temperature mimics high-light acclimation in Chlorella vulgaris. Plant Physiol 105:535–543
McMillan JR, Watson IA, Ali M, Jaafar W (2013) Evaluation and comparison of algal cell disruption methods: microwave, waterbath, blender, ultrasonic and laser treatment. Appl Energy 103:128–134
Osburn JO, Katz DL (1944) Structure as a variable in the application of diffusion theory to extraction. Trans Am Inst Chem Eng 40:511–531
Ryckebosch E, Bermúdez SPC, Termote-Verhalle R, Bruneel C, Muylaert K, Parra-Saldivar R, Foubert I (2014) Influence of extraction solvent system on the extractability of lipid components from the biomass of Nannochloropsis gaditana. J Appl Phycol 26:1501–1510
Shi J, Maguer M, Bryan M, Kakuda Y (2003) Kinetics of lycopene degradation in tomato puree by heat and light irradiation. J Food Process Eng 25:485–498
Singh D, Gupta A, Wilkens SL, Mathur AS, Tuli DK, Barrow CJ, Puri M (2015) Understanding response surface optimisation to the modeling of Astaxanthin extraction from a novel strain Thraustochytrium sp. S7. Algal Res 11:113–120
Soares AT, Lopes RG, Derner RB, Filho NRA (2016) Improvement of the extraction process for high commercial value pigments from Desmodesmus sp. Microalgae J Braz Chem Soc 6:1083–1093
Tao Y, Zhang Z, Sun DW (2014) Kinetic modeling of ultrasound-assisted extraction of phenolic compounds from grape marc: influence of acoustic energy density and temperature. Ultrason Sonochem 21:1461–1469
Taylor RL, Rand JD, Caldwell GS (2012) Treatment with algae extracts promotes flocculation, and enhances growth and neutral lipid content in Nannochloropsis oculata-a candidate for biofuel production. Mar Biotechnol 14:774–781
Wang T, Han J, Tian Y, Zhang D, Wang Y, Wu Y, Ni L (2016) Combined process of reaction, extraction, and purification of lutein in marigold flower by isopropanol–KOH aqueous two-phase system. Sep Sci Technol 6395:1–9
Acknowledgements
The NESRC Discovery Grant to Dr. Amarjeet Bassi [170124-2011-RGPIN] is gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Electronic supplementary material
ESM 1
(PDF 357 kb)
Rights and permissions
About this article

Cite this article
Gong, M., Wang, Y. & Bassi, A. Process analysis and modeling of a single-step lutein extraction method for wet microalgae. Appl Microbiol Biotechnol 101, 8089–8099 (2017). https://doi.org/10.1007/s00253-017-8496-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-017-8496-x