
Abstract
The heterotrophic microalga Crypthecodinium cohnii is well known for its lipid accumulation, with a high proportion of docosahexaenoic acid (DHA). In this study, we report a novel screening approach to obtain mutants of C. cohnii with high growth and lipid content using the acetyl-CoA carboxylase (ACCase) inhibitor sethoxydim. C. cohnii mutants were generated using atmospheric and room-temperature plasma (ARTP) and then screened for two rounds in media supplemented with sethoxydim. These efforts led to the identification of mutant M-1-2, which had 24.32% higher growth and 7.05% higher lipid content than the wild type, demonstrating the effectiveness of the sethoxydim-based screening. Consistently, the M-1-2 mutant displayed a 16.15% increase in ACCase enzymatic activity and 1.53-fold upregulation of its ACCase-encoding gene based on comparative ACCase activity analysis and transcriptomic analysis, respectively. In addition, transcriptomic analysis showed that transcripts involved in fatty acid biosynthesis, energy, central carbohydrate, and amino acid metabolism were upregulated in the mutant compared to the wild type.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
ω-3 long-chain polyunsaturated fatty acids (PUFAs) have attracted significant attention because of their relevance to human health (Glaser et al. 2011). Docosahexaenoic acid (DHA, 22:6, n-3), an important ω-3 PUFA, is an essential fatty acid component for brain development in infants (Kuratko and Salem 2013) and an important component in the membranes of human nervous and retinal tissues (Jiang et al. 1999). The traditional source of DHA is fish oil, as ocean fish can accumulate ω-3 PUFAs by consuming DHA-rich algae as food (Yongmanitchai and Ward 1989). However, in recent years, several issues have been raised with respect to the suitability and sustainability of fish oil as a source of DHA, such as its strong odor, unpleasant flavor, and heavy-metal contamination as well as a decrease in ocean fisheries (Jiang and Chen 1999). In addition, the presence of large quantities of saturated and other ω-3 fatty acids, e.g., eicosapentanoic acid (EPA), in fish oil also makes downstream separation and purification of DHA time-consuming and expensive (Jiang et al. 1999). As an alternative, methods to cultivate DHA-rich microalgae directly have been applied for commercial DHA production (Santos-Sánchez et al. 2016).
The heterotrophic marine microalga Crypthecodinium cohnii can accumulate DHA content up to 30–50% of its constituent fatty acids while accumulating a content of less than 1% of other types of PUFAs (Jiang et al. 1999; Yaakob et al. 2014). Due to these advantages, it has been widely used in industrial fermentation for algal DHA production in recent years (Jiang et al. 1999). Significant efforts have also been made to improve DHA productivity in C. cohnii. Fermentation parameters have been optimized in fed-batch experiments of C. cohnii; high levels of production of 109 g/L dry biomass, 61 g/L lipid, and 19 g/L DHA have been reported (De Swaaf et al. 2003). In addition, chemical modulators, such as n-dodecane (Lopes da Silva and Reis 2008), butylated hydroxyanisole (BHA) (Sui et al. 2014), and ethanolamine (ETA) (Li et al. 2015), were also applied to improve lipid accumulation and DHA production in C. cohnii. However, to date, the whole-genome sequence of C. cohnii has not been obtained, restricting the application of more radical genetic engineering approaches to produce C. cohnii mutants with better biological properties. However, high-yield mutants of C. cohnii can be obtained using suitable mutagenesis and high-efficiency screening.
Atmospheric and room-temperature plasma (hereafter ARTP) is a new mutagenesis technology that uses helium radiofrequency glow discharge plasma jets to change the microbial genome sequence, eventually leading to microbial mutation (Zhang et al. 2014). Compared to traditional mutation systems, ARTP possesses remarkable advantages, such as high efficiency, nontoxicity, zero pollution of the environment, and low cost (Wang et al. 2014). In recent years, ARTP has been successfully used for microbial breeding of bacteria (Ma et al. 2015a) and fungi (Qiang et al. 2014). ARTP was also recently applied to C. cohnii, leading to the identification of mutants with enhanced extracellular polysaccharides (EPS) (Liu et al. 2015a) and demonstrating the high mutagenesis efficiency of this technology. However, to obtain mutants with improved biological properties, it is still crucial to develop proper and efficient screening methods. Most conventional screening methods, such as phenotypic screening, are time- and labor-consuming (Gao et al. 2013). To address these issues, a new screening method was previously introduced for C. cohnii based on the colonial morphology, and the implementation of the method led to the identification of mutants with 33.85% higher EPS volumetric yield (Liu et al. 2015a). Nevertheless, to further increase growth and lipid content in C. cohnii, more effective screening methods must be developed.
Sethoxydim is a cyclohexanedione herbicide used to control grass weeds in dicotyledonous and small grain crops (Humburg et al. 1989). Early studies have found that sethoxydim targets acetyl-CoA carboxylase (ACCase) (Dotray et al. 1993), which is a key enzyme that catalyzes the first reaction in fatty acid synthesis: the conversion of acetyl-CoA to malonyl-CoA (Hu et al. 2008). The roles of ACCase in improving lipid accumulation were also previously demonstrated in plants and microalgae. For example, overexpression of the ACCase coding gene resulted in higher lipid content in potato tubers of Solanum tuberosum (Klaus et al. 2004). In addition, the expression patterns of ACCase coding gene were investigated in the microalgae Chlorella sorokiniana, and the results showed that the expression level of the ACCase gene was upregulated 2.1-fold following the growth time course from the exponential to stationary phases (Wan et al. 2011).
In this study, a high concentration of sethoxydim was found to significantly inhibit C. cohnii growth. We thus proposed to apply sethoxydim as a selective agent to screen for mutants with increased ACCase activity and possibly increased growth and lipid content as well. To this end, an integrated ARTP mutagenesis and sethoxydim-based screening protocol was applied to C. cohnii, leading to the identification of several mutants with high growth and high lipid content. In addition, comparative transcriptomic analysis was applied to explore the transcript changes associated with the increased growth and lipid content of the C. cohnii mutant, and the new information obtained could be useful for the better understanding of C. cohnii metabolism and for the engineering of high-yield C. cohnii strains in the future.
Materials and methods
Algae strains and cultivation
C. cohnii ATCC 30556 was obtained from the American Type Culture Collection (ATCC). It was grown on basal medium (pH 6.5) consisting of 9 g/L glucose, 2 g/L yeast extract (Oxoid, Basingstoke, UK), and 25 g/L sea salt (Sigma-Aldrich, St Louis, MO) (Li et al. 2015; Sui et al. 2014). The seed cultures were cultivated in 250-mL Erlenmeyer flasks each containing 50 mL of basal medium at 25 °C and shaken at 180 rpm. The seed cultures were cultivated for 2 days after reaching the exponential growth phase and were subsequently used to inoculate 100 mL of medium (pH 6.5) consisting of 27 g/L glucose, 6 g/L yeast extract, and 25 g/L sea salt in 500-mL Erlenmeyer flasks for 3–4 days. These cultures were used to inoculate bioreactors at an inoculum size of 10% (v/v).
Assay of C. cohnii sensitivity to sethoxydim
Fifty microliters of fresh C. cohnii culture in the exponential phase was spread on plates containing a concentration gradient of sethoxydim (i.e., 0, 5, 10, and 20 μM) and cultivated at 25 °C for up to 10 days. A minimal three replicate plates were used for each condition. Confirmation of initial selective concentration was conducted using six replicate plates.
ARTP mutagenesis of C. cohnii and screening
Cells were cultivated in the basal medium at 25 °C and 180 rpm for 36 h (Liu et al. 2015a), and the cell concentration was adjusted to 106–107 mL−1 with fresh basal medium. To protect cells from damage caused by water evaporation, glycerol (10% v/v) was added to the cell suspension. A 10-μL aliquot of the above suspension was applied to a sterilized sample plate. The sample plate was later exposed to ARTP mutagenesis treatment for a given time using an ARTP Breeding Mutagenesis Machine (a Model ARTP-M) (Yuanqingtianmu Biotechnol Inc., Wuxi, China). After mutagenesis, the plate was immediately placed into a sterilized tube containing 1 mL of basal medium. After the appropriate dilution, 50 μL of cell suspension from the tube was spread on basal medium plates and cultivated at 25 °C for up to 10 days. The lethal rate was determined to optimize the time treated with ARTP. Mutants appearing on the plates were picked and inoculated into 100-mL Erlenmeyer flasks containing 20 mL of basal medium and grown for 2 days. Then, 50 μL of cell culture was spread on fresh basal medium plates supplemented with sethoxydim at a suitable concentration, as described in the text. The plates were incubated for up to 10 days, and the mutants that appeared were again picked and inoculated into 250-mL Erlenmeyer flasks containing 50 mL of basal medium. The mutants with increased growth and lipid content were selected and used as the starting strain for the second round of ARTP mutagenesis and sethoxydim-based screening. The second round was conducted in the same manner as the first round, but with a higher concentration of sethoxydim. For comparative growth analysis on solid plates, the cell culture of the selected mutants and the wild type were diluted appropriately with the basal medium, and 2.5 μL of each dilution was spotted on basal medium plates supplemented with 10 or 20 μM sethoxydim.
Cultivation in 5-L bioreactors
Fed-batch fermentation experiments were performed in 5-L bioreactors (with an initial volume of 2.5 L fermentation medium) equipped with automatic controls for agitation, temperature, airflow rate, pH, and dissolved oxygen concentration (DO). The fermentation medium contains 27 g/L glucose, 6 g/L yeast extract, and 25 g/L sea salt. The temperature was maintained at 25 °C throughout the fermentation process. The DO was kept above 30% air saturation by automatically controlling agitation speed from 200 to 800 rpm and flushing (2.0 vvm) with filter-sterilized air. In addition, 0.1% (v/v) antifoam (Sigma-Aldrich, St Louis, MO) was added to suppress foam formation. A 70% (w/v) glucose solution was fed into the fermentation medium to maintain a glucose concentration between 10 and 27 g/L. In addition, a 50% (w/v) yeast extract solution was also fed according to a carbon nitrogen ratio (C/N) of 4:1 before 96 h. The pH was maintained at 6.5 ± 0.1, adjusted with 1.0 M H2SO4. Biomass was measured in terms of dry cell weight (DCW). The cell suspensions (3 mL) were centrifuged at 3500×g for 5 min. Cell pellets were freeze dried to a constant weight at −60 °C for 12 h. The glucose concentration in the culture was determined using a Glucose Assay Kit (Biosino, Beijing, China).
Determination of ACCase activity
Cells were harvested and suspended in 50 mM Tris-HCl buffer (pH 7.8) containing 3 mM dithioerythritol (DTE) and 0.2 mg DNase I per mL. HNX-2 cell disruptor (Honour, Tianjin, China) was used to rupture cells. The supernatant was collected by centrifugation at 10,000×g, 4 °C for 1 h and stored at −70 °C before use. ACCase activity was determined using a previously described method with slight modification (Guruvaiah et al. 2015). Briefly, the acetyl-CoA carboxylase activity was measured using a discontinuous spectrophotometric assay. The acetyl-CoA carboxylase reaction (reaction no. 1) was performed in a 150-μL reaction volume containing 100 mM potassium phosphate (pH 8.0), 15 mM KHCO3, 5 mM MnCl2, 5 mM ATP, 1 mg/mL bovine serum albumin (BSA), 1 mM acetyl-CoA, and 3 mg/L biotin. The reaction was initiated by adding 7 μL of the supernatant, after which the reaction mixture was incubated at 30 °C. At 2, 4, and 6 min, 50-μL aliquots were removed, transferred into microfuge tubes containing 3.3 μL 10% trifluoroacetic acid, and placed on ice. After all time points were measured, the 50-μL supernatant was transferred into a spectrophotometric cuvette containing 950 μL buffer composed of 100 mM KPO4 (pH 8.0), 0.1 mg/mL dithionitrobenzoic acid (DTNB), 20 mM oxaloacetate, and 1 mg/mL bovine serum albumin. The absorbance of the cuvettes at 412 nm was determined using a UV-1750 instrument (Shimadzu, Kyoto, Japan). Reaction no. 2 was initiated with the addition of 0.5 units of citrate synthase (Sigma-Aldrich, St Louis, MO). The change in absorption was recorded. These values correlate with the level of acetyl-CoA at the end of reaction no. 1. A standard curve was constructed each day by measuring the change in absorbance in reaction no. 2 with defined amounts of acetyl-CoA instead of supernatant and graphing the change in absorbance vs. concentration of acetyl-CoA. The protein concentration was determined using a NanoDrop 2000 instrument (Thermo, Waltham, MA) based on a previous publication (Wang et al. 2015).
Lipid extraction and measurement
The total lipids were extracted using a previous method with slight modification (Yang et al. 2009). Briefly, C. cohnii cells were collected by centrifugation (3550×g, 20 min) and freeze dried to generate a lyophilized algal powder. For lipid extraction, 15–25 mg of lyophilized algal powder was used for lipid extraction using a chloroform:methanol solution (2:1, v/v) with 0.01% butylated hydroxytoluene (BHT). The extraction process was repeated three to four times. The above extracts were washed with 1 mL of 1.0 M KCl followed by 1 mL of double-distilled water. The solvents were removed using a vacuum concentrator system (ZLS-1, Hunan, China). The DHA content and the lipid profile were analyzed using an Agilent 5975 MSD/7890 instrument (Agilent Corp, Santa Clara, CA) according to a previous publication (Xiong et al. 2008).
Flow cytometric analysis
Flow cytometric analysis was performed using a FACSCalibur instrument (Becton Dickinson, Sunnyvale, CA) equipped with a 488-nm argon laser using the following settings: forward scatter (FSC), E00 log; side scatter (SSC), 270 V; FL2, 350 V; FL3, 350 V; threshold, SSC 470. Cells were washed with 0.01 M phosphate buffer (pH 7.2–7.4), resuspended to a final OD490 of 1.0, and stained using Nile Red dye at a final concentration of 1 μg/mL for 10 min at 37 °C in darkness (Sui et al. 2014). A total of 200,000 cells were analyzed using CellQuest Pro software (Becton Dickinson, Sunnyvale, CA).
Transcriptomic analysis
Cells of the wild type and mutant M-1-2 were collected during the exponential phase of 36 h by centrifugation at 3500×g for 5 min at 4 °C and washed once with 0.01 M phosphate buffer (pH 7.2–7.4). The cell pellets were immediately frozen in liquid nitrogen and stored at −80 °C until use. Total RNA was isolated using an RNeasy plant Mini Kit reagent according to the manufacturer’s instruction (Qiagen, Hilden, Germany). The quantity and quality of the RNA was evaluated using an Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA). After removing the DNA in RNA samples using DNase I (New England Biolabs, Ipswich, MA), poly(A) messenger RNA (mRNA) was isolated from approximately 10 μg of total RNA using oligo-dT beads (Dynabeads mRNA Purification kit, Invitrogen, Carlsbad, CA). The isolated mRNA was fragmented into 200–250 bp with Fragment buffer (Ambion, Austin, TX). First strand complementary DNA (cDNA) was synthesized using First Strand Master Mix and Super Script II reverse transcription (Invitrogen, Carlsbad, CA). Second Strand Master Mix (Invitrogen, Carlsbad, CA) was added to synthesize the second strand cDNA at 16 °C for 2 h. The cDNA was purified using a QIAquick PCR Purification Kit (Qiagen, Hilden, Germany) and resolved with End Repair Mix for end repairing. A-Tailing Mix was added at 37 °C for 30 min for single nucleotide A (adenine) addition at the 3′ end. The cDNA was connected with adapters. The suitable fragments were selected for PCR amplification. A constructed cDNA library was qualified and quantified using Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA) and an ABI StepOnePlus Real-Time PCR System (Applied Biosystems, Foster City, CA), respectively. Sequencing of the libraries was performed using an Illumina HiSeq 4000 instrument (Illumina, San Diego, CA).
For data processing, the raw reads containing reads with low-quality sequences, adaptor-polluted sequences, or a high content of unknown nucleotides N were filtered to obtain clean reads using the internal software provided by Illumina. Clean reads were mapped to the reference C. cohnii transcriptome (NCBI TSA database with accession number GFIV00000000, available at: https://www.ncbi.nlm.nih.gov/nuccore/GFIV00000000) using Bowtie software (Langmead and Salzberg 2012). The transcript from RNA-seq was quantified using RESM software, which enables accurate transcript quantification by estimating maximum possible expression levels (Li and Dewey 2011). Transcriptome saturation curve analysis was performed according to a previous method with some modification (Robinson and Storey 2014). Subsequently, differential expression analysis (fold change) and related statistical significance between the mutant and the wild type were conducted using DEseq2 software (Love et al. 2014). Transcripts that were differentially expressed greater or less than 1.5-fold with false discovery-corrected statistical significance of p values below 0.05 were considered differentially expressed transcripts.
RNA-seq raw sequencing data accession information
The RNA-seq raw sequencing data of C. cohnii was deposited in the SRA database of NCBI with accession number SRP100483.
Validation of RNA-seq data by qRT-PCR
The RNA samples used in quantitative real-time PCR (qRT-PCR) were prepared from identical samples for transcriptomic analysis. Quantification of transcript expression was determined according to a standard process of qRT-PCR that used serial dilutions of known concentrations of chromosomal DNA as templates to construct a standard curve. Melting curve analyses showed that all target transcripts presented a single amplified product peak after primer optimization. The cDNA samples were prepared using oligo-dT primers. A total of 12 selected genes were selected for verification, and a housekeeping glyceraldehyde-3-phosphate dehydrogenase (GAPDH) gene was used as an internal control. Three biological replicates, each with four technical replicates, were analyzed for each transcript. Data analysis was performed using StepOnePlus analytical software (Applied Biosystems, Foster City, CA). Briefly, the amount of relative gene transcript was normalized using an internal control of GAPDH in each sample, and the data presented were ratios of the amount of normalized transcripts between different samples. The transcript IDs and their primer sequences used for qRT-PCR analysis are listed in Supplementary Table S1. The expression ratio of a given gene between samples was calculated according to a previous publication (Livak and Schmittgen 2001).
Comparison of ACCase gene sequences in the mutant and the wild type
The full-length sequence of ACCase gene including the coding sequence and 5′ up- and 3′ downstream untranslated region (UTR) was obtained through blasting analysis of the transcriptome data of the wild type (NCBI TSA database with accession number GFIV00000000). To identify whether the ACCase gene of the M-1-2 contains specific site mutations, first we used the full-length ACCase mRNA to map all mRNA sequencing data of the M-1-2 mutant using Bowtie software (Langmead and Salzberg 2012), and then determine any potential single nucleotide polymorphisms (SNP) and indel in the ACCase gene sequence of the M-1-2 mutant using SAMtools software (v. 0.1.19) (Li et al. 2009). In addition, the coding sequence and its 3′ and 5′ UTR regions were PCR amplified and sequenced. Briefly, the wild-type ACCase gene was separated into five overlapping regions, and then the specific primers were designed for PCR amplification using the cDNA of the wild type and the mutant M-1-2 as templates (Wang et al. 2015). The primer ID and sequences used for PCR are listed in Supplementary Table S2. After purification, the fragment was ligated into the pJET1.2 Blunting vector (Thermo, Waltham, MA) for sequencing. A full-length ACCase gene including the coding sequence, 5′ up- and 3′ downstream UTR, was assembled using the sequence of all amplicons from each strain. Subsequently, the sequence results of ACCase genes from both the wild type and the mutant were aligned by BlastN software to identify possible mutation.
Results
Mutagenesis with ARTP
In this study, ARTP mutagenesis was used to generate C. cohnii mutants with increased growth and lipid content. The working parameters of ARTP were set to 100 W, 10 SLM, and 2 mm for power, gas flow rate of helium gas, and distance between the plasma torch nozzle exit and the plate, respectively, according to a previous publication (Liu et al. 2015a). C. cohnii cells were treated with ARTP for varying durations (i.e., 0, 10, 20, 30, 40, 60, 80, or 100 s), and the optimal treatment time was determined to be 40 s with a lethal rate of 91.43–92.86% (data not shown). According to early studies, a high mutagenesis rate was typically achieved when the lethal rate was above 90% for ARTP (Guo et al. 2011).
Sethoxydim-based screening
To determine the sensitivity of the wild-type C. cohnii to sethoxydim, a concentration range of sethoxydim was added to the basal medium plates. The results showed that C. cohnii growth was significantly inhibited at 10 μM of sethoxydim, while less than 60% inhibition was observed when the sethoxydim concentration was 5 μM (Fig. 1). To confirm the results, the experiment was repeated with six replicate plates supplemented with 10 μM sethoxydim, and the results showed 90.66–94.16% inhibition (data not shown). For the initial mutant screening after the first round of ARTP mutagenesis, basal medium plates supplemented with 10 μM sethoxydim were used.

Sensitivity of wild-type C. cohnii on solid plates supplemented with sethoxydim
After the first round of ARTP, one mutant, named M-1, was selected from several positive colonies that appeared on the plates supplemented with 10 μM sethoxydim. Colonies were first selected based on colony size and growth rate on plates and then confirmed by comparative growth analysis of the mutants in flask cultures. No colony was observed on the plate supplemented with 10 μM sethoxydim for the wild type without ARTP treatment (Fig. 2). The M-1 mutant was then used as a starting strain for the second round of ARTP, and three highly resistant mutants were found on the plates with 20 μM sethoxydim, among which the mutant M-1-2 was selected according to colony size and growth rate both on solid plates and in flasks. No colony was found on the plate supplemented with 20 μM sethoxydim for either the wild type or the mutant M-1 without ARTP treatments (Fig. 2), demonstrating that the mutant M-1-2 indeed resulted from ARTP mutagenesis and sethoxydim-based screening. We further confirmed the sethoxydim tolerance of various C. cohnii mutants by cultivating them on plates supplemented with 10 or 20 μM sethoxydim. The results showed that only the mutant M-1-2 was able to survive on plates with 20 μM sethoxydim, and the growth of the mutant M-1 was only found on plates with 10 μM sethoxydim, while growth of the wild type was completely suppressed under both concentrations of sethoxydim (Fig. 3).

Screening for mutants using sethoxydim. a 10 μM sethoxydim. b 20 μM sethoxydim. WT, M-1, and M-1-2 refer to wild type, mutant M-1 after one round of ARTP, and mutant M-1-2 after two rounds of ARTP, respectively

Tolerance of the M-1 and M-1-2 mutants to sethoxydim at different concentrations. The wild type, which was sensitive to sethoxydim, was used as a control
Subsequently, comparative growth analysis of the mutant M-1, M-1-2, and the wild type was performed in media of varying glucose concentrations, ranging from 9 to 33 g/L. As shown in Fig. 4, at almost all glucose concentration levels, the maximum cell densities (OD490) of the M-1 and M-1-2 mutants were higher than that of the wild type. In addition, the growth increase was more significant at a high concentration of 27 g/L than the basal concentration of 9 g/L for both mutants, where the maximum cell densities (OD490) of M-1 and M-1-2 were 24.32 and 18.25% higher than that of the wild type at 96 h, respectively (Fig. 4b). The increased growth of the mutants was also confirmed using a statistical t test model with p values less than 0.05. However, the growth increase became less significant for both mutants when the glucose concentration was increased to 33 g/L (Fig. 4c).

Effects of the glucose concentration on the growth and on ACCase activity of the M-1 mutant, M-1-2 mutant, and wild type. a Growth at 9 g/L. b Growth at 27 g/L. c Growth at 33 g/L. d ACCase activity at 9 g/L. e ACCase activity at 27 g/L. f ACCase activity at 33 g/L. The data represent mean values ± SD of three individual experiments. Asterisks indicate significant difference between the control and stress treatments based on Student’s t tests (∗p < 0.05; ∗∗p < 0.01)
To confirm the effects of sethoxydim-based screening, ACCase activity in the mutant was determined relative to the wild type. The results showed that ACCase activity in the M-1-2 mutant was indeed higher than the wild type (Fig. 4d–f) on media supplemented with glucose of varying concentrations. The most significant increase in ACCase activity was found at a glucose concentration of 27 g/L, where the enzyme activity of ACCase was 16.15% higher than the wild type, consistent with the increased growth patterns discussed above (Fig. 4a–c).
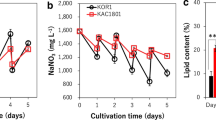
We also determined the lipid content in the M-1-2 mutant and the wild type at both the exponential and stationary phases at 27 g/L glucose. Cells were harvested at 36 and 96 h and subjected to lipid extraction. A clear increase in lipid accumulation was observed in the M-1-2 mutant at 36 h, with an average increase of approximately 7.05% (Fig. 5a). However, there was no apparent increase in lipid accumulation in the M-1-2 mutant at 96 h. In addition, we applied flow cytometric analysis to compare the lipid content in cells. Cells of the wild type and the M-1-2 mutant were first stained with Nile Red dye (Greenspan et al. 1985) and then examined for yellow-gold fluorescence in the FL2 channel (585 ± 42 nm) for neutral lipids and red fluorescence in the FL3 channel (670 nm LP) for polar lipids. Consistent with the results of lipid extraction analysis, the fluorescence results showed that the average content of both neutral and polar lipids was significantly enhanced in the mutant compared to the wild type at 36 h (Fig. 5b), while only neutral lipids were found at slightly increased levels in the M-1-2 mutant at 96 h (Fig. 5c). Finally, we also comparatively determined the DHA ratio in the lipids of the M-1-2 mutant and the wild type (Supplementary Table S3). However, no significant difference in the DHA ratio was observed. Additionally, we also compared the total fatty acid profiles between the wild type and the M-1-2 cells by GC-MS analysis; however, no significant difference was observed (Supplementary Table S3; Supplementary Fig. S1).

Lipid accumulation analysis in M-1-2 mutant and wild type. Lipid content in M-1-2 mutant and wild type is shown. The data represent mean values ± SD of three independent experiments. a 36 and 96 h. Flow cytometric analysis in M-1-2 mutant and wild type. b 36 h. c 96 h. M-1-2 mutant and the wild-type cells were stained with Nile Red dye and then examined for yellow-gold fluorescence in the FL2 channel (585 ± 42 nm) for neutral lipids and red fluorescence in the FL3 channel (670 nm LP) for polar lipids. Asterisks indicate significant difference between the control and stress treatments based on Student’s t tests (∗p < 0.05)
To determine whether the ACCase coding gene or its up- and downstream regions were changed during the ARTP mutagenesis, as described in the “Materials and methods,” we first mapped the full length of ACCase mRNA sequence of the wild type to RNA-seq data of the M-1-2 mutant and then determined whether there was any SNP and indel event (Li et al. 2009). However, the analysis showed no mutation in the mRNA sequence of the M-1-2 mutant. To further validate this result, we separated the full coding sequence of ACCase into five parts with overlapping regions. Five pairs of specific primers were designed for PCR amplification. However, sequencing, assembling, and comparison showed that no mutation was identified in the coding sequence and the 5′ up- and 3′ downstream UTR regions in the M-1-2 mutant.
Characterization of the M-1-2 mutant in a 5-L bioreactor
To further validate the improved properties of the M-1-2 mutant, fed-batch fermentation was performed in a 5-L bioreactor. To achieve the highest cell density during the fed-batch fermentation, the glucose feed was manually controlled to maintain a residual glucose concentration between 10 and 27 g/L. The fermentation curves of the M-1-2 mutant and the wild type are shown in Fig. 6a. During the first 24 h, glucose was slowly consumed and the cell growth of both the M-1-2 mutant and the wild type was relatively slow, consistent with the results from the flask experiments. The growth of both the M-1-2 mutant and the wild type entered exponential growth at 24 h when the rapid increase in biomass was observed along with rapid consumption of glucose. At this time, the growth of the M-1-2 mutant and the wild type was almost equal, whereas glucose consumption in the mutant M-1-2 was slightly slower than that in the wild type. After 84 h, the growth increase continued for the M-1-2 mutant, while growth of the wild type was significantly slowed, consistent with the faster glucose consumption rate in the M-1-2 mutant compared to the wild type (Fig. 6b). At 132 h, both strains reached their maximum cell growth. After 144 h of fermentation, the M-1-2 mutant yielded a maximum biomass concentration of 42.93 g/L, 29.40% higher than that of the wild type (32.41 g/L) under the same growth condition (Fig. 6a).

Comparison of the dry cell weight (DCW) (a) and glucose concentration (b) between the M-1-2 mutant and wild type in a fed-batch fermentation experiment. DCW refers to biomass, and glucose concentration is the residual glucose concentration in the fermentation broth before and after glucose feeding in the 5-L bioreactor
Comparative transcriptomic analysis using RNA-seq
A quantitative transcriptomic analysis was utilized to identify the transcripts that were differentially expressed between M-1-2 and the wild type. Samples of the M-1-2 mutant and wild type were harvested in the exponential growth phase at 36 h, each with two biological replicates for transcriptomic analysis. Based on a saturation curve analysis of a previous C. cohnii RNA-seq data (NCBI TSA database with accession number GFIV00000000), a total of 5G RNA-seq data for each sample was requested, resulting a total of 139 million raw sequencing reads for the four samples, with an average of 35 million reads per sample. After filtering low-quality and adaptor-polluted reads and reads with a high unknown base (N) content, a total of 120 million qualified mRNA-based sequence reads were taken for further analysis (Supplementary Table S4). The fragments per kilobase per million mapped fragments (FPKM) value was employed to normalize the raw reads and visualize the transcriptomic expression profiles. The analysis showed that using criteria of 1.5-fold change and p value less than 0.05 as cutoff, a total of 522 genes were upregulated and 598 genes were downregulated at 36 h in the M-1-2 mutant relative to the wild type. To validate the reliability of the RNA-seq data, a subset of 12 genes was randomly selected for qRT-PCR validation (primers listed in Supplementary Table S1). Comparative qRT-PCR analysis was conducted for the selected genes between the M-1-2 mutant and the wild-type samples. The results showed very good correlation between qRT-RCR and RNA-seq transcriptomic data in terms of the ratio changes (Table 1), suggesting high reliability in our RNA-seq analysis.
To reveal metabolic changes in the M-1-2 mutant, differentially expressed transcripts were categorized into different cellular functional groups (Table 2). Based on the number of differentially regulated transcripts identified in each functional category, the most frequently detected functional categories were “translation” and “global and overview maps” representing 25.09 and 18.41% of all differentially regulated transcripts, respectively. In addition, “carbohydrate metabolism,” “transport and catabolism,” “amino acid metabolism,” “lipid metabolism,” and “energy metabolism” represented 7.76, 6.50, 4.33, 3.97, and 2.71% of all differentially regulated transcripts, respectively, suggesting that global changes in almost every aspects of C. cohnii cellular metabolism occurred in the M-1-2 mutant.
As ACCase activity was increased in the C. cohnii mutants, it was expected that the expression of the ACCase gene might also be upregulated in the ACCase inhibitor-tolerant mutant. The RNA-seq analysis identified only one putative ACCase gene in the C. cohnii genome (TR24065|c0_g1_i1 with E-value less than 1e-148). The gene was upregulated by 1.53-fold at 36 h, consistent with the increased lipid content in the M-1-2 mutant at 36 h. Additionally, the transcriptomic analysis results showed that two transcripts of putative ATP citrate lyase (ACL, TR37608|c1_g7_i2 and TR37608|c1_g8_i3) were upregulated by 1.67- and 1.88-fold, respectively.
The transcriptomic analysis results showed that many transcripts related to energy and central carbohydrate metabolism were differentially regulated in the M-1-2 mutant. Our analysis showed that transcripts related to “glycolysis” and “tricarboxylic acid cycle (TCA cycle)” were upregulated. Specifically, transcripts of putative 6-phosphofructokinase (PFK) (i.e., TR21050|c0_g1_i1 and TR63019|c0_g1_i1) were upregulated 1.97- and 2.02-fold, respectively. Transcripts of putative pyruvate kinase (PK) (i.e., TR55138|c0_g1_i7, TR87974|c0_g1_i1, and TR81292|c1_g1_i4) were also significantly upregulated by 11.70-, 4.74-, and 2.72-fold, respectively. PFK and PK were known to be the rate-limiting steps in the glycolytic pathway. In addition, transcripts related to putative pyruvate carboxylase (PYC) (TR18842|c0_g1_i1), citrate synthase (CS) (TR84552|c0_g3_i2), and a malic enzyme (ME) transcript (TR89670|c1_g1_i1) were upregulated by 1.70-, 1.57- and 1.72-fold in the C. cohnii mutant, respectively. In addition, an NADH dehydrogenase (TR6179|c0_g2_i1) transcript and a phosphoenolpyruvate carboxykinase (PEPCK) transcript (TR16282|c4_g1_i1) were also upregulated by 3.06- and 1.75-fold, respectively.
The results showed that transcripts related to amino acid metabolism were also differentially regulated. Transcripts of glutamate kinase (TR55074|c0_g1_i2) and glutamate receptor (TR16260|c3_g4_i2) were upregulated 1.80- and 2.25-fold, respectively. In addition, a proline synthase transcript (TR32691|c0_g1_i1) was upregulated by 1.88-fold in the M-1-2 mutant. Moreover, transcripts of aspartate kinase (TR25821|c0_g1_i1) and aspartate aminotransferase (TR51804|c0_g1_i1) were upregulated by 1.67- and 2.08-fold, respectively, showing that asparagine metabolism might also be increased in the C. cohnii mutant. Aspartate could then be used to synthesize asparagine and methionine. Consistently, transcripts of asparagine synthetase (TR34174|c0_g1_i1) and methionine synthase (TR10288|c0_g1_i2) were also upregulated by 1.51- and 2.86-fold, respectively. An aminotransferase transcript (TR33310|c0_g1_i1) was also upregulated 1.56-fold in the mutant. Together, the results showed that amino acid metabolism was enhanced in the C. cohnii mutant.
Discussion
Previous studies have demonstrated that ARTP is an effective mutagenesis approach for many microorganisms (Ma et al. 2015a; Qiang et al. 2014). In C. cohnii, mutants with high EPS content were successfully generated by ARTP (Liu et al. 2015a). Metabolic inhibitors are widely used for screening mutants with high metabolite yields (Liu et al. 2015b). Previous studies have shown that sethoxydim was able to interfere with lipid metabolism in cells by directly inhibiting ACCase activity in grass (Dotray et al. 1993). In this study, C. cohnii was subjected to ARTP mutagenesis coupled with a novel screening method using the ACCase inhibitor sethoxydim for high-growth and high-lipid-content mutants. We found that the growth of C. cohnii was restrained by 10 μM sethoxydim and the percentage inhibition was 90.66–94.16%. Compared to other reported species, C. cohnii appears to be more sensitive to the ACCase inhibitor than other microalgae reported, as an early study showed that 50% reduction of the microalgae Dunaliella primolecta growth rates and cell density was observed upon treatment with 0.27 mM sethoxydim (Santin-Montanya et al. 2007). In another study, the growth of Schizochytrium limanium was suppressed by another ACCase inhibitor quizalofop at a concentration of 90 μM (Pora and Zhou 2012). After two rounds of mutation and screening, an M-1-2 mutant was obtained with 24.32 and 7.05% higher growth and lipid content than those of the wild type, respectively. However, no significant difference in the DHA ratio and overall fatty acid profiles was observed between the mutants and the wild type, which may be because malonyl-CoA was a universal precursor for all major types of fatty acids in cells (Huang et al. 2010). Consistently, the activity assay and transcriptomic analysis confirmed that the ACCase enzymatic activity and transcriptional level of the ACCase-encoding gene were both upregulated in the M-1-2 mutant. The roles of ACCase in fatty acid biosynthesis have been well investigated previously, and it has been proposed that increased ACCase activity is a feasible method to accumulate lipids in Chlorella (Hsieh and Wu 2009). In one study, the expression pattern of the ACCase gene was studied in Chlorella sorokiniana, and the results showed that along with increased ACCase activity, the lipid content also increased (Wan et al. 2011). Our study using the ACCase inhibitor sethoxydim to screen high-growth and high-lipid-content mutants provides further lines of evidence that ACCase plays key roles in cellular growth and lipid accumulation in C. cohnii, and demonstrated that ARTP mutagenesis coupled with a sethoxydim-based screening strategy could be an effective approach for C. cohnii mutants with increased growth and lipid content. Although more proof is needed, the approach could also be applied to other microorganisms with slight modification. It was also reported that no significant increase in lipid accumulation was observed with increased expression of the ACCase gene and enzymatic activity in Cyclotella cryptica and Navicula saprophila (Dunahay et al. 1995, 1996), which may be because ACCase was less of a rate-limiting step in these microbial species. Furthermore, the ACCase genes and its up- and downstream regions of the wild type and the M-1-2 mutant were PCR amplified and resequenced. Sequence comparison showed no mutation within the coding sequence of the ACCase gene or its up- and downstream regions in the M-1-2 mutant, suggesting that the upregulation of the ACCase transcription could be due to possible other changes in cells, such as mutation of the relevant regulatory proteins acting on the ACCase gene rather than direct mutation in the gene itself. Such a speculation is consistent with previous results that the ACCase level was regulated by multiple cellular factors in plant (Sasaki and Nagano 2004). Those changes may not be necessarily within the ACCase coding region or its up- or downstream regions. Nevertheless, it is worthy of further investigation for the detailed mechanism in the future.
A comparative transcriptomic analysis showed that transcripts involved in fatty acid biosynthesis were upregulated in the high-growth and high-lipid-content mutant. According to a previous study, ACCase was significantly upregulated during the fatty acid biosynthesis phase under cold stress in Aurantiochytrium, which could provide more precursor malonyl-CoA for fatty acid biosynthesis (Ma et al. 2015b). ACL is a key enzyme related to the formation of cytoplasmic acetyl-CoA that is essential for fatty acid production (Hynes and Murray 2010). Another previous study also indicated that ACL is required for normal growth and development in plants (Fatland et al. 2005), which is likely also due to its relevance to all essential fatty acids. The increased consumption of acetyl-CoA due to enhanced ACCase activity could increase demand for more acetyl-CoA generated by ACL.
Previous studies have demonstrated that central carbohydrate metabolism is crucial for growth and lipid accumulation in C. cohnii (Pleissner and Eriksen 2012). The analysis also showed that transcripts involved in energy and central carbohydrate metabolism were upregulated in the high-growth and high-lipid-content mutant, including PYC, CS, and ME. Consistent with this finding, a previous study showed that the genes of the TCA cycle had a high degree of co-expression with genes related to fatty acid metabolism in the species of Chromista (Mühlroth et al. 2013), and it is well known that ME can catalyze the oxidative decarboxylation of malate to pyruvate, thus plays important roles in providing the necessary reducing power from NADPH for the fatty acid biosynthesis pathway (Xue et al. 2016). In the oleaginous fungus Moritierella alpina, overexpression of ME resulted in a significant increase in fatty acid content (by 30%) (Hao et al. 2014), while the overexpression of an ME gene from Mucor circinelloides increased lipid accumulation from 18.74% of the biomass to 39.35% in the oleaginous yeast Rhodotorula glutinis (Li et al. 2013). In addition, the upregulation of NADH dehydrogenase indicates that the C. cohnii mutant could produce more ATP, which may lead to increases in cell growth (Schneider and Gourse 2004). Moreover, upregulation of a PEPCK transcript may increase the carboxylation of phosphoenolpyruvate (PEP) or the reversible decarboxylation of oxaloacetate (OAA) (He et al. 2013; Reiskind and Bowes 1991). In bacteria, yeast, and plants, PEPCK plays a central role in gluconeogenesis, nitrogenous assimilates, anaplerotic reactions, and amino acid metabolism (He et al. 2013). In our previous metabolomic study of C. cohnii induced by chemicals, such as ETA, salicylic acid (SA), and naphthoxyacetic acid (BNOA), increased lipid content was accompanied by downregulation of PEP and upregulation of oxaloacetic acid (OXA) (Li et al. 2015).
This analysis showed that transcripts involved in amino acid biosynthesis were upregulated in the high-growth and high-lipid-content mutant. Consistent with this finding, a previous study showed that a mutant of Aurantiochytrium sp. cultured in fermentation medium was able to accumulate 21.3% more DHA, and transcriptomic results showed that amino acid biosynthesis was upregulated in the mutant strain compared to the parent strain Aurantiochytrium sp. (Qi et al. 2016). The new information obtained from the mutant analysis could be useful for rational engineering of C. cohnii for increased lipid accumulation and DHA production in the future.
References
De Swaaf ME, Sijtsma L, Pronk JT (2003) High-cell-density fed-batch cultivation of the docosahexaenoic acid producing marine alga Crypthecodinium cohnii. Biotechnol Bioeng 81(6):666–672. doi:10.1002/bit.10513
Dotray PA, DiTomaso JM, Gronwald JW, Wyse DL, Kochian LV (1993) Effects of acetyl-coenzyme A carboxylase inhibitors on root cell transmembrane electric potentials in graminicide-tolerant and -susceptible corn (Zea mays L.) Plant Physiol 103(3):919–924. doi:10.1104/pp.103.3.919
Dunahay TG, Jarvis EE, Dais SS, Roessler PG (1996) Manipulation of microalgal lipid production using genetic engineering. Appl Biochem Biotech 57–58(1):223–231. doi:10.1007/BF02941703
Dunahay TG, Jarvis EE, Roessler PG (1995) Genetic transformation of the diatoms Cyclotella cryptica and Navicula saprophila. J Phycol 31(6):1004–1012. doi:10.1111/j.0022-3646.1995.01004.x
Fatland BL, Nikolau BJ, Wurtele ES (2005) Reverse genetic characterization of cytosolic acetyl-CoA generation by ATP-citrate lyase in Arabidopsis. Plant Cell 17(1):182–203. doi:10.1105/tpc.104.026211
Gao M, Song X, Feng Y, Li W, Cui Q (2013) Isolation and characterization of Aurantiochytrium species: high docosahexaenoic acid (DHA) production by the newly isolated microalga, Aurantiochytrium sp. SD116. J Oleo Sci 62(3):143–151. doi:10.5650/jos.62.143
Glaser C, Lattka E, Rzehak P, Steer C, Koletzko B (2011) Genetic variation in polyunsaturated fatty acid metabolism and its potential relevance for human development and health. Matern Child Nut 7(Suppl 2):27–40. doi:10.1111/j.1740-8709.2011.00319.x
Greenspan P, Mayer EP, Fowler SD (1985) Nile red: a selective fluorescent stain for intracellular lipid droplets. J Cell Biol 100(100):965–973. doi:10.1083/jcb.100.3.965
Guo T, Tang Y, Xi YL, He AY, Sun BJ, Wu H, Liang DF, Jiang M, Ouyang P-K (2011) Clostridium beijerinckii mutant obtained by atmospheric pressure glow discharge producing high proportions of butanol and solvent yields. Biotechnol Lett 33(12):2379–2383. doi:10.1007/s10529-011-0702-9
Guruvaiah M, Narra M, Dixit G, Karawadia P, Shah D (2015) Isolation, screening and optimization of estuary region (Khambhat, Gujarat) microalgae for lipid/oil production. Int J Appl Sci Biotechnol 3(2):197–201. doi:10.3126/ijasbt.v3i2.12534
Hao G, Chen H, Wang L, Gu Z, Song Y, Zhang H, Chen W, Chen YQ (2014) Role of malic enzyme during fatty acid synthesis in the oleaginous fungus Mortierella alpina. Appl Environ Microbiol 80(9):2672–2678. doi:10.1128/AEM.00140-14
He LW, Zhang XJ, Wang G (2013) Expression analysis of phosphoenolpyruvate carboxykinase in Porphyra haitanensis (Rhodophyta) sporophytes and gametophytes. Phycol Res 61(3):172–179. doi:10.1111/pre.12005
Hsieh CH, Wu WT (2009) Cultivation of microalgae for oil production with a cultivation strategy of urea limitation. Bioresour Technol 100(17):3921–3926. doi:10.1016/j.biortech.2009.03.019
Hu Q, Sommerfeld M, Jarvis E, Ghirardi M, Posewitz M, Seibert M, Darzins A (2008) Microalgal triacylglycerols as feedstocks for biofuel production: perspectives and advances. Plant J 54(4):621–639. doi:10.1111/j.1365-313X.2008.03492.x
Huang GH, Chen F, Dong W, Zhang XW, Chen G (2010) Biodiesel production by microalgal biotechnology. Appl Energ 87(1):38–46. doi:10.1016/j.apenergy.2009.06.016
Humburg NE, Colby SR, Hill ER, Kitchen LM, Lym RG, McAvoy WJ, Prasad R (1989) Herbicide handbook of the Weed Science Society of America, ed 6 edn. Weed Science Society of America, Champaign-Urbana
Hynes MJ, Murray SL (2010) ATP-citrate lyase is required for production of cytosolic acetyl coenzyme A and development in Aspergillus nidulans. Eukaryot Cell 9(7):1039–1048. doi:10.1128/EC.00080-10
Jiang Y, Chen F (1999) Effects of salinity on cell growth and docosahexaenoic acid content of the heterotrophic marine microalga Crypthecodinium cohnii. J Ind Microbiol Biot 23(6):508–513. doi:10.1038/sj.jim.2900759
Jiang Y, Chen F, Liang SZ (1999) Production potential of docosahexaenoic acid by the heterotrophic marine dinoflagellate Crypthecodinium cohnii. Process Biochem 34(6–7):633–637. doi:10.1016/S0032-9592(98)00134-4
Klaus D, Ohlrogge JB, Neuhaus HE, Dormann P (2004) Increased fatty acid production in potato by engineering of acetyl-CoA carboxylase. Planta 219(3):389–396. doi:10.1007/s00425-004-1236-3
Kuratko CN, Salem N (2013) Docosahexaenoic acid from algal oil. Eur J Lipid Sci Tech 115(9):965–976. doi:10.1002/ejlt.201300060
Langmead B, Salzberg SL (2012) Fast gapped-read alignment with Bowtie 2. Nat Methods 9(4):357–359. doi:10.1038/nmeth.1923
Li B, Dewey CN (2011) RSEM: accurate transcript quantification from RNA-seq data with or without a reference genome. BMC Bioinformatics 12(1):93–99. doi:10.1186/1471-2105-12-323
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R (2009) The sequence alignment/map format and SAMtools. Bioinformatics 25(16):2078–2079. doi:10.1093/bioinformatics/btp352
Li J, Niu X, Pei G, Sui X, Zhang X, Chen L, Zhang W (2015) Identification and metabolomic analysis of chemical modulators for lipid accumulation in Crypthecodinium cohnii. Bioresour Technol 191:362–368. doi:10.1016/j.biortech.2015.03.068
Li Z, Sun H, Mo X, Li X, Xu B, Tian P (2013) Overexpression of malic enzyme (ME) of Mucor circinelloides improved lipid accumulation in engineered Rhodotorula glutinis. Appl Microbiol Biotechnol 97(11):4927–4936. doi:10.1007/s00253-012-4571-5
Liu B, Sun Z, Ma X, Yang B, Jiang Y, Wei D, Chen F (2015a) Mutation breeding of extracellular polysaccharide-producing microalga Crypthecodinium cohnii by a novel mutagenesis with atmospheric and room temperature plasma. Int J Mol Sci 16(4):8201–8212. doi:10.3390/ijms16048201
Liu C, Zhang X, Rao ZM, Shao ML, Zhang LL, Wu D, Xu ZH, Li H (2015b) Mutation breeding of high 4-androstene-3,17-dione-producing Mycobacterium neoaurum ZADF-4 by atmospheric and room temperature plasma treatment. J Zhejiang Univ Sci B 16(4):286–295. doi:10.1631/jzus.B1400274
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25(4):402–408. doi:10.1006/meth.2001.1262
Lopes da Silva T, Reis A (2008) The use of multi-parameter flow cytometry to study the impact of n-dodecane additions to marine dinoflagellate microalga Crypthecodinium cohnii batch fermentations and DHA production. J Ind Microbiol Biotechnol 35(8):875–887. doi:10.1007/s10295-008-0360-7
Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15(12):550. doi:10.1186/s13059-014-0550-8
Mühlroth A, Li K, Røkke G, Winge P, Olsen Y, Hohmann-Marriott MF, Vadstein O, Bones AM (2013) Pathways of lipid metabolism in marine algae, co-expression network, bottlenecks and candidate genes for enhanced production of EPA and DHA in species of Chromista. Mar Drugs 11(11):4662–4697. doi:10.3309/md11114662
Ma Y, Yang H, Chen X, Sun B, Du G, Zhou Z, Song J, Fan Y, Shen W (2015a) Significantly improving the yield of recombinant proteins in Bacillus subtilis by a novel powerful mutagenesis tool (ARTP): alkaline α-amylase as a case study. Protein Expres Purif 114:82–88. doi:10.1016/j.pep.2015.06.016
Ma Z, Tan Y, Cui G, Feng Y, Cui Q, Song X (2015b) Transcriptome and gene expression analysis of DHA producer Aurantiochytrium under low temperature conditions. Sci Rep 5:14446. doi:10.1038/srep14446
Pleissner D, Eriksen NT (2012) Effects of phosphorous, nitrogen, and carbon limitation on biomass composition in batch and continuous flow cultures of the heterotrophic dinoflagellate Crypthecodinium cohnii. Biotechnol Bioeng 109(8):2005–2016. doi:10.1002/bit.24470
Pora B, Zhou J (2012) Methods of mutagenesis of Schizochytrium sp. and variant strains produced thereof, WO, WO/2012/175027 A1
Qi F, Zhang M, Chen Y, Jiang X, Lin J, Cao X, Huang J (2016) A lignocellulosic hydrolysate-tolerant Aurantiochytrium sp. mutant strain for docosahexaenoic acid production. Bioresour Technol 227:221–226. doi:10.1016/j.biortech.2016.12.011
Qiang W, Ling-ran F, Luo W, Han-guang L, Lin W, Ya Z, Xiao-bin Y (2014) Mutation breeding of lycopene-producing strain Blakeslea trispora by a novel atmospheric and room temperature plasma (ARTP). Appl Biochem Biotechnol 174(1):452–460. doi:10.1007/s12010-014-0998-8
Reiskind JB, Bowes G (1991) The role of phosphoenolpyruvate carboxykinase in a marine macroalga with C4-like photosynthetic characteristics. Proc Natl Acad Sci U S A 88(7):2883–2887. doi:10.1073/pnas.88.7.2883
Robinson DG, Storey JD (2014) subSeq: determining appropriate sequencing depth through efficient read subsampling. Bioinformatics 30(23):3424–3426. doi:10.1093/bioinformatics/btu552
Santin-Montanya I, Sandin-Espana P, Garcia Baudin JM, Coll-Morales J (2007) Optimal growth of Dunaliella primolecta in axenic conditions to assay herbicides. Chemosphere 66(7):1315–1322. doi:10.1016/j.chemosphere.2006.07.019
Santos-Sánchez NF, Valadez-Blanco R, Hernández-Carlos B, Torres-Ariño A, Guadarrama-Mendoza PC, Salas-Coronado R (2016) Lipids rich in ω-3 polyunsaturated fatty acids from microalgae. Appl Microbiol Biotechnol 100(20):8667–8884. doi:10.1007/s00253-016-7818-8
Sasaki Y, Nagano Y (2004) Plant acetyl-CoA carboxylase: structure, biosynthesis, regulation, and gene manipulation for plant breeding. Biosci Biotechnol Biochem 68(6):1175–1184. doi:10.1271/bbb.68.1175
Schneider DA, Gourse RL (2004) Relationship between growth rate and ATP concentration in Escherichia coli: a bioassay for available cellular ATP. J Biol Chem 279(9):8262. doi:10.1074/jbc.M311996200
Sui X, Niu X, Shi M, Pei G, Li J, Chen L, Wang J, Zhang W (2014) Metabolomic analysis reveals mechanism of antioxidant butylated hydroxyanisole on lipid accumulation in Crypthecodinium cohnii. J Agr Food Chem 62(51):12477–12484. doi:10.1021/jf503671m
Wan M, Liu P, Xia J, Rosenberg JN, Oyler GA, Betenbaugh MJ, Nie Z, Qiu Q (2011) The effect of mixotrophy on microalgal growth, lipid content, and expression levels of three pathway genes in Chlorella sorokiniana. Appl Microbiol Biotechnol 91(3):835–844. doi:10.1007/s00253-011-3399-8
Wang X, Lu M, Wang S, Fang Y, Wang D, Ren W, Zhao G (2014) The atmospheric and room-temperature plasma (ARTP) method on the dextranase activity and structure. Int J Biol Macromol 70:284–291. doi:10.1016/j.ijbiomac.2014.07.006
Wang Y, Tao S, Gao X, Shi M, Wu L, Lei C, Zhang W (2015) Biosynthesis of platform chemical 3-hydroxypropionic acid (3-HP) directly from CO2 in cyanobacterium Synechocystis sp. PCC 6803. Metab Eng 34:60–70. doi:10.1016/j.ymben.2015.10.008
Xiong W, Li X, Xiang J, Wu Q (2008) High-density fermentation of microalga Chlorella protothecoides in bioreactor for microbio-diesel production. Appl Microbiol Biotechnol 78(1):29–36. doi:10.1007/s00253-007-1285-1
Xue J, Wang L, Zhang L, Balamurugan S, Li DW, Zeng H, Yang WD, Liu JS, Li HY (2016) The pivotal role of malic enzyme in enhancing oil accumulation in green microalga Chlorella pyrenoidosa. Microb Cell Factories 15(1):120. doi:10.1186/s12934-016-0519-2
Yaakob Z, Ali E, Zainal A, Mohamad M, Takriff MS (2014) An overview: biomolecules from microalgae for animal feed and aquaculture. J Biol Res-Thessalon 21(1):6. doi:10.1186/2241-5793-21-6
Yang S, Lu SH, Yuan YJ (2009) Cerium elicitor-induced phosphatidic acid triggers apoptotic signaling development in Taxus cuspidata cell suspension cultures. Chem Phys Lipids 159(1):13–20. doi:10.1016/j.chemphyslip.2009.02.004
Yongmanitchai W, Ward OP (1989) Omega-3 fatty acids: alternative sources of production. Process Biochem 24(12):117–125
Zhang X, Zhang XF, Li HP, Wang LY, Zhang C, Xing XH, Bao CY (2014) Atmospheric and room temperature plasma (ARTP) as a new powerful mutagenesis tool. Appl Microbiol Biotechnol 98(12):5387–5396. doi:10.1007/s00253-014-5755-y
Acknowledgments
The authors would like to thank Prof. Hao Song for the help with the ARTP experiments. This study was funded by grants from the National High-tech R&D Program (“863” program, project No. 2012AA02A707), the National Basic Research Program of China (“973” program, project No. 2014CB745101), the Doctoral Program of Higher Education of China (Nos. 20120032110020 and 20130032120022), the National Natural Science Foundation of China (No. 21621004), the Tianjin Municipal Science and Technology Commission (No. 15JCZDJC32500), and Zaoneng Biotechnology Inc.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human participants performed by any of the authors.
Electronic supplementary material
ESM 1
(PDF 321 kb)
Rights and permissions
About this article

Cite this article
Liu, J., Pei, G., Diao, J. et al. Screening and transcriptomic analysis of Crypthecodinium cohnii mutants with high growth and lipid content using the acetyl-CoA carboxylase inhibitor sethoxydim. Appl Microbiol Biotechnol 101, 6179–6191 (2017). https://doi.org/10.1007/s00253-017-8397-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-017-8397-z