
Abstract
DNA methylation is an important epigenetic mark in mammals, plants, and fungi and depends on multiple genetic pathways involving de novo and maintenance DNA methyltransferases (DNMTases). Metarhizium robertsii, a model system for investigating insect-fungus interactions, has been used as an environmentally friendly alternative to chemical insecticides. However, little is known concerning the molecular basis for DNA methylation. Here, we report on the roles of two DNMTases (MrRID and MrDIM-2) by characterizing ΔMrRID, ΔMrDIM-2, and ΔRID/ΔDIM-2 mutants. The results showed that approximately 71, 10, and 8% of mC sites remained in the ΔMrRID, ΔMrDIM-2, and ΔRID/ΔDIM-2 strains, respectively, compared with the wild-type (WT) strain. Further analysis showed that MrRID regulates the specificity of DNA methylation and MrDIM-2 is responsible for most DNA methylation, implying an interaction or cooperation between MrRID and MrDIM-2 for DNA methylation. Moreover, the ΔMrDIM-2 and ΔRID/ΔDIM-2 strains showed more defects in radial growth and conidial production compared to the WT. Under ultraviolet (UV) irradiation or heat stress, an obvious reduction in spore viability was observed for all the mutant strains compared to the WT. The spore median lethal times (LT50s) for the ΔMrDIM-2 and ΔRID/ΔDIM-2 strains in the greater wax moth, Galleria mellonella, were decreased by 47.7 and 65.9%, respectively, which showed that MrDIM-2 is required for full fungal virulence. Our data advances the understanding of the function of DNMTase in entomopathogenic fungi, which should contribute to future epigenetic investigations in fungi.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In eukaryotes, DNA methylation describes the transfer of a methyl group (CH3) to the fifth position of a cytosine residue and occurs almost exclusively in association with transposable elements and repeat sequences (Jeon et al. 2015; Suzuki and Bird 2008). In the nucleus, cytosine methylation cooperates with N-terminal histone modifications to establish a silenced chromatin structure, thus regulating nuclear gene expression (Shock et al. 2011). Methylation patterns in animals are established by two de novo DNA methyltransferases (DNMTases), DNMTase 3a (DNMT3a) and DNMTase 3b (DNMT3b), and are maintained by DNMTase 1 (DNMT1). In plants, domains that are rearranged by methyltransferase 2 (DRM2) establish the DNA methylation pattern maintained by methyltransferase 1 (MET1) and chromo-methylase 3 (CMT3) (Cao and Jacobsen 2002; Law and Jacobsen 2010; Li 2002). These genes function within an ancient regulatory mechanism shared by eukaryotes, serving in diverse roles often related to the repression of gene expression (Feng et al. 2010; Jurkowski and Jeltsch 2011; Nanty et al. 2011; Zemach et al. 2010).
The DNMTases in fungi such as Neurospora crassa, Ascobolus immerses, and Magnaporthe oryzae have been characterized. The methylation mechanism of DNMTases in N. crassa has been well studied. Repeat-induced point (RIP) defective (RID) is essential for RIP mutation, and DIM-2 is responsible for all known cytosine methylation events; mutation of either of these DNMTases causes no obvious defect in the phenotype (Freitag et al. 2002; Kouzminova and Selker 2001). In M. oryzae, DIM-2 is responsible for DNA methylation and RID regulates the methylation specificity of the genome. Abnormalities in colony morphology, radial growth, and sporulation have been observed in a mutant with a deletion of the DIM-2 gene (Jeon et al. 2015). In A. immersus, the Masc1 mutation has no obvious defect in the phenotype during vegetative growth but blocks the methylation that is induced premeiotically (methylation induced premeiotically (MIP)) and confers sterility in Masc1 homozygous dikaryons (Selker 1997).
Most fungi have low levels of DNA methylation, ranging from imperceptible to barely detectable (Liu et al. 2012; Martienssen and Colot 2001; Selker et al. 2003). Approximately 1.5% of cytosines are methylated in the genome of N. crassa, while <0.1% of methylcytosines were found to be methylated in Schizosaccharomyces pombe and Aspergillus nidulans (Antequera et al. 1984; Foss et al. 1993; Selker and Stevens 1987). The variation in genome methylation levels suggests that DNA methylation is not strictly conserved among different fungal species. In N. crassa and Candida albicans, DNA methylation silences transposable elements and repetitive DNA sequences as a genome defense mechanism (Mishra et al. 2011; Selker et al. 2003). However, DNA methylation plays a role in fungal development throughout the asexual life cycle of M. oryzae (Jeon et al. 2015).
Metarhizium robertsii, an arthropod pathogen, saprophyte, and beneficial colonizer of rhizospheres, has been used as an environmentally friendly alternative to chemical insecticides (Gao et al. 2011; Roberts and St Leger 2004). M. robertsii is also well suited for use as a representative species for simultaneously studying several major lifestyles, including parasitism, saprophytism, and symbiotism, in a way that is not matched by the “model” fungi Saccharomyces cerevisiae and N. crassa (Bidochka et al. 2001; Fang and St Leger 2010). Despite the considerable progress that has been made with regards to unraveling and understanding the genetic pathways that govern morphogenesis and pathogenicity, there have been no surveys of the impact of DNMTases on important economic characteristics such as conidiation, stress tolerance, or virulence in entomopathogenic fungi (Aramayo and Selker 2013; Wang and St Leger 2014).
To investigate DNMTases in M. robertsii, we selected two putative DNMTases, MrRID (accession no. EFZ01240.1) and MrDIM-2 (accession no. EFZ00016.1), by performing a BLAST search using the amino acid sequences of known DNMTases as queries. Genetic manipulation and traditional bisulfite sequencing (BS-PCR) were used to understand the mechanism of DNMTase on DNA methylation. We further investigated whether disrupting DNMTases impacted the M. robertsii life cycle by examining any phenotypic changes in the mutants. Our data suggests a wider role for DNMTases in DNA methylation and the development of entomopathogenic fungi, which should contribute to future epigenetic investigations in fungi.
Materials and methods
Fungal strains and growth conditions
The wild-type (WT) M. robertsii strain ARSEF 23 (ATCC no. MYA-3075) was generously provided by Dr. Chengshu Wang (Gao et al. 2011). Stock cultures were grown on potato dextrose agar (PDA; 20% potato, 2% dextrose, and 2% agar, w/v) in the dark at 28 °C for 12 days to obtain conidia. Conidia were harvested in a 0.05% Tween 80 aqueous solution, and the conidial suspension was filtered through sterile non-woven fabric to remove mycelia, which were then washed in sterilized water. The final spore concentrations were determined by direct counting using a hemocytometer. Unless otherwise noted, for phenotypic assays, conidial suspensions (1-μl suspension, 1 × 106 conidia ml−1) of each strain were spotted onto various media.
Genetic manipulation and fungal transformation
The binary vectors pDHt-SK-bar (conferring resistance against ammonium glufosinate) and pDHt-SK-ben (conferring resistance against benomyl) and the Agrobacterium strain AGL-1 were kindly provided by Dr. Chengshu Wang (Gao et al. 2013). Target genes were deleted using an Agrobacterium-mediated transformation method as described previously (Duan et al. 2013). In brief, the 5′ and 3′ flanking regions of MrRID and MrDIM-2 were amplified using WT genomic DNA as a template and the forward and reverse primer pairs MrRID 5′ flanking, MrRID 3′ flanking, MrDIM-2 5′ flanking, and MrDIM-2 3′ flanking, respectively (Table 1). To construct the MrRID deletion vector, the products of the 5′ flanking regions were digested with XhoI and BamHI (TaKaRa, Dalian, China), while the products of the 3′ flanking regions were digested with SpeI and XbaI (TaKaRa, Dalian, China) and then inserted into the corresponding sites of the binary vector pDHt-SK-bar to produce the binary vector pbar-RID (Fig. 1a). To construct the MrDIM-2 deletion structure, the 5′ and 3′ flanking region products were digested with PstI and SpeI (TaKaRa, Dalian, China), respectively, and then inserted into the corresponding sites of the binary vector pDHt-SK-ben to produce the binary vector pben-DIM-2 (Fig. 1b). The mutants (deletions of MrRID, MrDIM-2, and RID/DIM-2) were constructed by means of Agrobacterium-mediated fungal transformation (ATMT) and replacements by homologous recombination as previously described (Duan et al. 2009).

Deletion of genes encoding DNMTases in Metarhizium robertsii. a The disruption and complementation plasmids of MrRID. b The disruption and complementation plasmids of MrDIM-2. c Confirmation by PCR shows the disruption of the genes of the mutants. Blue, genomic DNA; light green, cDNA; M, marker; 1, PCR conducted with the primers a and b; 2, PCR conducted with the primers g and h; 3, PCR conducted with the primers c and d; 4, PCR conducted with the primers e and f; 5, PCR conducted with primers for the glyceraldehyde 3-phosphate dehydrogenase (GAPDH) gene as an internal control for each sample. d Southern blotting hybridization with amplified probes with MrRID (p1) and MrDIM-2 (p2) ORF fragments, respectively. Further information on primers can be found in Table 1
The complementation vector Com-pben-RID was constructed using the entire MrRID gene plus 1.3 and 0.2 kb of the upstream and downstream sequences, respectively (Fig. 1a). The DNA was amplified from WT genomic DNA with primers i and j (Table 1). Then, the products were digested with PstI (TaKaRa, Dalian, China) and inserted into the corresponding sites of the binary vector pDHt-SK-ben (Fig. 1a). The complementation vector Com-pbar-DIM-2 was constructed using the entire MrDIM-2 gene plus 0.9 and 0.2 kb of the upstream and downstream sequences, respectively (Fig. 1b). The DNA was amplified from WT genomic DNA with primers k and l (Table 1). Then, the products were digested with BamHI (TaKaRa, Dalian, China) and inserted into the corresponding sites of the binary vector pDHt-SK-bar (Fig. 1b). The plasmids Com-pben-RID and Com-pbar-DIM-2 were mobilized into AGL-1 and then transformed into different mutants (with deletions of MrRID or MrDIM-2). All the primers used are listed in Table 1.
Confirmation of mutants
Single conidia from the gene deletion mutants were isolated by streaking individual colonies on selection plates and confirmed with PCR and reverse transcription PCR (RT-PCR). The genomic DNA was extracted with a Genomic DNA Extraction Kit (Qiagen, Hilden, Germany). Total RNA was extracted using TRIzol Reagent (Invitrogen, Carlsbad, CA, USA), and first-strand cDNA was synthesized using a PrimeScript™ II 1st Strand cDNA Synthesis Kit (TaKaRa) according to the manufacturer’s instructions. The glyceraldehyde 3-phosphate dehydrogenase (GAPDH) gene (MAA_07675) was used as an internal control. All the primers used are listed in Table 1. For Southern blotting, 30 μg samples of genomic DNA extracted from the PDA colonies of WT and each mutant were digested with XhoI, separated by electrophoresis in 0.7% agarose gel, and then transferred to Hybond-N+ membrane (Amersham, Piscataway, USA). The MrRID and MrDIM-2 ORF fragments used as probes were, respectively, labelled with the PCR DIG Labelling Mix Kit (Roche, Penzberg, Germany). Probe preparation, membrane hybridization, and visualization were according to the manufacturer’s instructions (DIG High Prime DNA Labelling and Detection Starter Kit II, Roche).
BS-PCR for selected segments in different mutants
Ten methylated regions, based on genome-wide DNA methylation in M. robertsii, deposited in the NCBI GEO database with the accession number GSE78019, were randomly selected, following the categories of high (>66%), medium, and low (<33%) methylated patterns, to examine the methylation changes in different mutants. BS-PCR was performed according to our earlier reports (Wang et al. 2015). One-hundred-microliter aliquots of a conidial suspension from either WT or mutant colonies were spread on PDA plates and incubated in the dark at 28 °C for 12 days. DNAs from these cultures, including both mycelium and conidia, of different strains were used for BS-PCR, and DNA samples per strain were extracted from three independent culture experiments. DNA from these cultures of the different strains was used for BS-PCR. Briefly, DNA (1 μg) was bisulfite converted as previously described (Espada et al. 2014). Two-hundred nanograms of bisulfite-converted DNA was PCR-amplified, and the purified amplicons were cloned into a pMD18-T vector (TaKaRa) and sequenced. An average of 15 clones were randomly chosen to sequence for each segment. All experiments were performed in triplicate. All the primers used are listed in Table 1.
Phenotypic assays
Vegetative growth. PDA plates were spotted in the center with aliquots of conidia harvested from WT and mutant cultures growing on PDA plates. The radial growth (colony diameter) of the vegetative mycelia was measured daily at 28 °C.
Chemical stress. Fungal colony disks (5-mm diameter) were cut from cultures of WT and mutants that were grown for 3 days at 28 °C on PDA plates smeared with 100-μl aliquots of a conidial suspension to initiate the cultures. The disks were gently placed in the center of (9-cm diameter) PDA plates containing different chemical reagents, including osmotic (0.5 M NaCl), oxidative (2 mM H2O2), cell wall (3 mg/ml−1 Congo red and 2.5 μg/ml−1 SDS), and fungicidal (2 μg/mL carbendazim) stressors. Radial growth was measured as described earlier, and PDA plates (without any modifications) were used as the control. All experiments were performed in triplicate.
Conidial yield. To assess the sporulation capacity of the WT and that of each mutant, 100-μl aliquots of a conidial suspension were spread on PDA plates and incubated for 20 days at 28 °C. From day 3 onwards, colony disks (5-mm diameter) were excised at random from the plates daily. The conidia on each disk were washed off into 2 ml of 0.05% Tween 80 by subjecting each disk to 10 min of supersonic vibration. The concentration of the conidial suspension was determined by counting the conidia using a hemocytometer under a microscope.
Conidial viability assays. To assay the fresh conidial germination rates of the strains, 30-μl aliquots of a conidial suspension were spread onto PDA plates (9-cm diameter), followed by incubation at 28 °C for 48 h. From 12 h onwards, the percentage of germinated conidia on each plate was assessed every 4 h by counting the number of germinated conidia that could be seen under the microscope. Three counts were performed for each plate. The morphological development of the germ tubes and the growing hyphae were also examined under a microscope.
Thermotolerance assays. Each mutant strain was assayed using a method described elsewhere (Wang et al. 2014). Briefly, 1-ml aliquots of conidial suspensions in 1.5-ml Eppendorf tubes were placed in a water bath at 42 °C for up to 90 min, and then, PDA plates were inoculated with 100 μl of the conidial suspension from each tube. After 24 h of incubation, conidial germination on each plate was observed under the microscope; conidia with visible germ tubes were considered to have germinated.
UV irradiation assays. The conidial tolerance of each mutant was assayed using a previous method (Yao et al. 2010). Briefly, 10-μl aliquots of conidia were centrally spotted onto the marked area (10-mm diameter) of glass slides and then exposed to weighted UV-B irradiation at a wavelength of 312 nm (280–320 nm) at gradient doses of 0.1–0.8 J/cm−2 in a Bio-Sun++ chamber (Vilber Lourmat, Marne-la-Vallée, France). After exposure, conidial germination on each plate was observed as mentioned above.
All experiments were performed in triplicate, and all data were analyzed as previously reported; the growth inhibition (GI) rate (%) was calculated as (C − S)/C × 100, where C is the growth rate of the control and S is the growth rate under a given stress (Ying et al. 2013). Residue viability was assessed by determining the percentage of germination, which was calculated using the counts of the number of germinated and ungerminated conidia.
Bioassays for fungal virulence
WT and M. robertsii mutants were bioassayed using Galleria mellonella as reported previously (Fang et al. 2009). Conidia were applied topically by immersing larvae for 20 s in an aqueous suspension containing 1 × 107 conidia ml−1. Each treatment had three replicates with 15 insects in each replicate. The experiments were performed in triplicate. Mortality was recorded every 12 h. The median lethal time (LT50) was estimated and compared by performing a Kaplan-Meier analysis using SPSS (version 13.0) (Wang and St Leger 2007).
BS-PCR and real-time PCR for five selected genes
The genes for tuberin (MAA_09983) and an autophagy-related protein (MAA_03501), a DNA repair protein (MAA_06876), cysteine proteinase (MAA_05503), and chitinase (MAA_00157) were selected to examine the effect of DNA methylation on gene transcription. Primers overlapping with the gene promoters (1 kb upstream of the gene-coding regions) and with the gene-coding regions were used for amplification, and BS-PCR was performed as mentioned above (Table 1). Gene expression was detected by RT-PCR, and the specific primers are listed in Table 1; three replicates were performed independently for each gene tested.
Results
Identification of genes encoding putative DNMTases
To investigate the function of DNMTases in the entomopathogenic fungus M. robertsii, we sought to identify and characterize genes encoding putative DNMTases in the fungal genome. By performing a BLAST search using the amino acid sequences of known DNMTases as queries, we found two genes (MrRID and MrDIM-2) which encode proteins containing a DNMTase domain (MAA_03836 and MAA_04944). Phylogenetic analysis showed that MrRID and MrDIM-2 are closely related to RID and DIM-2 in N. crassa. RID is responsible for the specificity of DNA methylation and RIP mutation, and DIM-2 is capable of both de novo and maintenance DNA methylation (Fig. 2a). According to further analysis of the domain architecture, MrDIM-2, which is similar to DIM-2 in M. oryzae, is typical of DIM-2-type fungal DMTases that have a long N-terminal extension containing a bromo-adjacent homology (BAH) domain and a C-terminal extension (Fig. 2b) (Jeon et al. 2015).

DNMTases in diverse organisms. a Phylogenetic analysis of DNMTases. The phylogenetic tree was constructed by a neighbor-joining method using the DNMTase domains as representative amino acid sequences of DNMTases. Af Aspergillus flavus, Ai Ascobolus immersus, At Arabidopsis thaliana, Cm Cordyceps militaris, Ec Escherichia coli, Mo Magnaporthe oryzae, Mr Metarhizium robertsii, Nc Neurospora crassa, Ur Uncinocarpus reesii. Bootstrap values are adjacent to each internal node, representing the percentage of 1000 bootstrap replicates. b Domain architecture of representative DNMTases. Light green, BAH (bromo-adjacent homology) domain; red, DNMTase domain
Construction and validation of DNMTase mutants
To investigate the role of DNMTases in M. robertsii, three targeted insertion mutants were constructed based on the identified sequences. Strains ΔMrRID and ΔMrDIM-2 were constructed by replacing the coding regions of MrRID and MrDIM-2 with the bar and ben gene cassettes inserted with flanking fragments of MrRID and MrDim-2 in vectors pbar-RID and pben-DIM-2, respectively (Fig. 1a, b). ΔRID/ΔDIM-2 was constructed by replacing the coding region of MrRID in ΔMrDIM-2 with the bar cassette in vector pbar-RID. Complementation strains (ΔRID/RID and ΔDIM-2/DIM-2) were obtained by transforming Com-pben-RID and Com-pbar-DIM-2 into strains ΔMrRID and ΔMrDIM-2, respectively (Fig. 1a, b).
All the mutant strains were confirmed by PCR using genomic DNA as a template. PCR analysis indicated that a partial MrRID fragment (528 bp) was present in the WT and ΔRID/RID but not in strains ΔMrRID and ΔRID/ΔDIM-2 (Fig. 1c). In addition, a partial MrDIM-2 fragment (599 bp) was present in the WT and ΔDIM-2/DIM-2 but not in strains ΔMrDIM-2 and ΔRID/ΔDIM-2 (Fig. 1c). A 434-bp fragment corresponding to the bar gene was present in strains ΔMrRID, ΔRID/RID, ΔDIM-2/DIM-2, and ΔRID/ΔDIM-2, and a 328-bp fragment corresponding to the ben gene was present in strains ΔRID/RID, ΔMrDIM-2, ΔDIM-2/DIM-2, and ΔRID/ΔDIM-2 (Fig. 1c). RT-PCR was used to further confirm expression of genes in the mutants, using the GAPDH gene (EFY96862) as an internal control for each sample (Fig. 1c). Southern blotting further validated the homologous recombination in the deletion mutants. As expected, the MrRID ORF fragment probe detected a 5.4-kb XhoI band in the WT and a XhoI band of 4.1 kb in the complemented strain ΔRID/RID, respectively, but not in strains ΔMrRID and ΔRID/ΔDIM-2 (Fig. 1d). The shorter fragment of 4.1 kb indicates that the complemented strain carries the transformed MrRID gene inserted at another site than at the homologous place. However, the probe did not detect any band in strains ΔMrRID and ΔRID/ΔDIM-2, further confirming the loss of the MrRID gene in these transformants. The MrDIM-2 ORF fragment probe detected a 6.8-kb XhoI band in the WT and a 7.9-kb XhoI band in the complemented ΔDIM-2/DIM-2 transformant, respectively (Fig. 1d). The size of the 7.9 kb in the ΔDIM-2/DIM-2 transformant suggests that the transformed MrDIM-2 DNA inserted at an ectopic place into the genome of mutant ΔMrDIM-2. In contrast, no signal was detected in strains ΔMrDIM-2 and ΔRID/ΔDIM-2, confirming that in these strains, the MrDIM-2 gene was deleted.
Functional analysis of the role of DNMTases in DNA methylation
To test genetically whether the two putative DNMTase genes are involved in DNA methylation, we checked the DNA methylation status of ten known methylated regions using BS-PCR; 71% (±2.4%), 10% (±0.6%), and 8% (±0.4%) of the mC sites remained in ΔMrRID, ΔMrDIM-2, and ΔRID/ΔDIM-2, respectively, compared with the WT strain (Figs. 3 and 4). Alignments of the WT strain and the different mutant revealed that 35% (±0.3%) of the remaining mC sites in ΔMrRID did not overlap with those in the WT strain. In ΔRID/ΔDIM-2, because of the deletions of MrRID in ΔMrDIM-2, more than half of the remaining mC sites did not overlap with the mC sites in ΔMrDIM-2. Taken together, MrRID in M. robertsii seems to play a role in regulating methylation specificity. Importantly, the mC sites that disappeared in ΔMrRID were a subset of the missing sites in ΔMrDIM-2, which suggests that the methylation of those cytosine sites is accomplished by both MrRID and MrDIM-2 (Figs. 3 and 4). These results are consistent with previous studies in M. oryzae, which suggests that DNMTases in different fungi have highly conserved functions (Jeon et al. 2015).

Venn diagrams show overlapping and non-overlapping mCs in ten selected regions of the different strains. The red, green, blue, and yellow ellipses refer to the WT, ΔMrRID, ΔMrDIM-2, and ΔRID/ΔDIM-2 strains, respectively

Model for DNA methylation in the WT, ΔMrRID, ΔMrDIM-2, and ΔRID/ΔDIM-2 strains. The schematic diagram describes the DNA methylation in and around genes within the nuclear genomes
Phenotypic analyses of the wild type and mutants
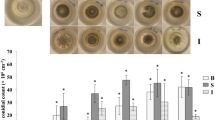
The effect of disrupting different DNMTases on vegetative growth was examined by incubating WT and mutant cultures at 28 °C on PDA medium. There was little difference in the growth rate between the ΔMrRID (2.95 mm/day) and WT strains (3.07 mm/day), while the growth rates of the ΔMrDIM-2 (1.89 mm/day) and ΔRID/ΔDIM-2 (1.81 mm/day) strains were significantly reduced compared with that of the WT strain and the complemented strains (Fig. 5a). Consequently, the number of conidia produced by the colonies differed significantly among these strains (Fig. 5b). Conidiation was more defective in ΔRID/ΔDIM-2 and ΔMrDIM-2 than in ΔMrRID; the conidial yields obtained from these mutant cultures on day 7 were reduced by 57.82, 49.76, and 6.97%, respectively, compared with the yield obtained from the control strains on PDA medium (Fig. 5b). These data indicate that both MrRID and MrDIM-2 affected the normal growth and sporulation of M. robertsii but that MrDIM-2 was more influential.

Growth and conidial production of the wild type, ΔMrRID, ΔMrRID/RID, ΔMrDIM-2, ΔMrDIM-2/DIM-2, and ΔRID/ΔDIM-2. a Growth of WT M. robertsii and different mutants. b Conidial production was measured for the WT and different mutants. Error bars SD of the mean from three replicate assays
The effects of osmotic (NaCl), oxidative (H2O2), cell wall (Congo Red and SDS), and fungicidal (carbendazim) stressors were examined on plates containing the indicated chemical reagents with the data presented as the percentage of GI. Contrary to our expectations, the DNMTase mutants showed a similar growth capability against stressful chemicals to that shown by the control strains (Fig. 6a).

Effect of chemical stress reagents on growth and conidial viability of WT M. robertsii and different mutants. a The GI calculated after incubation of the indicated strains in the presence of different chemical stress reagents. b Conidial viability of WT M. robertsii and different mutants under different conditions. Error bars SD of the mean from three replicate assays
The conidial tolerance of the WT and DNMTase mutants to UV irradiation and heat shock changed dramatically 24 h after germination. The conidial survival indices were reduced to approximately 42, 17, and 16% for ΔMrRID, ΔMrDIM-2, and ΔRID/ΔDIM-2, respectively, while the conidial survival index for the WT strain decreased to 50% after UV-B irradiation. Similar findings were observed for conidial tolerance to the high temperature of 42 °C; 57% of the WT conidia germinated, while only 43, 15, and 14% of the ΔMrRID, ΔMrDIM-2, and ΔRID/ΔDIM-2 germinated, respectively (Fig. 6b). These data indicated that MrRID and MrDIM-2, and of the two especially MrDIM-2, play important roles in conidial tolerance to both UV irradiation and thermal stress.
Virulence of the wild type and mutants
Insect bioassays using the greater wax moth G. mellonella were employed to assess the consequences of the loss of MrRID and MrDIM-2 on insect virulence. Insects were infected topically, representing the natural route of infection, and mortality was monitored daily over a 12-day period. ΔMrDIM-2 and ΔRID/ΔDIM-2 insect groups displayed mean lethal times to death (LT50) of 6.5 ± 0.9 and 7.3 ± 1.2 days, with a significant (P < 0.05) attenuation of virulence, while ΔMrRID and the control groups displayed LT50s of 4.6 ± 0.8 and 4.4 ± 0.5 days, respectively. Their LT50s against G. mellonella were decreased by 47.7 and 65.9% for ΔMrDIM-2 and ΔRID/ΔDIM-2, respectively, but were not significant for ΔMrRID (P > 0.05) (Fig. 7a). Deletion of MrDIM-2 delayed not only fungal development and mycosis on an insect cadaver but also sporulation on insect cuticles. Dead larvae were selected and incubated at 28 °C after surface sterilization. Those killed by the ΔMrRID and the control strains were mycosed with a heavy layer of conidia, whereas only a fine layer of conidia could be seen on larvae killed by the ΔMrDIM-2 and ΔRID/ΔDIM-2 strains (Fig. 7b). These results indicate that the deletion of MrDIM-2 impaired fungal virulence and conidiogenesis.

Insect bioassays. a Mortality of larvae of the greater wax moth G. mellonella were treated topically with conidia from the WT and different mutants. Control larvae were treated with sterile distilled H2O. b Representative images of the insect cadavers treated with the different strains. W WT, 1 ΔMrRID, 2 ΔMrRID/RID, 3 ΔMrDIM-2, 4 ΔMrDIM-2/DIM-2, 5 ΔRID/ΔDIM-2. Error bars SD of the mean from three replicate assays
Effect of DNA methylation on gene transcription
To clarify the relationship between DNA methylation and the phenotypes mentioned above, some genes (regulation of cellular process, conidia production, response to stress, and virulence) were selected to detect the expression levels. Genes for tuberin (MAA_09983, regulation of cellular process), an autophagy-related protein (MAA_03501, conidia production) and a DNA repair protein (MAA_06876, response to stress), were downregulated in ΔMrDIM-2 (Goldman et al. 2002; Kikuma et al. 2007; Ying et al. 2013). However, DNA methylation in those gene regions dropped to an average of ∼10% compared with the wild type (Fig. S1 in the Supplementary Material). Two important virulence genes, cysteine proteinase (MAA_05503) and chitinase (MAA_00157), were selected to detect the relationship between the gene expression levels and DNA methylation patterns (Duan et al. 2013; Niassy et al. 2013). Virulence genes were downregulated in ΔMrDIM-2, but in the gene regions, DNA methylation dropped to an average of ∼10% compared with the wild type, which is consistent with the results seen in the above genes (Fig. S1 in the Supplementary Material).
Discussion
DNA methylation is an epigenetic marker that serves as a bridge between genetic components and the environment and plays a vital role in the regulation of gene expression in eukaryotes. DNA methylation was maintained or induced by several DNMTases in different fungal species (Jeon et al. 2015; Kouzminova and Selker 2001; Malagnac et al. 1999). Two putative DNMTases (MrRID and MrDIM-2) were found in M. robertsii.
In N. crassa, RID is required for RIP, in which most relics of transposons inactivated by RIP are methylated in the genome. RIP introduces C:G-to-T:A transition mutations, creates targets for subsequent DNA methylation, and is always reported to take place during the sexual phase of the life cycle. However, RIP has been reported to occur in at least two asexual fungi (Clutterbuck 2011; Freitag et al. 2002). Genome data demonstrated that M. robertsii might have undergone RIP at some stage in its evolution, which suggests that MrRID may be a RIP-defective gene (Gao et al. 2011). Our phylogenetic and functional analyses showed that MrRID is closely related to RID and regulates methylation specificity in the genome. Thus, MrRID likely participated in RIP in M. robertsii. The comparison in this study of methylation patterns in the WT strain and different mutants supports that MrRID plays a role in regulating methylation specificity. However, MrRID-dependent mCs showed no sequence preference, which is different from RID in M. oryzae (Jeon et al. 2015). Therefore, such specificity is dependent on the chromatin structures, likely modified by the methyltransferase MrRID, without which, new non-specific methylated sites can be produced by MrDIM-2.
The other protein, MrDIM-2, is closely related to DIM-2 from N. crassa, which is responsible for all detected cytosine methylation (Kouzminova and Selker 2001). A remaining ∼10% of methylated cytosines were found in ΔMrDIM-2, which is consistent with previous studies in M. oryzae, which suggested that DIM-2 is a major DNMTase in the fungus (Jeon et al. 2015). Approximately 8% of methylated cytosines were found in ΔRID/ΔDIM-2, which shows that the deletion of both MrRID and MrDIM-2 has an additive effect on DNA methylation in M. robertsii. In plants, RNA-directed DNA methylation (RdDM) is an important de novo DNA methylation pathway, in which a 24-nucleotide small interfering RNA (siRNA) guides cytosine methylation (Lewsey et al. 2016; Matzke and Mosher 2014). Previous research has reported that there are many 24-nt siRNAs in M. robertsii, suggesting that RdDM may be responsible for DNA methylation in ΔRID/ΔDIM-2, which is a target for future study (Zhou et al. 2012).
The impact of deleting DNMTases on the life cycle of M. robertsii was verified by examining the phenotypic changes and stress tolerance of mutants. We observed that the radial growth and conidial production in ΔMrDIM-2 and ΔRID/ΔDIM-2 were more defective when compared to those in the wild type than in ΔMrRID, which is similar to observations in M. oryzae (Jeon et al. 2015). When comparing the mutant strains to the wild type under UV irradiation or heat, spore viability was noticeably decreased, especially for ΔMrDIM-2 and ΔRID/ΔDIM-2. This leads to the conclusion that there is a direct link between the loss of DNA methylation and radial growth, conidial production, or viability because MrDIM-2 is responsible for the vast majority of DNA methylation in M. robertsii. The genes for tuberin (regulation of cellular process), an autophagy-related protein (conidia production) and a DNA repair protein (response to stress), were downregulated in ΔMrDIM-2. However, DNA methylation in those gene regions dropped to an average of ∼10% compared with the wild type, which conflicts with the traditional view that DNA methylation inhibits gene expression (Fig. S1 in the Supplementary Material). We speculate that DNA methylation patterns in M. robertsii may have unknown roles in gene expression, and more experiments are needed to explore those patterns.
M. robertsii is a well-known insect pathogen and is actively being studied as a microbial means of pest control (Gao et al. 2011). In this study, insect bioassays using a topical application of fungal conidia revealed severe attenuation of infection in ΔMrDIM-2 and ΔRID/ΔDIM-2, along with subsequent poor sporulation in the infected larvae. DNA methylation is correlated with infection-related morphogenesis in M. oryzae, which leads to decreased virulence in its mutants (Jeon et al. 2015). However, previous studies could not explain the functional mechanism of DNA methylation in virulence from the perspective of molecular biology. Virulence genes, for cysteine proteinase and chitinase, were downregulated in ΔMrDIM-2, but in the gene regions, DNA methylation dropped to an average of ∼10% compared with the wild type, which is consistent with the results seen in the above genes (Fig. S1 in the Supplementary Material). Therefore, more work is needed to explore the DNA methylation patterns in M. robertsii. There is no difference in virulence between ΔMrRID and the WT parent, suggesting that ∼25% of the mCs methylated by MrRID do not influence the normal infection of insects.
In conclusion, our data indicate that MrDIM-2 is responsible for almost all DNA methylation, and that MrRID regulates the methylation specificity of the genome in M. robertsii, similar to N. crassa. DNMTases play an important role in the development, stress tolerance, and virulence of the pathogenic insect fungus M. robertsii. Data from this study advance our understanding of the function of DNMTase in entomopathogenic fungi, which should contribute to future epigenetic investigations in fungi.
References
Antequera F, Tamame M, Villanuevaz JR, SantosQ T (1984) DNA methylation in the fungi. J Biol Chem 259:8033–8036
Aramayo R, Selker EU (2013) Neurospora crassa, a model system for epigenetics research. Cold Spring Harb Perspect Biol 5(10):a017921. doi:10.1101/cshperspect.a017921
Bidochka MJ, Kamp AM, Lavender TM, Dekoning J, De Croos JN (2001) Habitat association in two genetic groups of the insect-pathogenic fungus Metarhizium anisopliae: uncovering cryptic species? Appl Environ Microbiol 67(3):1335–1342. doi:10.1128/AEM.67.3.1335-1342
Cao XF, Jacobsen SE (2002) Role of the Arabidopsis DRM methyltransferases in de novo DNA methylation and gene silencing. Curr Biol 12(13):1138–1144
Clutterbuck AJ (2011) Genomic evidence of repeat-induced point mutation (RIP) in filamentous ascomycetes. Fungal Genet Biol 48(3):306–326. doi:10.1016/j.fgb.2010.09.002
Duan Z, Chen Y, Huang W, Shang Y, Chen P, Wang C (2013) Linkage of autophagy to fungal development, lipid storage and virulence in Metarhizium robertsii. Autophagy 9(4):538–549. doi:10.4161/auto.23575
Duan Z, Shang Y, Gao Q, Zheng P, Wang C (2009) A phosphoketolase Mpk1 of bacterial origin is adaptively required for full virulence in the insect-pathogenic fungus Metarhizium anisopliae. Environ Microbiol 11(9):2351–2360. doi:10.1111/j.1462-2920.2009.01961.x
Espada J, Carrasco E, Calvo MI (2014) Standard DNA methylation analysis in mouse epidermis: bisulfite sequencing, methylation-specific PCR, and 5-methyl-cytosine (5mC) immunological detection. Methods Mol Biol 1094:221–231. doi:10.1007/978-1-62703-706-8_17
Fang W, Pava-ripoll M, Wang S, St. Leger RJ (2009) Protein kinase A regulates production of virulence determinants by the entomopathogenic fungus, Metarhizium anisopliae. Fungal Genet Biol 46(3):277–285. doi:10.1016/j.fgb.2008.12.001
Fang W, St. Leger RJ (2010) RNA binding proteins mediate the ability of a fungus to adapt to the cold. Environ Microbiol 12(3):810–820. doi:10.1111/j.1462-2920.2009.02127.x
Feng SH, Cokus SJ, Zhang XY, Chen PY, Bostick M, Goll MG, Hetzel J, Jain J, Strauss SH, Halpern ME, Ukomadu C, Sadler KC, Pradhan S, Pellegrini M, Jacobsen SE (2010) Conservation and divergence of methylation patterning in plants and animals. PNAS 107(19):8689–8694. doi:10.1073/pnas.1002720107
Foss HM, Roberts CJ, Claeys KM, Selkert EU (1993) Abnormal chromosome behavior in Neurospora mutants defective in DNA methylation. Science 262(5140):1737–1741
Freitag M, Williams RL, Kothe GO, Selker EU (2002) A cytosine methyltransferase homologue is essential for repeat-induced point mutation in Neurospora crassa. Proc Natl Acad Sci U S A 99(13):8802–8807. doi:10.1073/pnas.132212899
Gao Q, Jin K, Ying S-H, Zhang Y, Xiao G, Shang Y, Duan Z, Hu X, Xie XQ, Zhou G, Peng U, Luo Z, Huang W, Wang B, Fang W, Wang S, Zhong Y, Ma LJ, Leger RJS, Zhao GP, Pei Y, Feng MG, Xia Y, Wang C (2011) Genome sequencing and comparative transcriptomics of the model entomopathogenic fungi Metarhizium anisopliae and M. acridum. PLoS Genet 7(1):e1001264. doi:10.1371/journal.pgen.1001264.g001
Gao Q, Shang Y, Huang W, Wang C (2013) Glycerol-3-phosphate acyltransferase contributes to triacylglycerol biosynthesis, lipid droplet formation, and host invasion in Metarhizium robertsii. Appl Environ Microbiol 79(24):7646–7653
Goldman GH, McGuire SL, Harris SD (2002) The DNA damage response in filamentous fungi. Fungal Genet Biol 35(3):183–195. doi:10.1006/fgbi.2002.1344
Jeon J, Choi J, Lee G-W, Park S-Y, Huh A, Dean RA, Lee Y-H (2015) Genome-wide profiling of DNA methylation provides insights into epigenetic regulation of fungal development in a plant pathogenic fungus, Magnaporthe oryzae. Sci Rep 5:8567. doi:10.1038/srep08567
Jurkowski TP, Jeltsch A (2011) On the evolutionary origin of eukaryotic DNA methyltransferases and Dnmt2. PLoS One 6(11):e28104. doi:10.1371/journal.pone.0028104.g001
Kikuma T, Arioka M, Kitamoto K (2007) Autophagy during conidiation and conidial germination in filamentous fungi. Autophagy 3(2):128–129
Kouzminova E, Selker EU (2001) dim-2 encodes a DNA methyltransferase responsible for all known cytosine methylation in Neurospora. EMBO J 20(15):4309–4323
Law JA, Jacobsen SE (2010) Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat Rev Genet 11(3):204–220. doi:10.1038/nrg2719
Lewsey MG, Hardcastle TJ, Melnyk CW, Molnar A, Valli A, Urich MA, Nery JR, Baulcombe DC, Ecker JR (2016) Mobile small RNAs regulate genome-wide DNA methylation. PNAS 113(6):E801–E810. doi:10.1073/pnas.1515072113
Li E (2002) Chromatin modification and epigenetic reprogramming in mammalian development. Nat Rev Genet 3(9):662–673. doi:10.1038/nrg887
Liu SY, Lin JQ, Wu HL, Wang CC, Huang SJ, Luo YF, Sun JH, Zhou JX, Yan SJ, He JG, Wang J, He ZM (2012) Bisulfite sequencing reveals that Aspergillus flavus holds a hollow in DNA methylation. PLoS One 7(1):e30349. doi:10.1371/journal.pone.0030349
Malagnac F, Grégoire A, Goyon C, Rossignol J-L, Faugeron G (1999) Masc2, a gene from Ascobolus encoding a protein with a DNA-methyltransferase activity in vitro, is dispensable for in vivo methylation. Mol Microbiol 31(1):331–338
Martienssen RA, Colot V (2001) DNA methylation and epigenetic inheritance in plants and filamentous fungi. Science 293(5532):1070–1074. doi:10.1126/science.293.5532.1070
Matzke MA, Mosher RA (2014) RNA-directed DNA methylation: an epigenetic pathway of increasing complexity. Nat Rev Genet 15(6):394–408. doi:10.1038/nrg3683
Mishra PK, Baum M, Carbon J (2011) DNA methylation regulates phenotype-dependent transcriptional activity in Candida albicans. PNAS 108(29):11965–11970. doi:10.1073/pnas.1109631108
Nanty L, Carbajosa G, Heap GA, Ratnieks F, van Heel DA, Down TA, Rakyan VK (2011) Comparative methylomics reveals gene-body H3K36me3 in Drosophila predicts DNA methylation and CpG landscapes in other invertebrates. Genome Res 21(11):1841–1850. doi:10.1101/gr.121640.111
Niassy S, Subramanian S, Ekesi S, Bargul JL, Villinger J, Maniania NK (2013) Use of Metarhizium anisopliae chitinase genes for genotyping and virulence characterization. Biomed Res Int doi. doi:10.1155/2013/465213
Roberts DW, Leger RJS (2004) Metarhizium spp., cosmopolitan insect-pathogenic fungi: mycological aspects. Adv Appl Microbiol 54:1–70
Selker EU, Stevens JN (1987) Signal for DNA methylation associated with tandem duplication in Neurospora crassa. Mol Cell Biol 7(3):1032–1038
Selker EU, Tountas NA, Cross SH, Margolin BS, Murphy JG, Bird AP, Freitag M (2003) The methylated component of the Neurospora crassa genome. Nature 422:893–897
Shock LS, Thakkar PV, Peterson EJ, Moran RG, Taylor SM (2011) DNA methyltransferase 1, cytosine methylation, and cytosine hydroxymethylation in mammalian mitochondria. Proc Natl Acad Sci U S A 108(9):3630–3635
Suzuki MM, Bird A (2008) DNA methylation landscapes: provocative insights from epigenomics. Nat Rev Genet 9(6):465–476. doi:10.1038/Nrg2341
Selker UE (1997) Epigenetic phenomena in filamentous fungi useful paradigms or repeat-induced confusion? Trends Genet 13(8):296–301
Wang C, St. Leger RJ (2007) A scorpion neurotoxin increases the potency of a fungal insecticide. Nat Biotechnol 25(12):1455–1456. doi:10.1038/nbt1357
Wang C, St. Leger RJ (2014) Genomics of entomopathogenic fungi. In: Martin F (ed) The ecological genomics of fungi. Hoboken, NJ, pp 243–260
Wang YL, Wang ZX, Liu C, Wang SB, Huang B (2015) Genome-wide analysis of DNA methylation in the sexual stage of the insect pathogenic fungus Cordyceps militaris. Fungal Biol 119(12):1246–1254. doi:10.1016/j.funbio.2015.08.017
Wang ZX, Zhou XZ, Meng HM, Liu YJ, Zhou Q, Huang B (2014) Comparative transcriptomic analysis of the heat stress response in the filamentous fungus Metarhizium anisopliae using RNA-Seq. Appl Microbiol Biotechnol 98(12):5589–5597. doi:10.1007/s00253-014-5763-y
Yao SL, Ying SH, Feng MG, Hatting JL (2010) In vitro and in vivo responses of fungal biocontrol agents to gradient doses of UV-B and UV-A irradiation. BioControl 55(3):413–422. doi:10.1007/s10526-009-9265-2
Ying SH, Feng MG, Keyhani NO (2013) A carbon responsive G-protein coupled receptor modulates broad developmental and genetic networks in the entomopathogenic fungus, Beauveria bassiana. Environ Microbiol 15(11):2902–2921. doi:10.1111/1462-2920.12169
Zemach A, McDaniel IE, Silva P, Zilberman D (2010) Genome-wide evolutionary analysis of eukaryotic DNA methylation. Science 328(5980):916–919. doi:10.1126/science.1186366
Zhou Q, Wang Z, Zhang J, Meng H, Huang B (2012) Genome-wide identification and profiling of microRNA-like RNAs from Metarhizium anisopliae during development. Fungal Biol 116(11):1156–1162. doi:10.1016/j.funbio.2012.09.001
Acknowledgement
This work was supported by the National Natural Science Foundation of China (Grant Nos. 31272096, 31471821, and 31572060).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Human and animal rights
This article does not contain any studies with human participants or animal subjects performed by any of the authors.
Electronic supplementary material
.
ESM 1
(PDF 314 kb)
Rights and permissions
About this article

Cite this article
Wang, Y., Wang, T., Qiao, L. et al. DNA methyltransferases contribute to the fungal development, stress tolerance and virulence of the entomopathogenic fungus Metarhizium robertsii . Appl Microbiol Biotechnol 101, 4215–4226 (2017). https://doi.org/10.1007/s00253-017-8197-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-017-8197-5