
Abstract
Clostridium acetobutylicum is an important industrial microorganism for n-butanol bioproduction, and its transcription factor AbrB0310 regulates various important cellular processes. However, the roles of two abrB homologues, abrB1941 and abrB3647, have not been determined because they appear inactive during transcription. Here, we performed a detailed investigation into the function of abrB1941 and abrB3647 in C. acetobutylicum. Interestingly, we observed that AbrB3647 exerts an important influence on biphasic fermentation that opposes the influence of AbrB0310, while AbrB1941 might not be essential. When abrB3647 was disrupted using the Targetron system, a greatly improved cellular growth occurred. The following analysis shows that all three AbrBs participated in metabolically regulating acidogenesis, solventogenesis, and a two-phase transition in C. acetobutylicum, but the AbrB0310 and AbrB3647 functions were the most important. Moreover, the target genes subject to AbrB0310 and AbrB3647 regulation closely overlap. Based on these results, we will better understand the roles of the three AbrBs in regulating solventogenic clostridia cell physiology.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Fermentation based on solventogenic clostridia is currently the most robust biological process for n-butanol production (Durre 2008, 2011; Green 2011; Lee et al. 2008). n-butanol is not only a bulk chemical but also an excellent fuel with better characteristics than ethanol (Durre 2007). The solventogenic clostridia life cycle can be divided into three typical phases: acidogenesis, the transition state, and solventogenesis (Grupe and Gottschalk 1992). Normally, acetic and butyric acid are formed and reassimilated in the acidogenic and transition phases, respectively; the target products, namely the ABE solvents (acetone, n-butanol, and ethanol), are then produced during the solventogenic phase.
Throughout the fermentation process, the transition state is a “switch” that controls the flux from acid formation towards solvents production. Comprehensively dissecting the regulatory mechanisms underlying the above three stages, especially the transition stage, would aid in developing a more efficient ABE solvent production process (Amador-Noguez et al. 2011; Jang et al. 2014; Kumar et al. 2013; Millat et al. 2014). However, to date, our understanding on how clostridia regulate such biphasic fermentation is limited, and very few relevant regulators have been identified, such as Spo0A (Harris et al. 2002), CcpA (Ren et al. 2012), Rex (Wietzke and Bahl 2012), and AbrB (Scotcher et al. 2005).
AbrB is a global regulator that regulates many cellular processes and has been mostly reported in Bacillus species (Gone et al. 2014; Olson et al. 2012; Phillips and Strauch 2002). AbrB and its orthologs aid in controlling sporulation (Shafikhani and Leighton 2004), antibiotic biosynthesis (Park et al. 2012), biofilm formation (Murray et al. 2009), and cyanotoxin production (Shalev-Malul et al. 2008), among other processes. For solventogenic clostridia, the AbrB regulator has only been characterized in the typical species Clostridium acetobutylicum by Scotcher, et al. (Scotcher et al. 2005). Among the three abrB genes (CAC0310, CAC1941, and CAC3647) in C. acetobutylicum, only abrB0310 (CAC0310) was transcriptionally active in the catP reporter assay. Furthermore, lower abrB0310 expression upon using an abrB0310 antisense construct produced clear phenotypic effects, which indicates the importance of abrB0310 in regulating acid and solvent formation as well as the two-phase transition in C. acetobutylicum (Scotcher et al. 2005). However, interesting questions include whether the other two AbrBs also play a role in C. acetobutylicum despite no detected expression and how these three AbrBs coordinate their functions are not clear.
In this study, we performed a detailed investigation to clarify the function of these three C. acetobutylicum abrBs. Interestingly, abrB3647 (CAC3647) exerted an important influence on cellular performance that opposes abrB0310’s influence, while abrB1941 played a slight role. Compared with abrB0310, abrB3647 disruption enhanced fermentation efficiency, which was demonstrated by a significantly increased growth rate. The following analysis using quantitative reverse transcription PCR (qRT-PCR) and electrophoretic mobility shift assays (EMSA) further confirms that both AbrB0310 and AbrB3647 play a major role in regulating acidogenesis, solventogenesis, and the two-phase transition in C. acetobutylicum.
Materials
Strains, plasmids, media, and cultivation conditions
The bacterial strains and plasmids used in this work are listed in Table S1. Top10 and ER2275 were grown in Luria–Bertani (LB) medium or on LB agar and used for gene cloning. Rosetta (DE3) was used for protein purification. The appropriate quantity of antibiotics (100 μg/ml ampicillin, 50 μg/ml kanamycin, 25 μg/ml chloramphenicol, and 100 μg/ml spectinomycin) was added to the LB medium when necessary. The plasmids used in C. acetobutylicum ATCC 824 were first methylated by ER2275 and then added to C. acetobutylicum through electroporation. For inoculum preparation, CGM medium (Wiesenborn et al. 1988) was inoculated with C. acetobutylicum, which was grown anaerobically (Thermo Forma Inc., Waltham, MA) at 37 °C. We used P2 (Baer et al. 1987) medium for solvent production. Erythromycin (10 μg/ml) and thiamphenicol (8 μg/ml) were added to the P2 medium when necessary.
Plasmid construction
The primers used in this work are listed in Table S2. The Targetron plasmid pWJ1 (Sun et al. 2015) was used to disrupt the genes in C. acetobutylicum. In this work, we used the Targetron system to disrupt CAC0310 and CAC3647. The 350 bp Targetron fragment used for CAC0310 disruption was amplified using PCR and the following primers: the EBS universal primer, CAC0310-109S-IBS, CAC0310-109S-EBS1d, and CAC0310-109S-EBS2. The 350-bp Targetron fragment used for CAC3647 disruption was PCR-amplified using the EBS universal primer, CAC3647-109S-IBS, CAC3647-109S-EBS1d, and CAC3647-109S-EBS2 according to the protocol of the Targetron™ Gene Knockout System Kit (Sigma-Aldrich, St Louis, MO, USA). The 350-bp fragments were digested with Xhol and BsrGI and cloned into pWJ1 (Sun et al. 2015) to construct the plasmids pWJ1-abrB0310 and pWJ1-abrB3647. CAC1941 was deleted from the C. acetobutylicum genome using double crossover homologous recombination technology (Cartman et al. 2012). The codA gene was amplified from the vector pMTL-SC7315 using PCR and then cloned into pWJ1, yielding the plasmid pSJ1. To delete CAC1941, the 1.2-kb and 500-bp sequences in both the up- and down-stream regions flanking CAC1941 were amplified using PCR and cloned into pSJ1, yielding pSJ1-abrB1941. Next, CAC1941 was deleted from the C. acetobutylicum genome using pSJ1-abrB1941 according to the double crossover homologous recombination technology (Cartman et al. 2012). The abrB-overexpression plasmids pIMP1-P thl -AbrB0310, pIMP1-P thl -AbrB1941, and pIMP1-P thl -AbrB3647 were constructed using the primer pairs CAC0310-for/CAC0310-rev, CAC1941-for/CAC1941-rev, and CAC3647-for/CAC3647-rev, respectively. After PCR-amplification, the abrB genes were digested with XbaI and BamHI and then cloned into pIMP1-P thl , yielding the plasmids pIMP1-P thl -AbrB0310, pIMP1-P thl -AbrB1941, and pIMP1-P thl -AbrB3647. To purify the proteins AbrB0310 and AbrB3647, the abrB0310 and abrB3647 fragments were first PCR-amplified by using the primer pairs CAC0310-ex-for/CAC0310-ex-rev and CAC3647-ex-for/CAC3647-ex-rev, respectively. The two fragments were then digested with XhoI and BamHI and cloned into pET28a (Novagen, Madison, WI), yielding the plasmids pET-28a-abrB0310 and pET-28a-abrB3647, respectively.
Fermentation
P2 medium with D-glucose (70 g/l) as the sole carbon source was used to perform the batch fermentation. Erythromycin (10 μg/ml) was added to the medium to sustain the plasmid. To begin fermentation, the frozen C. acetobutylicum cell stock was first used to inoculate CGM liquid medium, which was incubated at 37 °C for the first preparation. After growing for 12 h (OD600 = 0.8–1.0), 5 ml of the cells was transferred to 50 ml CGM liquid medium and grown for 6 h at 37 °C. Finally, 2 ml of the cells (OD600 = 0.8–1.0) was used to inoculate 28 ml P2 medium for fermentation. The samples were removed and stored at −20 °C at different time points.
Analytical methods
Cell growth (OD600) was monitored using a spectrophotometer (DU730, Beckman Coulter). The fermentation products (acetone, acetic acid, butyric acid, butanol, and ethanol) were assayed using gas chromatography (7890 A, Agilent, Wilmington, DE, USA), during which isobutyl alcohol and isobutyric acid were used as internal standards. The glucose concentration was assayed using a high-pressure liquid chromatography system (model 1200, Agilent).
Southern blot
Southern blot was performed using a digoxigenin (DIG) High-Prime DNA labeling and detection kit (Roche, Mannheim, Germany) as instructed by the manufacturer. Model 785 Vacuum Blotter (Bio-Rad, California, USA) was used to transfer DNA from an agarose gel onto a nylon membrane.
Protein purification
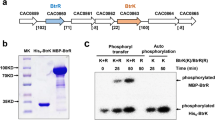
To purify the AbrB proteins, the plasmids pET-28a-abrB0310 and pET-28a-abrB3647 were used to transform the E. coli Rosetta (DE3) strain. These two strains were grown in 200 ml LB medium. When the OD600 reached 0.8, lactose was added to the medium at 10 mg/ml to induce AbrB expression. After growing at 16 °C for 24 h, the cells were harvested and suspended with a buffer containing 20 mM Tris (pH 7.9), 500 mM KCl, 10 % (v/v) glycerol, and 10 mM imidazole. The suspended cells were then disrupted using a French press (Constant Systems Limited, UK), and the cell debris and membrane fractions were removed using centrifugation (1 h, 11,400×g, 4 °C). The His-tagged AbrB dissolved in the supernatant was purified using a column filled with Ni Sepharose™ 6 fast flow agarose (GE Healthcare, Waukesha, WI) in accordance with the manufacturer’s instructions and was finally stored at −80 °C.
Electrophoretic mobility shift assay
The probes used for the electrophoretic mobility shift assay were firstly amplified from the C. acetobutylicum genome. Next, a Cy5-labeled universal primer (5′-AGCCAGTGGCGATAAG-3′) was used to label the EMSA probes. The Cy5-labeled DNA probe (0.04 pmol) was incubated with the indicated quantity of His6-AbrB in the binding buffer containing 20 mM Tris-HCl (pH 7.9), 40 ng/ml bovine serum albumin (BSA), 5 % glycerol, 10 mM MgCl2, 40 mM KCl, 0.25 mM DTT, and 50 ng/μl fish sperm DNA (Wu et al. 2015). After incubating at 25 °C for 20 min, the DNA-protein complexes were loaded onto a pre-runed 5 % polyacrylamide gel (acrylamide: bis-acrylamide = 80:1) and run for 70 min in an ice bath. Finally, the gel was scanned using a FLA-9000 phosphorimager (FujiFilm, Japan) for visualization.
Quantitative real-time PCR
For qRT-PCR analyses, cells were grown in 1 l P2 medium supplemented with erythromycin (10 μg/ml) and D-glucose (70 g/l) as the sole carbon source. The cells grown for 20 h (acidogenic phase) and 44 h (solventogenic phase) were harvested and used for qRT-PCR analyses. RNA was isolated through TRIzol (Invitrogen, Carlsbad, USA) extraction in accordance with the manufacturer’s instructions. The PrimeScript RT reagent kit (Takara, Otsu, Japan) was used to synthesize cDNA. qRT-PCR was performed by using CAC2679 (pullulanase) as the internal control and conducted in a MyiQ2 two-color real-time PCR detection system (Bio-Rad, Singapore) using iQ™ SYBR Green Supermix (Bio-Rad, Singapore). The primers used for qRT-PCR are listed in Table S2.
Results
Both AbrB0310 and AbrB3647 play major regulatory roles in C. acetobutylicum
We first attempted to inactivate these three abrB genes in vivo to observe the resulting phenotypic effects. Using the Targetron system (Shao et al. 2007), we disrupted abrB0310 and abrB3647; however, this method did not successfully disrupt abrB1941. Therefore, we used homologous recombination and the newly constructed plasmid pSJ1 to delete abrB1941. As shown in Fig. S1, abrB1941 was successfully deleted, while abrB0310 and abrB3647 were intact. Besides, Southern blot was further performed to rule out the potential unintended deletions in the chromosome using Targetron system (Fig. S2).
The resulting three mutants, 824m-0310, 824m-1941, and 824m-3647, were cultivated to examine the phenotypic changes compared with the wild-type strain (824WT). As shown in Fig. 1 and Table 1, except for similar growth, sugar consumption, acid reassimilation and ABE solvent formation in the strain 824m-0310 were inferior to the strain 824WT. This result was largely consistent with the previous finding from Scotcher, et al. (Scotcher et al. 2005), in which the abrB0310 transcript levels decreased using an antisense construct. In contrast, the strain 824m-3647 exhibited much better growth and sugar consumption, which thereby enabled faster acid reassimilation and solvent production than the strain 824WT (Fig. 1). The strain 824m-3647 reached the highest cell density (OD600 = 14.01) and total solvents (23.45 g/l) within 53 h, whereas those of the strain 824WT were only 8.55 (OD600) and 14.27 g/l at this time point. However, during the subsequent fermentation period, this mutated strain quickly died, and thus, no further increases in the concentration of solvents were observed (Fig. 1). These results suggest that abrB3647 is a pivotal regulator involved in growth of C. acetobutylicum.

Comparison of growth, sugar consumption, and solvent and acid production between the 824WT (Clostridium acetobutylicum wild-type strain, denoted as an open triangle), 824m-0310 (the abrB0310-inactivated strain, denoted as an open circle), 824m-1941 (the abrB1941-inactivated strain, denoted as a filled triangle), and 824m-3647 (the abrB3647-inactivated strain, denoted as a filled circle) strains in fermenting D-glucose. The fermentation experiments were performed in triplicate
The plasmid-based abrB3647 gene restored the behavior of the strain 824m-3647 to a similar level as the wild-type strain (Fig. S3), which demonstrates that the phenotypes of the strain 824m-3647 was due to the disrupted abrB3647 gene. The performance of the strain 824m-1941 during fermentation was similar to the strain 824WT (Fig. 1), which indicates that abrB1941 did not play a major regulatory role in the C. acetobutylicum life cycle.
We also examined the phenotypic effects of overexpression of abrB genes. However, as shown in Fig. S4 and Table S3, the reinforcement of the expressional level of each abrB gene in C. acetobutylicum did not cause obvious phenotypic changes.
The roles of the three AbrBs in regulating genes involved in the acidogenic, solventogenic, and transition states
To explain the phenotypic effects from disrupting abrB at the molecular level, we examined transcription changes in the relevant 19 genes that compose the pathways for phase transition as well as acid and solvent formation (Fig. 2a). Here, a two-fold change in the transcription level was regarded as a significant difference.

Quantitative RT-PCR analysis of the transcription changes for the genes involved in solvent formation after abrB was inactivated in Clostridium acetobutylicum. a Schematic of the genes related to solvent formation in C. acetobutylicum. b qRT-PCR of solvent formation-related genes in the abrB0310-inactivated strain (824m-0310) at 20 and 44 h. c qRT-PCR of solvent formation-related genes in the abrB1941-inactivated strain (824m-1941) at 20 and 44 h. d qRT-PCR of solvent formation-related genes in the abrB3647-inactivated strain (824m-3647) at 20 and 44 h. Gene expression is represented as the fold difference normalized to wild-type C. acetobutylicum. Error bars indicate the standard deviation for two independent biological replicates
As shown in Fig. 2, compared with abrB1941, disrupting abrB0310 and abrB3647 produced greatly altered transcription levels in more genes. Notably, after disrupting abrB3647, all eight genes (adhE2, ctfA-ctfB-adhE1, bdhA, bdhB, edh, and adc) responsible for ABE solvent formation exhibited an over two-fold increase in transcription at the 44 h time point (within the solventogenic phase) (Fig. 2d). This increase was especially observed for adhE2 and ctfA-ctfB-adhE1, which exhibited an approximately 124.67- and 9.78-fold increase, respectively. The significantly increase mRNA level of adhE2 might correlate with the higher ethanol production by the strain 824m-3647.
In contrast, out of the above eight genes responsible for solvents production, only adhE2 and adc showed a significant transcriptional change within the solventogenic phase (44 h) after inactivating abrB0310 (2.44- and 4.98-fold, respectively) (Fig. 2b); more genes, adhE2, bdhA, bdhB, edh, and adc, showed a significant increase in transcription during the acidogenic phase (20 h) (Fig. 2b). However, although adc was upregulated after abrB0310 inactivation, no obvious increase was observed in acetone production (Fig. 1), which may be due to the relatively lower mRMA level of ctfAB, another key gene encoding CoA-transferase for acetone formation (Fig. 2a). In addition, the bukII gene maintained an increased transcription level at both the acidogenic and solventogenic phase when abrB0310 was disrupted (Fig. 2b).
Inactivating abrB1941 only produced a clear transcription change in three genes (adhE2, adc, and bukII) at different time points, a 2.92-fold upregulation for adhE2 at 20 h, 2.67-fold upregulation for adc at 44 h, and 4.17-fold downregulation for bukII at 44 h (Fig. 2c). No significant change was observed for ctfA-ctfB-adhE1, which is the important gene cluster responsible for acid reassimilation and solvent production in C. acetobutylicum. These data may partly explain why abrB1941 disruption does not confer a clear phenotypic effect.
Disrupting abrB0310 and abrB3647 influenced transcription of most genes involved in acid and solvent formation as well as the two-phase transition (Fig. 2b, 2d). Therefore, we next attempted to identify the direct targets of AbrB0310 or AbrB3647 among these genes. The EMSA results show that, among the 19 candidates (Fig. 3a), AbrB0310 bound to the promoter region of ten genes (Fig. 3b and S5a). In addition, eight genes were subject to AbrB3647 (Fig. 3c and S5b). Therefore, these data further demonstrated that AbrB0310 and AbrB3647 directly regulate many genes located in the solvent-forming pathways.

EMSAs of AbrB0310 and AbrB3647 binding to probes derived from the promoter region of solvent-formation genes in Clostridium acetobutylicum. a The probes used in the EMSA analyses. b EMSAs of AbrB0310 binding to seven probes. We used probes derived from the promoter regions of CAP0162, CAP0035, CAP0059, CAC3298, CAC1660, CAP0165, and CAC1742. c EMSAs of AbrB3647 binding to six probes. We used probes derived from the promoter regions of CAP0162, CAP0035, CAP0059, CAC3298, CAC1660, and CAC3299. We used 0–0.6 μM His6-AbrB
Discussion
The C. acetobutylicum genome sequence provides three putative abrB genes (Nolling et al. 2001). These three clostridial AbrBs (AbrB0310, AbrB1941, and AbrB3647) show a similar amino acid sequence identity to the Bacillus subtilis AbrB (BSU00370) (65.79, 63.51, and 64.47 %, respectively). In the previous report, abrB0310 was identified as a functional copy of the abrBs in C. acetobutyliucm, and abrB1941 and abrB3647 were proposed as pseudogenes due to undetectable expression levels (Scotcher et al. 2005). However, the results here show that abrB3647 and abrB1941 are also involved in regulating genes responsible for acidogenesis and solventogenesis as well as the two-phase transition in C. acetobutylicum. Especially for abrB3647, its disruption yields a significant effect on growth, which opposes abrB0310’s effect, suggesting that abrB3647 also plays a major regulatory role.
In fact, our microarray data (unpublished results) show that both abrB3647 and abrB1941 are transcribed in C. acetobutylicum. The transcription level of abrB3647 was even higher than abrB0310 at both the acidogenic and solventogenic stages; in stark contrast, abrB1941 transcription was over ten-fold lower than abrB0310, which is consistent with previous results (Scotcher et al. 2005). These data also suggest that abrB3647 is an active copy of abrBs in C. acetobutylicum. The conflicting conclusions for abrB3647’s behavior in C. acetobutylicum in this study and the previous report may be attributed to the different growth conditions used. Compared with Bacillus subtilis, multiple active AbrBs in C. acetobutylicum may produce more diverse regulation in this anaerobe during biphasic fermentation.
Based on the EMSA results (Fig. 3), many genes responsible for acetogenesis, solventogenesis, and the two-phase transition are directly regulated by both AbrB0310 and AbrB3647, which demonstrates that the regulatory networks for these two AbrBs overlap. This may explain the result where certain genes that bind multiple AbrBs did not show significant transcription changes. However, simultaneously, a comparative transcription analysis showed different genes with greatly altered transcriptional levels after knocking out abrB0310 or abrB3647 (Fig. 2b, 2d), which suggests that AbrB0310 and AbrB3647 feature their own regulatory emphases. Notably, adhE1 and adhE2, two aldehyde/alcohol dehydrogenase-encoding genes, were markedly upregulated at 44 h (during the solventogenic phase) in 824m-3647 mutant. adhE2 exhibited especially upregulated transcription levels with an over 120-fold increase at 44 h after disrupting abrB3647 (Fig. 2d), which may contribute to the high ethanol production in 824m-3647. It should be noted that, although adhE2 encodes an active aldehyde/alcohol dehydrogenase (Millat et al. 2013; Yu et al. 2011), adhE1 is more important than adhE2 for solvent synthesis in C. acetobutylicum. Its knockout yielded little solvent production, and no obvious phenotypic changes were observed upon inactivating adhE2 (Cooksley et al. 2012). Thus, significant upregulation of the sol operon (adhE1-ctfA-ctfB) in the abrB3647-disrupted strain may be a major factor in the improved solvent-producing capacity of the mutated strain 824m-3647 (Fig. 1). Further, in the previous report, a comparison of the proteomic profiles between wild-type C. acetobutylicum and a mutated non-solventogenic strain (M5) also linked high expression of AdhE1 and its co-expressed product CtfB to the capacity for solvent production (Jang et al. 2014).
In contrast to the significant phenotypic effects caused by knocking out abrB0310 or abrB3647, overexpression of these two abrB genes only slightly influenced the cellular performance of C. acetobutylicum. This result indicates that the expression levels of the abrBs in C. acetobutylicum may be sufficient for exerting regulatory roles.
In summary, here, we discerned the regulatory roles of the three abrB genes in C. acetobutylicum. We show that abrB1941 and abrB3647, two previously proposed pseudogenes, function in C. acetotutylicum. Especially abrB3647, it plays a major role in regulating biphasic fermentation of C. acetobutylicum, while abrB1941 might not be essential. Furthermore, we observed an overlap in the target genes subject to direct regulation of AbrB0310 and AbrB3647. Elucidating the regulatory roles of AbrBs will expand our understanding of the mechanisms underlying biphasic fermentation of C. acetobutylicum.
References
Amador-Noguez D, Brasg IA, Feng XJ, Roquet N, Rabinowitz JD (2011) Metabolome remodeling during the acidogenic-solventogenic transition in Clostridium acetobutylicum. Appl Environ Microbiol 77(22):7984–7997
Baer SH, Blaschek HP, Smith TL (1987) Effect of butanol challenge and temperature on lipid composition and membrane fluidity of butanol-tolerant Clostridium acetobutylicum. Appl Environ Microbiol 53(12):2854–2861
Cartman ST, Kelly ML, Heeg D, Heap JT, Minton NP (2012) Precise manipulation of the Clostridium difficile chromosome reveals a lack of association between the tcdC genotype and toxin production. Appl Environ Microbiol 78(13):4683–4690
Cooksley CM, Zhang Y, Wang H, Redl S, Winzer K, Minton NP (2012) Targeted mutagenesis of the Clostridium acetobutylicum acetone-butanol-ethanol fermentation pathway. Metab Eng 14(6):630–641
Durre P (2007) Biobutanol: an attractive biofuel. Biotechnol J 2(12):1525–1534
Durre P (2008) Fermentative butanol production—bulk chemical and biofuel. Ann N Y Acad Sci 1125:353–362
Durre P (2011) Fermentative production of butanol—the academic perspective. Curr Opin Biotechnol 22(3):331–336
Gone AML, Giron JDT, Montejo FEJ, Hidalgo-Lara ME, Lopez VELY (2014) Behavior of transition state regulator AbrB in batch cultures of Bacillus thuringiensis. Curr Microbiol 69(5):725–732
Grupe H, Gottschalk G (1992) Physiological events in Clostridium acetobutylicum during the shift from acidogenesis to solventogenesis in continuous culture and presentation of a model for shift induction. Appl Environ Microbiol 58(12):3896–3902
Green EM (2011) Fermentative production of butanol - the industrial perspective. Curr Opin Biotechnol 22(3):337–343
Harris LM, Welker NE, Papoutsakis ET (2002) Northern, morphological, and fermentation analysis of spo0A inactivation and overexpression in Clostridium acetobutylicum ATCC 824. J Bacteriol 184(13):3586–3597
Jang YS, Han MJ, Lee J, Im JA, Lee YH, Papoutsakis ET, Bennett G, Lee SY (2014) Proteomic analyses of the phase transition from acidogenesis to solventogenesis using solventogenic and non-solventogenic Clostridium acetobutylicum strains. Appl Microbiol Biotechnol 98(11):5105–5115
Kumar M, Gayen K, Saini S (2013) Role of extracellular cues to trigger the metabolic phase shifting from acidogenesis to solventogenesis in Clostridium acetobutylicum. Bioresour Technol 138:55–62
Lee SY, Park JH, Jang SH, Nielsen LK, Kim J, Jung KS (2008) Fermentative butanol production by clostridia. Biotechnol Bioeng 101(2):209–228
Millat T, Janssen H, Thorn GJ, King JR, Bahl H, Fischer RJ, Wolkenhauer O (2013) A shift in the dominant phenotype governs the pH-induced metabolic switch of Clostridium acetobutylicumin phosphate-limited continuous cultures. Appl Microbiol Biotechnol 97(14):6451–6466
Millat T, Voigt C, Janssen H, Cooksley CM, Winzer K, Minton NP, Bahl H, Fischer RJ, Wolkenhauer O (2014) Coenzyme A-transferase-independent butyrate re-assimilation in Clostridium acetobutylicum—evidence from a mathematical model. Appl Microbiol Biotechnol 98(21):9059–9072
Murray EJ, Strauch MA, Stanley-Wall NR (2009) SigmaX is involved in controlling Bacillus subtilis biofilm architecture through the AbrB homologue Abh. J Bacteriol 191(22):6822–6832
Nolling J, Breton G, Omelchenko MV, Makarova KS, Zeng Q, Gibson R, Lee HM, Dubois J, Qiu D, Hitti J, Wolf YI, Tatusov RL, Sabathe F, Doucette-Stamm L, Soucaille P, Daly MJ, Bennett GN, Koonin EV, Smith DR (2001) Genome sequence and comparative analysis of the solvent-producing bacterium Clostridium acetobutylicum. J Bacteriol 183(16):4823–4838
Olson AL, Bobay BG, Melander C, Cavanagh J (2012) H-1, C-13, and N-15 resonance assignments and secondary structure prediction of the full-length transition state regulator AbrB from Bacillus anthracis. Biomol NMR Assign 6(1):95–98
Park SY, Choi SK, Kim J, Oh TK, Park SH (2012) Efficient production of polymyxin in the surrogate host Bacillus subtilis by introducing a foreign ectB gene and disrupting the abrB gene. Appl Environ Microbiol 78(12):4194–4199
Phillips ZE, Strauch MA (2002) Bacillus subtilis sporulation and stationary phase gene expression. Cell Mol Life Sci 59(3):392–402
Ren C, Gu Y, Wu Y, Zhang WW, Yang C, Yang S, Jiang WH (2012) Pleiotropic functions of catabolite control protein CcpA in butanol-producing Clostridium acetobutylicum. BMC Genomics 13:349
Scotcher MC, Rudolph FB, Bennett GN (2005) Expression of abrB310 and sinR, and effects of decreased abrB310 expression on the transition from acidogenesis to solventogenesis, in Clostridium acetobutylicum ATCC 824. Appl Environ Microbiol 71(4):1987–1995
Shafikhani SH, Leighton T (2004) AbrB and Spo0E control the proper timing of sporulation in Bacillus subtilis. Curr Microbiol 48(4):262–269
Shalev-Malul G, Lieman-Hurwitz J, Viner-Mozzini Y, Sukenik A, Gaathon A, Lebendiker M, Kaplan A (2008) An AbrB-like protein might be involved in the regulation of cylindrospermopsin production by Aphanizomenon ovalisporum. Environ Microbiol 10(4):988–999
Shao L, Hu S, Yang Y, Gu Y, Chen J, Yang Y, Jiang W, Yang S (2007) Targeted gene disruption by use of a group II intron (targetron) vector in Clostridium acetobutylicum. Cell Res 17(11):963–965
Sun Z, Chen YX, Yang C, Yang S, Gu Y, Jiang WH (2015) A novel three-component system-based regulatory model for D-xylose sensing and transport in Clostridium beijerinckii. Mol Microbiol 95(4):576–589
Wiesenborn DP, Rudolph FB, Papoutsakis ET (1988) Thiolase from Clostridium acetobutylicum ATCC 824 and its role in the synthesis of acids and solvents. Appl Environ Microbiol 54(11):2717–2722
Wietzke M, Bahl H (2012) The redox-sensing protein Rex, a transcriptional regulator of solventogenesis in Clostridium acetobutylicum. Appl Microbiol Biotechnol 96(3):749–761
Wu Y, Yang YP, Ren C, Yang C, Yang S, Gu Y, Jiang WH (2015) Molecular modulation of pleiotropic regulator CcpA for glucose and xylose coutilization by solvent-producing Clostridium acetobutylicum. Metab Eng 28:169–179
Yu M, Zhang Y, Tang IC, Yang ST (2011) Metabolic engineering of Clostridium tyrobutyricum for n-butanol production. Metab Eng 13(4):373–382
Acknowledgments
This work was supported by the National Natural Science Foundation of China (31570043, 31370133, and 31421061), National High-tech Research and Development Program of China (2011AA02A208 and 2015AA020202), and Youth Innovation Promotion Association CAS.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Additional information
Y. Y and Q. X. contributed equally to this study.
Electronic supplementary material
ESM 1
(DOC 1.58 mb)
Rights and permissions
About this article

Cite this article
Xue, Q., Yang, Y., Chen, J. et al. Roles of three AbrBs in regulating two-phase Clostridium acetobutylicum fermentation. Appl Microbiol Biotechnol 100, 9081–9089 (2016). https://doi.org/10.1007/s00253-016-7638-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-016-7638-x