
Abstract
In spite of their involvement in foodborne illness, the epidemiological relevance of staphylococcal enterotoxin C (SEC) subtypes is poorly documented may be due to high sequence similarity. Among subtypes, SEC1, SEC2, and SEC3 exhibit more than 97 % homology because of which specific detection tools are seldom available to identify and differentiate them. In this study, a SYBR Green-based RT-PCR followed by melt curve analysis was developed for differentiation of entC1 from entC2/entC3 using a single primer pair. Nucleotide sequences of all three subtypes were analyzed using Clustal Omega program and the region with significant sequence variation/heterogeneity (where utmost SNPs were closely located and accessible for RT-PCR) was selected for amplification by designing a single primer pair that could amplify all three subtypes. In spite of same amplicon size, entC1 showed distinct melt peak at 76 °C. However, due to high similarity between entC2 and entC3, the developed format was deficient to discriminate between them and both showed melt peak at 82 °C. Reliability of developed RT-PCR was evaluated using various naturally contaminated samples and 91 food and clinical Staphylococcus aureus isolates where satisfactory results were obtained in comparison with commercial immunoassay kit and conventional PCRs using validated primers. To the best of our knowledge, this is the first method being reported to differentiate entC1 from entC2/entC3 using single primer pair which is unachievable by conventional PCR due to same amplicon size. As benefits, the method is sensitive, rapid, and inexpensive with no requirement of fluorescent probes, multiple primers, and post-PCR procedures. Thus, the assay might find its utility as a detection tool in epidemiological survey of foodborne outbreaks for simultaneous identification and differentiation of entC1 from entC2/entC3.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Heat-labile Staphylococcus aureus is known to produce heat-stable enterotoxins, the ingestion of which leads to awful food poisoning consequences (Balaban and Rasooly 2000). More than 21 types of SEs have been identified including SEA-SEE, which are designated as classical enterotoxins; SEG-SEU, which are known as recently described enterotoxins and staphylococcal enterotoxin-like proteins (SEls). Among the group, SEC exhibits more heat tolerance followed by SEB and SEA (Tibana et al. 1987). In addition, the member SEC is divided into molecular variants such as SEC1, SEC2, and SEC3 as well as SECbovine, SECcanine, and SECovine which are highly conserved proteins. The subtypes SEC1, SEC2, and SEC3 are encoded by entC1, entC2, and entC3, respectively, and are most immunologically related. More than 97 % homology has been encountered among three subtypes both in nucleotide and protein sequences (Betley et al. 1992). The C-terminal domain is highly conserved and variation is mainly confined to N-terminal region. The gene entC1 shares 97.9 % homology with entC2 and 97.4 % homology with entC3, whereas entC2 and entC3 share 98.0 % homology. As mature toxins, SEC2 and SEC3 share the highest homology with only four amino acid difference (98.3 % homology). SEC1 differs from SEC2 by seven amino acid residues (97.1 % homology) and from SEC3 by nine amino acids (96.2 % homology) (Hovde et al. 1990; Marrack and Kappler 1990).
In spite of high degree of sequence homology, each subtype differ biologically and physicochemically demonstrating unique properties. They exhibit distinct epitopes, different isoelectric points (8.6, 7.8, and 8.2 respectively for SEC1, SEC2, and SEC3), and ability to stimulate divergent T cell populations (Reiser et al. 1984; Deringer et al. 1996). Enterotoxin C has been proven for its superantigenic activity that induces enormous immune response leading to toxic shock (Bohach et al. 1990, 1997; Marrack and Kappler 1990). In fact, the ability of SEC to induce massive cytokine release has been studied for its effect to inhibit tumor formation. Consequently, the subtype SEC2 and its mutants have been reported for its antitumor activity (Ming-Kai and Cheng-Gang 2006; Wang et al. 2009; Zhou et al. 2013) and have been used for tumor therapy in China for decades (Chen 2001). On the other hand, the presence of other subtype SEC1 has been demonstrated in synovial fluid of arthritis patients (Ataee et al. 2014) and it was also found to be involved in sudden infant death syndrome (Zorgani et al. 1999). Thus, these structurally related toxins differ in their mode of action/biological properties also.
As a potential gastrointestinal toxin, SEC is involved in foremost foodborne outbreaks and the gene sec is very frequently encountered in myriad disease cases (Mizukane et al. 1998; Normanno et al. 2005). Particularly, S. aureus strains isolated from animals affected with mastitis produced SEC as a major toxin (Da Silva et al. 2005; Chu et al. 2012). However, information about the involvement of particular subtype is not available in majority of suspected cases. Although these subtypes and their respective genes were described in the middle of 1980s, their incidence in any foodborne outbreak is rarely reported may be due to lack of reliable detection systems which can detect and discriminate subtypes. Furthermore, SEC is commonly isolated from human and food sources and sequence heterogeneity among subtypes is associated with their survival in respective host (Bohach and Schlievert 1989; Marr et al. 1993). This suggests that, investigation of a specific subtype coupled with differentiation enables one to anticipate host specificity and possible consequences of particular toxin.
Commercial immunoassay kits such as RIDASCREEN, SET-RPLA, SET-EIA, VIDAS, and TECRA, though available for detection of SEA-SEE toxins, are unable to detect SEC subtypes. Further, immunoassay-based detection approaches have been reported for the detection of SEC1 and SEC2 (Gao et al. 2000; Dong et al. 2001; Hun and Zhujun 2007; Huang et al. 2015). But the methods usually identify only single subtype at a time. Previous attempts for molecular based detection of subtype-specific SEC include application of PCR and PCR-RFLP (Chen et al. 2001; Hsiao et al. 2003; Wang et al. 2003) which necessitates post-PCR methods such as gel electrophoresis that involve carcinogenic reagents for result interpretation, time-consuming and may often lead to carry over contamination. Further in available methods, differentiation is not possible in a single reaction.
In this article, we report our study on the development of an SYBR Green-based RT-PCR followed by melt curve analysis to differentiate entC1 from entC2/entC3 using a single set of unlabelled primers. N-terminal sequence showing significant sequence variation/heterogeneity was opted, and a suitable primer pair was designed which can amplify all three subtypes. Application of the developed RT-PCR aided simple, rapid, and real-time detection and differentiation of entC1 from entC2/entC3 in a single, closed tube format.
Materials and methods
Bacterial strains and cultivation
S. aureus and other non-staphylococcal bacterial reference strains used in this study are listed in Table 1. In addition, 91 S. aureus strains isolated from various food and clinical sources were also used in the study (Table 3). These isolates were biochemically confirmed using tests that are specific to S. aureus in our previous study (Nagaraj et al. 2014). The bacterial strains were grown in Brain Heart Infusion (BHI) broth (HiMedia, Mumbai, India) for DNA extraction. Briefly, one loopful of bacterial culture was inoculated into 5 ml BHI broth and incubated in 37 °C shaking incubator for 18–24 h. For enterotoxin production, S. aureus strains were grown in Tryptic Soy Broth (TSB) (HiMedia, Mumbai, India) for 24 h. The cultures were stored as 15 % glycerol stocks at −80 °C for future use.
DNA extraction
Genomic DNA of bacterial strains was extracted by phenol:chloroform liquid-liquid extraction method (Sambrook and Russell 1989) with slight modification. Briefly, cells harvested from an overnight culture in BHI broth were pelleted and resuspended in 1× TE buffer (10 mM Tris-HCl, 1 mM EDTA, pH 8.0) containing 20 μL lysostaphin (Sigma). After incubation at 37 °C for 1 h, 20 μL proteinase K (20 mg/mL) (Sigma) was added followed by incubation at 55 °C to get a clear lysate. This was followed by phenol:chloroform:iso-amyloalcohol in the ratio of 25:24:1. The aqueous phase collected in the previous step was precipitated by two volumes of absolute alcohol and washed by ice-cold 70 % ethanol. The residual ethanol was dried and DNA pellet was resuspended in 50 mM Tris-HCl buffer (pH 7.5). The purity as well as concentration of purified DNA was analyzed using NanoDrop 2000 (Thermo Scientific, Waltham, MA, USA).
In silico analysis of entC1, entC2, and entC3 gene sequence
Clustal Omega, a multiple sequence alignment tool available on EMBL-EBI website (http://www.ebi.ac.uk/Tools/msa/clustalo/) was used for the conservation and variation analysis of entC1 (Accession No. X05815.1), entC2 (Accession No. GQ461752.1), and entC3 (Accession No. X51661.1) genes. N-terminal sequence where the subtypes show many SNPs was considered to differentiate the subtypes.
RT-PCR primer design
Based on in silico analysis, a small sequence of 125-bp length was selected to facilitate effectual amplification in RT-PCR. A single set of primer (entC) was designed for the amplification of selected region using the conserved region of all three variants, so that the primer could amplify entC1, entC2, and entC3 (Table 2). Gene Runner version 3.01 software (Hastings Software Inc. Hastings on Hudson, NY, USA) was employed for primer designing.
Conventional PCR and sequencing analysis
A conventional singleplex PCR was performed to ensure the ability of entC primers to amplify all three subtypes. DNA from the standard strains ATCC 19095 (SEC1), FRI 361 (SEC2), and ATCC 700699 (SEC3) were used as reference templates for respective subtype specific gene amplification. The PCR was achieved in 20 μL reaction mixture with 1× PCR buffer, 2.0 mMMgCl2, 0.2 mM dNTPs (Fermentas GMBH, Saint Leon-Rot, Germany), 0.6 μM primers, 50 ng of template DNA, and 1.25 U Taq polymerase (Sigma). The amplification conditions included an initial denaturation at 94 °C for 3 min, followed by 30 cycles of denaturation at 94 °C for 10 s, annealing at 55 °C for 10 s, and extension at 72 °C for 10 s. This was followed by a final extension at 72 °C for 5 min. Thermal cycling was carried out in Master Cycler-Pro thermal cycler (Eppendorf, Hamburg, Germany). The PCR product was resolved on 1.5 % agarose (Seakem LE) gel in 0.5× Tris-borate EDTA buffer containing 0.5 μg/mL ethidium bromide at 150 V for 15 min. The amplicons were examined and documented using GelDoc system (Syngene, UK). The amplicons were purified by NucleoSpin, PCR clean-up, and gel extraction kit (Macherey-Nagel, Duren, Germany) and sequenced at Pentavalent Biosciences Pvt. Ltd., Bangalore. The sequencing result was analyzed by BLAST program.
RT-PCR conditions
Genomic DNA from ATCC 19095, FRI 361, and ATCC 700699 were used as reference template to optimize RT-PCR for entC1, entC2, and entC3, respectively. Each PCR assay was performed in 20 μL final volumes containing 1× SYBR Green PCR master mix (TaKaRa), 600 nM of entC primers, and 10 ng of respective reference DNA templates. Cycling was performed as follows: initial denaturation step at 98 °C for 2 min, 40 cycles of 98 °C for 10 s, 55 °C for 10 s, and 72 °C for 10 s. The fluorescence acquisition was done at the end of each extension step. Finally, melt curve analysis was performed by cooling the PCR products to 70 °C followed by heating at 95 °C at a rate of 0.5 °C per second with data acquisition every 1 s. Reaction containing all components except DNA was used as no template control (NTC). RT-PCR assays were performed on a fluorometric thermal cycler CFX 96™ Real-Time PCR Detection System. The data were collected and processed using the software Bio-Rad CFX-Manager Software v2.0 (Bio-Rad Laboratories, Inc., Hercules, CA). The threshold cycle (C T) was defined as the cycle number at which fluorescence of the sample crossed threshold point. Melt curves were later converted into melt peaks (T m) based on initial fluorescence curves (RFU/T) by plotting negative derivative of fluorescence versus temperature (−d(RFU)/dT versus T). The T m value was defined as the peak of the melt curve. The assay was performed in duplicates for reproducibility.
Sensitivity
The sensitivity of the developed assay was determined using purified genomic DNA as well as culture from bacterial strain ATCC 19095. In case of purified genomic DNA, 10-fold serially diluted template DNA ranging from 10 ng to 100 fg was subjected to RT-PCR and analyzed for melt curve peaks. In case of bacterial cell load, target bacteria were grown overnight at 37 °C in BHI broth, turbidity was adjusted to 0.5 McFarland standard followed by 10-fold serial dilutions from 106 to 100 CFU/mL. Template was prepared from each dilution by boil-lysate technique (Theron et al. 2000) and 1 μL of obtained template was used to perform RT-PCR as mentioned earlier. Parallely, 100 μL aliquot from each dilution was spread on plate count agar (HiMedia, Mumbai, India) plate and incubated at 37 °C for overnight and the grown bacterial colonies were counted in order to determine the bacterial load. In both cases, an NTC was also included in the reaction. Based on the C T values obtained in RT-PCR, limit of detection was determined which is defined as the least concentration of DNA where detectable florescence was achieved. The experiment was performed in duplicates for reproducibility.
Reliability
For determination of specificity of the developed assay, staphylococcal as well as non-staphylococcal bacterial strains listed in Tables 1 and 3 were used. We investigated 91 S. aureus strains isolated from various food and clinical samples by RT-PCR to detect the presence of subtype specific genes and differentiate among them based on melt curve analysis. The clinical isolates (CI) were obtained from diarrhea patients with suspected foodborne cases. Briefly, genomic DNA was extracted from bacterial strains and 10 ng of DNA was used as template to carry out RT-PCR as mentioned earlier. Parallely, the assay was compared with PCR tests for the presence of sec as well as subtype specific genes (entC1, entC2, and entC3) using reported primers (Schmitz et al. 1998; Chen et al. 2001) (Table 2). Additionally, immunoassay was also performed with a commercial kit for the production of SEC toxin. The reaction volume of conventional PCR was 20 μL containing 1× PCR buffer (Sigma), 2.0 mM MgCl2, 0.2 mM dNTPs (Fermentas GMBH, Saint Leon-Rot, Germany), 1 μM primer, 50 ng of template DNA, and 1.25 U Taq polymerase (Sigma). The amplification conditions included an initial denaturation at 94 °C for 4 min, followed by 30 cycles of denaturation at 94 °C for 1 min, annealing at 58 °C for 1 min, and extension at 72 °C for 30 s. This was followed by a final extension at 72 °C for 5 min. Thermal cycling and gel analysis were performed as mentioned earlier. Tecra™ Staph Enterotoxin Visual Immunoassay kit (TECRA International Pvt. Ltd., Frenchs Forest, NSW, Australia) was used to detect the enterotoxin production and Tecra™ Staph Enterotoxins ID Test was used to identify specific type of the toxin (SEA, SEB, SEC, SED, and SEE) following the manufacturer’s instructions.
Naturally contaminated samples
In order to evaluate the real-time application of RT-PCR to detect and differentiate target genes, numerous (n = 89) naturally contaminated samples were tested. Samples were processed and template was prepared as per Nagaraj et al. (2014) following pre-enrichment. Briefly, 10 g of each sample was homogenized and transferred to 90 mL modified MSB containing 0.4 % sodium pyruvate. Following an overnight enrichment, 1 mL aliquot of sample was processed by boil-lysate technique for conventional PCR to assess the presence of sec and RT-PCR for the detection of entC1/entC2/entC3. Concurrently, same samples were subjected to immunoassay using Tecra™ kit following the manufacturer’s instructions.
Result
Sequence analysis and primer designing
The major objective of the study was to identify polymorphic sequence and to design a single primer pair that could amplify and differentiate all three subtypes. Due to availability of partial gene sequence of entC2 in NCBI database (81 bases are not available towards N-terminal with respect to entC1 and entC3 sequences) (Fig. S1), Clustal Omega multiple sequence alignment was considered from 82nd position so that the alignment contained sequence from all three subtypes. Thus, performed in silico comparative analysis of the partial nucleotide sequences of entC1, entC2, and entC3 revealed that the bases are highly conserved in the C-terminal region and divergence in a short stretch was found near N-terminal region. To explain, though at nucleotide level entC1 shares 97.9 % homology with entC2 and 97.4 % homology with entC3, Clustal Omega multiple sequence analysis revealed the presence of 19 SNPs among three variants (from 82nd position) (Table 4). Of the identified SNPs, 15 were closely located between 87 and 197 position of nucleotides, whereas four SNPs were distantly located at 255, 296, 402, and 478 positions on entC1 (Table 4). On the other hand, overall, only five SNPs were documented between entC2 and entC3 variants and were found to be individually dispersed throughout the gene stretch (Fig. S1). Accordingly, a 125-bp region of entC1, entC2, and entC3 was selected for primer designing (Fig. 1). Since the aim was to design a single set of primers to amplify all three subtypes, care was taken to include all possible SNPs within the selected 125-bp region. As a result, within the selected region, all 15 closely located SNPs were present in case of entC1, whereas only three of five SNPs in case of entC2 and entC3 (Fig. 1). Thus, designed pair of primer was comprised of a 19-mer forward primer spanning from 82 to 100 positions and an 18-mer reverse primer spanning from 189 to 206 positions with respect to entC1 sequence. The forward primer was 100 % identical in case of entC2 and entC3, but possessed an SNP at 6th position from 5′ end in case of entC1. Similarly, in case of reverse primer, all three subtypes had an SNP at 9th position from 5′ end. However, these SNPs were found to be highly conserved in case of each subtype.

The Clustal Omega multiple sequence alignment data showing conserved and polymorphic regions between SEC subtype nucleotide sequences. The sequence alignment was color coded for easy identification (blue = entC primer sequence, black = identical, red = SNPs). Alignment was considered from 82nd position with respect to entC1 and entC2 database sequences. a Alignment result between entC1, entC2, and entC3. b Alignment result between entC2 and entC3
Conventional PCR
To assess the efficiency of designed primer to hybridize and amplify all three subtypes, DNA from respective S. aureus reference strains was amplified by conventional PCR and the products were sequenced. The primer could amplify all three subtypes with a band length of 125 bp as interpreted by gel electrophoresis, irrespective of different templates (Fig. 2b). Further, BLAST analysis of sequencing results confirmed the presence of respective nucleotide sequences with conserved and polymorphic regions specific to each subtype (data not shown).

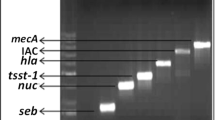
An example of PCR amplification using primers employed in the present study. a Agarose gel electrophoresis showing 270-bp product amplified using sec specific primers. b Agarose gel electrophoresis showing 125-bp product amplified using entC primers. Lanes: 1, DNA from S. aureus ATCC 19095 (SEC1); 2, DNA from S. aureus FRI 361 (SEC2); 3, DNA from S. aureus ATCC 700699 (SEC3); L, standard 1-kb ladder; M, standard 100-bp DNA ladder
RT-PCR
RT-PCR was carried out using entC primers with three different subtype-specific DNA as templates. Post-PCR melt curve analysis revealed that two distinct melt peaks were obtained even though a single primer pair was used for amplification. entC1 showed peak at 76 °C (Fig. 3a) whereas entC2 and entC3 showed peak at 82 °C (Fig. 3b). Also, a primer dimer was observed as a low melt peak at around 70 °C that could be distinguished from the intended product since they melt at a lower temperature (Fig. 3).

Fluorescence melt curves of SEC1 and SEC2 subtypes with distinct melt peaks after SYBR Green RT-PCR amplification using entC primers. PD—a non-specific peak at relatively low temperature (around 70 °C) represents primer dimer. a Characteristic melt peak at 76 °C in presence of DNA from SEC1-positive S. aureus ATCC 19095 strain. b Characteristic melt peak at 82 °C in presence of DNA from SEC2-positive S. aureus FRI 361. Similar peak was obtained for SEC3-positive S. aureus ATCC 700699 strain. c Comparison of C1 and C2 peaks by overlapping results obtained
Detection limit
Various DNA concentrations and cell load were tested. The C T values increased with decreasing DNA concentration and the limit of detection was obtained at a concentration of 100 fg of template (Fig. 4), and the result was found to be reproducible. Similarly the RT-PCR could detect 10 CFU/mL of bacterial cell load (data not shown).

Determination of sensitivity of RT-PCR in presence of different concentrations of DNA from S. aureus strain ATCC19095 using entC primers. NTC represents no template control containing all PCR components except DNA. Tenfold serial dilutions of DNA were used ranging from 10 ng to 100 fg. The C T values increased with decreasing DNA concentration and the limit of detection was obtained at a concentration of 100 fg of template DNA
Specificity of the assay
The assay was found to be specific to amplify DNA from staphylococcal strains only. There was no amplification plot or increased fluorescence observed in presence of non-staphylococcal DNA. Further, RT-PCR generated amplification plot only with SEC S. aureus strains which showed positive result for conventional PCR and Tecra™ immunoassay kit as well. Of the 91 isolates, 56 were found to be positive for conventional PCR with an amplicon of 270-bp length (Fig. 2a) and also for SEC toxin. These sec positive isolates showed positive result for RT-PCR also (entC primers) along with subtype-specific differentiation (Table 3). The RT-PCR results obtained were compared with conventional PCR results derived from validated subtype-specific primers. Among the 91 isolates tested, 12 isolates yielded distinct melt peaks at 76 °C corresponding to entC1, indicating that they were SEC1-positive strains. These isolates showed amplification for ENTC1 primers also. On the other hand, 27 among the 91 isolates yielded melt peaks at 82 °C corresponding to entC2/entC3. But when PCR was performed using validated subtype-specific primers for these isolates, 7 strains were found positive for entC2 and 20 were found positive for entC3 genes. The results obtained indicated the efficiency of developed assay to differentiate SEC1 from entC2/entC3 and also its inadequacy to differentiate between C2 and C3 subtypes. The remaining SEC-positive strains (17 strains) among 56 isolates could belong to other SEC subtypes such as SECbovine, SECcanine, and SECovine.
Evaluation
Among 89 different naturally contaminated samples tested, 11 samples were found to be positive for SEC as analyzed by conventional PCR and Tecra™ kit. Of these, the developed RT-PCR was able to detect three and seven samples as entC1 and entC2/entC3 positive respectively based on melt curve analysis. Thus, results obtained highlighted the application of developed assay directly on naturally contaminated samples (Table 5).
Discussion
During the past decade, RT-PCR has been established as a sensitive detection method in the field of molecular diagnostics (Beuret 2004; Richards et al. 2004). SYBR Green chemistry combined with melt curve analysis has revolutionized the application of RT-PCR by reducing the cost since expensive probes are not needed and yet retain the sensitivity (Varga and James 2005; Hosmillo et al. 2010). The method is simple, rapid, reproducible, and easy to perform as it requires no post-PCR steps such as gel electrophoresis which often entail carcinogenic reagents. Contamination is also precluded in the method since protocol and result interpretation can be done in a closed reaction tube.
Staphylococcal enterotoxin C is known to have highly conserved homologous and immunologically cross-reactive subtype variants. Of these, SEC1, SEC2, and SEC3 toxins are known to possess a high degree of similarity and there is a pressing need for a highly specific detection tool that would sensitively identify and differentiate these subtypes. The high degree of homology in terms of nucleotide as well as amino acid sequences stands as a major drawback in this case. Though some of the immunological assay-based methods are available and are potent of differentiating subtypes based on specific epitopes, the presence of the toxin at detectable concentration in the analyte is a pre-requisite. Thus, the possibility of false-negative results in these cases (absence of toxin but presence of enterotoxigenic strain) may not be neglected. As an alternative strategy, we attempted to aptly utilize the technique of SYBR Green-based RT-PCR followed by melt curve analysis to specifically and differentially detect the formerly mentioned subtypes.
The major objective of the study was to determine regions of variation/heterogeneity among entC1, entC2, and entC3 and employ the promising variation/heterogeneity in differentiation of the three toxin subtypes using SYBR Green-based RT-PCR combined with melt curve analysis. This was challenging, due to the high degree of homology exhibited by these variants. With the aid of multiple sequence alignment, highly conserved and gene-specific SNPs among the three subtypes were identified. Based on these results, the work was designed to develop a single novel primer pair that amplifies the gene stretch of all three variants comprising of SNPs followed by melt curve analysis. Since shape and peak location of the melt curve varies with GC/AT ratio, length, and sequence of the fragment, melt peaks will be generated at different temperatures, thus allowing differentiation of closely related amplicons based on melt curve (Ririe et al. 1997; Wehrle et al. 2009). The same strategy was followed to achieve subtype differentiation.
Taking into consideration that short amplicons yield the most consistent results in RT-PCR technique, a 125-bp region with considerable variation/heterogeneity was selected for primer designing. Thus, designed primer pair was found to encompass one or two SNPs near to 5′ end in both forward and reverse primer sequences. Though studies have reported the detrimental effects of primer mismatches in terms of reduced sensitivity and efficiency of PCR, the effect is more when the mismatch is towards 3′ end (Guy et al. 2004; Bru et al. 2008). However, in our study, mismatch was away from 3′ end (6th position in forward primer and 9th position in reverse primer). Hence, when amplification was carried out, the oligos were able to hybridize and amplify all three subtypes regardless of SNPs as confirmed further by sequencing analysis. But, a difference of two logs was observed in C T value (loss of sensitivity) even when equal concentration of template DNA was used for amplification (Fig. S2). Optimization of RT-PCR assay was successful using the designed primer pair. Post-RT-PCR melt curve analysis revealed that, based on the melt peaks, entC1 was successfully differentiable from entC2/entC3. However, with dispersed and limited number of SNPs within the selected 125-bp gene stretch in the case of entC2 and entC3, both genes resulted in identical melt peak. Hence, the developed RT-PCR coupled with melt curve analysis failed to distinguish between entC2 and entC3. Further, since priority was given to design a single primer pair containing maximum number of SNPs, parameters such as primer dimer formation was not taken into consideration. As a result, a small non-specific peak was observed in melt curve analysis. However, since it melts at a low temperature than that of actual amplicon, it was clearly distinguished from the intended product and thus could be ignored.
According to the best of our knowledge, this is the first report of unambiguously discriminating entC1 from entC2/entC3 using a single primer pair. However, due to the high degree of homology, the developed assay was inadequate to discriminate between entC2 and entC3 genes. Perhaps, sensitive methods such as high-resolution melt (HRM) curve analysis can be exploited coupled with the developed assay to utilize the limited number of SNPs encountered between entC2 and entC3 and thus achieve reliable differentiation. High detection sensitivity was achieved which enable amplification even in presence of low number of DNA copies/bacterial cell load. Thus, in addition to rapid and sensitive detection of sec in any sample, the assay developed facilitates molecular discrimination of its subtypes in epidemiological surveillance of type C staphylococcal enterotoxin relevant to food poisoning cases. Moreover, the differentiation ability on naturally contaminated samples emphasized its real-time application on naturally contaminated food samples. With the benefit of short amplicon size, the assay protocol and result interpretation requires minimal time. To conclude, the SYBR Green-based real-time PCR assay described in this study can be implemented as an excellent tool for rapid, sensitive detection and differentiation of entC1 from entC2/entC3 with a single primer pair using inexpensive RT-PCR approach. The assay needs to be evaluated for its detection potential from different sample analytes so as to understand the possibility of employing the method in the diagnosis of SEC-mediated food poisoning cases.
References
Ataee RA, Mohammad HA, Gholam HA, Davoud E (2014) Staphylococcal enterotoxin C in synovial fluid of patients with rheumatoid arthritis. Iran Red Crescent Med J 16(10):e16075
Balaban N, Rasooly A (2000) Staphylococcal enterotoxins. Int J Food Microbiol 61(1):1–10
Betley MJ, Borst DW, Regassa LB (1992) Staphylococcal enterotoxins, toxic shock syndrome toxin and streptococcal pyrogenic exotoxins: a comparative study of their molecular biology (part 1 of 2). Chem Immunol 55:1–18
Beuret C (2004) Simultaneous detection of enteric viruses by multiplex real-time RT-PCR. J Virol Methods 115(1):1–8
Bohach GA, Dinges MM, Mitchell DT, Ohlendorf DH, Schlievert PM (1997) Exotoxins. The staphylococci in human disease. Churchill Livingstone, New York, pp. 83–111
Bohach GA, Fast DJ, Nelson RD, Schlievert PM (1990) Staphylococcal and streptococcal pyrogenic toxins involved in toxic shock syndrome and related illnesses. Crit Rev Microbiol 17(4):251–272
Bohach GA, Schlievert PM (1989) Conservation of the biologically active portions of staphylococcal enterotoxins C1 and C2. Infect Immun 57(7):2249–2252
Bru D, Martin-Laurent F, Philippot L (2008) Quantification of the detrimental effect of a single primer-template mismatch by real-time PCR using the 16S rRNA gene as an example. Appl Environ Microbiol 74(5):1660–1663
Chen TZ (2001) The exploitation of HAS and its application in tumor therapy. Prog Microbiol Immun China 29:63–69
Chen TR, Hsiao MH, Chiou CS, Tsen HY (2001) Development and use of PCR primers for the investigation of C1, C2 and C3 enterotoxin types of Staphylococcus aureus strains isolated from food-borne outbreaks. Int J Food Microbiol 71(1):63–70
Chu C, Yu C, Lee Y, Su Y (2012) Genetically divergent methicillin-resistant Staphylococcus aureus and sec-dependent mastitis of dairy goats in Taiwan. BMC Vet Res 8(1):1
Da Silva ER, do Carmo LS, Da Silva N (2005) Detection of the enterotoxins A, B, and C genes in Staphylococcus aureus from goat and bovine mastitis in Brazilian dairy herds. Vet Microbiol 106(1):103–107
Deringer JR, Ely RJ, Stauffacher CV, Bohach GA (1996) Subtype-specific interactions of type C staphylococcal enterotoxins with the T-cell receptor. Mol Microbiol 22(3):523–534
Dong S, Luo G, Feng J, Li Q, Gao H (2001) Immunoassay of staphylococcal enterotoxin C1 by FTIR spectroscopy and electrochemical gold electrode. Electroanalysis 13(1):30–33
Gao Z, Chao F, Chao Z, Li G (2000) Detection of staphylococcal enterotoxin C2 employing a piezoelectric crystal immunosensor. Sensors Actuators B Chem 66(1):193–196
Guy RA, Xiao C, Horgen PA (2004) Real-time PCR assay for detection and genotype differentiation of Giardia lamblia in stool specimens. J Clin Microbiol 42(7):3317–3320
Hsiao MH, Chen TR, Tsen HY (2003) Novel PCR primers for specific detection of C1, C2 and C3 enterotoxin genes in Staphylococcus aureus. J Food Drug Anal 11:239–245
Hosmillo MD, Jeong YJ, Kim HJ, Collantes TM, Alfajaro MM, Park JG, Kim HH, Kwon HJ, Park SJ, Kang MI, Park SI (2010) Development of universal SYBR Green real-time RT-PCR for the rapid detection and quantitation of bovine and porcine toroviruses. J Virol Methods 168(1):212–217
Hovde CJ, Hackett SP, Bohach GA (1990) Nucleotide sequence of the staphylococcal enterotoxin C3 gene sequence comparison of all three type C staphylococcal enterotoxins. Mol Gen Genomics 220:329–333
Huang Y, Chen X, Duan N, Wu S, Wang Z, Wei X, Wang Y (2015) Selection and characterization of DNA aptamers against Staphylococcus aureus enterotoxin C1. Food Chem 166:623–629
Hun X, Zhujun Z (2007) A novel sensitive staphylococcal enterotoxin C1 fluoroimmunoassay based on functionalized fluorescent core-shell nanoparticle labels. Food Chem 105:1623–1629
Marr JC, Lyon JD, Roberson JR, Lupher M, Davis WC, Bohach GA (1993) Characterization of novel type C staphylococcal enterotoxins: biological and evolutionary implications. Infect Immun 61(10):4254–4262
Marrack P, Kappler J (1990) The staphylococcal enterotoxins and their relatives. Science 248(4956):705–711
Ming-Kai X, Cheng-Gang Z (2006) Gene expression and function study of fusion immunotoxin anti-Her-2-scFv—SEC2 in Escherichia coli. Appl Microbiol Biotechnol 70(1):78–84
Mizukane R, Nakatomi M, Futsuki Y, Araki J, Asai S, Sawatari K, Hirakata Y, Maesaki S, Tomono K, Kohno S (1998) A study of virulence factors produced by MRSA strains isolated from blood samples. Kansesshogaku Zasshi Jansenshogaku Zasshi 72(8):788–793
Nagaraj S, Ramlal S, Sripathy MH, Batra HV (2014) Development and evaluation of a novel combinatorial selective enrichment and multiplex PCR technique for molecular detection of major virulence-associated genes of enterotoxigenic Staphylococcus aureus in food samples. J Appl Microbiol 116(2):435–446
Normanno G, Firinu A, Virgilio S, Mula G, Dambrosio A, Poggiu A, Decastelli L, Mioni R, Scuota S, Bolzoni G, Di Giannatale E (2005) Coagulase-positive staphylococci and Staphylococcus aureus in food products marketed in Italy. Int J Food Microbiol 98(1):73–79
Reiser RF, Robbins RN, Noleto AL, Khoe GP, Bergdoll MS (1984) Identification, purification, and some physicochemical properties of staphylococcal enterotoxin C3. Infect Immun 45(3):625–630
Richards GP, Watson MA, Kingsley DH (2004) A SYBR Green, real-time RT-PCR method to detect and quantitate Norwalk virus in stools. J Virol Methods 116(1):63–70
Ririe KM, Rasmussen RP, Wittwer CT (1997) Product differentiation by analysis of DNA melting curves during the polymerase chain reaction. Anal Biochem 245(2):154–160
Sambrook J, Russell DW (1989) Molecular cloning: a laboratory manual, vol 3. Cold Spring Harbor Laboratory Press, Cold Spring Harbor
Schmitz FJ, Steiert M, Hofmann B, Verhoef J, Hadding U, Heinz HP, Köhrer K (1998) Development of a multiplex-PCR for direct detection of the genes for enterotoxin B and C, and toxic shock syndrome toxin-1 in Staphylococcus aureus isolates. J Med Microbiol 47(4):335–340
Theron J, Cilliers J, Du Preez M, Brözel VS, Venter SN (2000) Detection of toxigenic Vibrio cholerae from environmental water samples by an enrichment broth cultivation–pit-stop semi-nested PCR procedure. J Appl Microbiol 89(3):539–546
Tibana A, Rayman K, Akhtar M, Szabo R (1987) Thermal stability of staphylococcal enterotoxins A, B and C in a buffered system. J Food Prot 50(3):239–242
Varga A, James D (2005) Detection and differentiation of plum pox virus using real-time multiplex PCR with SYBR Green and melting curve analysis: a rapid method for strain typing. J Virol Methods 123(2):213–220
Wang JY, Huang TP, Chang YC, Shih DYC (2003) Subtyping of enterotoxin C strains isolated from food poisoning outbreaks in Taiwan. J Food Drug Anal 11(3):239–245
Wang X, Xu M, Zhang H, Liu J, Li X, Zhang C (2009) Enhancement of superantigen activity and antitumor response of staphylococcal enterotoxin C2 by site-directed mutagenesis. Cancer Immunol Immunother 58 (5):677–686
Wehrle E, Didier A, Moravek M, Dietrich R, Martlbauer E (2009) Detection of Bacillus cereus with enteropathogenic potential by multiplex real-time PCR based on SYBR Green I. Mol Cell Probes 24(3):124–130
Zhou J, Liu L, Xu M, Zhang H, Zhang Y, Zhang C (2013) T-cell proliferation and antitumour activities of a truncated mutant of staphylococcal enterotoxin C2 with decreased cytokine secretion. J Med Microbiol 62(3):451–456
Zorgani A, Essery SD, Madani O, Bentley AJ, James VS, MacKenzie DA, Keeling JW, Rambaud C, Hilton J, Blackwell CC, Weir DM (1999) Detection of pyrogenic toxins of Staphylococcus aureus in sudden infant death syndrome. FEMS Immunol Med Microbiol 25(1–2):103–108
Acknowledgments
The authors are grateful to DRDO for providing the necessary facilities to carry out this work.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethics statement
This article does not contain any studies with human participants or animals performed by any of the authors.
Electronic supplementary material
ESM 1
(PDF 274 kb)
Rights and permissions
About this article

Cite this article
Nagaraj, S., Ramlal, S., Venkataswamachari, B.P. et al. Differentiation of entC1 from entC2/entC3 with a single primer pair using simple and rapid SYBR Green-based RT-PCR melt curve analysis. Appl Microbiol Biotechnol 100, 8495–8506 (2016). https://doi.org/10.1007/s00253-016-7637-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-016-7637-y