
Abstract
The persistent problem of antibiotic resistance has created a strong demand for new methods for therapy and disinfection. Photodynamic inactivation (PDI) of microbes has demonstrated promising results for eradication of antibiotic-resistant strains. PDI is based on the use of a photosensitive compound (photosensitizer, PS), which upon illumination with visible light generates reactive species capable of damaging and killing microorganisms. Since photogenerated reactive species are short lived, damage is limited to close proximity of the PS. It is reasonable to expect that the larger the number of damaged targets is and the greater their variety is, the higher the efficiency of PDI is and the lower the chances for development of resistance are. Exact molecular mechanisms and specific targets whose damage is essential for microbial inactivation have not been unequivocally established. Two main cellular components, DNA and plasma membrane, are regarded as the most important PDI targets. Using Zn porphyrin-based PSs and Escherichia coli as a model Gram-negative microorganism, we demonstrate that efficient photoinactivation of bacteria can be achieved without detectable DNA modification. Among the cellular components which are modified early during illumination and constitute key PDI targets are cytosolic enzymes, membrane-bound protein complexes, and the plasma membrane. As a result, membrane barrier function is lost, and energy and reducing equivalent production is disrupted, which in turn compromises cell defense mechanisms, thus augmenting the photoinduced oxidative injury. In conclusion, high PDI antimicrobial effectiveness does not necessarily require impairment of a specific critical cellular component and can be achieved by inducing damage to multiple cellular targets.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Antimicrobial resistance poses a danger for the treatment of common infections that were easily cured with available antibiotics (WHO 2014). Among the reasons for increasing resistance is the widespread and often unnecessary use of antibiotic drugs (Leung et al. 2011; Levy and Marshall 2004). One way to prevent resistance is to limit bacterial exposure to conventional antibiotics by developing other means of bacterial eradication. Photodynamic inactivation (PDI) has proven efficient against various classes of microorganisms without triggering resistance (for details, see Alves et al. 2014b; Jori et al. 2006; Maisch 2015; Sharma et al. 2012; Wainwright 2014; Yin and Hamblin 2015). PDI is based on generation of reactive species, mainly singlet oxygen, by a non-toxic photosensitizer (PS) and visible light (Benov 2015; Vatansever et al. 2013). The short life of the photogenerated reactive species and consequently the small distance they can travel in a biological environment (Ogilby 2010) imply that localization of the PS is a key factor for its photodynamic efficiency. Even though PSs have been successfully used for treatment of infectious diseases (Denis et al. 2011; Hamblin and Hasan 2004), for disinfection of water and surfaces (Almeida et al. 2014; Jori et al. 2011), for treatment of hospital waste, and for other practical purposes (Ballatore et al. 2015; Craig et al. 2015; Jori and Brown 2004), PDI has not seen widespread use. Among the reasons for this is a lack of PSs specifically designed for particular applications. Such design requires thorough understanding of molecular properties of a PS that determine antimicrobial photoefficiency, specificity, selectivity, and microbial targets of PS action. A good strategy to avoid development of resistance is to design PSs that hit multiple targets (Alves et al. 2014a). Molecular mechanisms leading to PDI-induced bacterial death have not been fully elucidated; it has been proposed that oxidative damage of DNA and/or plasma membrane components are key targets responsible for PDI inactivation of bacteria.
The aim of this study was to identify microbial components that are potential targets for microbial photoinactivation.
Previous research has shown that porphyrin-based PSs kill Gram-positive and Gram-negative bacteria (Jori et al. 2006) and are efficient against antibiotic-resistant strains (Benov et al. 2002; Thomas et al. 2015). Due to the specific structure of the outer membrane, Gram-negative microorganisms are much more resistant to PDI than Gram-positive microbes. Using Escherichia coli as a model for a Gram-negative organism and cationic Zn porphyrins that differ by lipophilicity, we demonstrate that rapid photoinduced loss of viability upon illumination using fluorescent light tubes with emission properties similar to those used for domestic and workplace lighting is associated with loss of plasma membrane barrier function, inactivation of respiratory complexes and metabolic enzymes, but not with gross DNA damage.
Methods
Photosensitizers
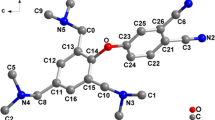
Synthesis and characterization of Zn(II) meso-tetrakis(N-alkylpyridinium-2-yl)porphyrins was performed as previously described (Ezzeddine et al. 2013). Structures of ZnPs used in this study are shown in Fig. 1.

Structures of ZnPs explored in this study
Bacterial culture media
E. coli strain GC4468 (genotype F-Δlac U169 rpsL, CGSC strain no. 7100) was used as a model system for a Gram-negative organism. Overnight cultures were grown in liquid Luria-Bertani (LB) medium. When agar plates were needed, 15 g of agar was added to 1 l of liquid LB medium.
Experimental cultures were grown in M9CA medium. The medium consisted of M9 salts, prepared by dissolving 6 g Na2HPO4, 3 g K2HPO4, 1 g NH4Cl, and 0.5 g NaCl in 1 l of distilled water. To 100 ml of M9 salts, 1 ml of each 0.2 M MgSO4, 20 % glucose and 20 % casamino acids, and 50 μl of 0.2 M CaCl2 autoclaved separately were added. Immediately before use, filter-sterilized solutions of pantothenic acid and thiamine were added to a final concentration of 3 mg/l.
Growth conditions
The cells in LB medium were grown overnight in a shaking water bath at 200 rpm and 37 °C. For experiments, the overnight cultures were diluted 200-fold in M9CA medium and grown to A 600 nm~0.5–1.0.
For investigation of the effect of ZnPs on cell proliferation, 96-well plates were used (Tovmasyan et al. 2014). Portions (100 μl/well) of diluted M9CA culture were transferred into quadruplicate wells, and ZnPs were added to give the desired final concentrations. The plates were illuminated on a shaker at 37 °C and 200 rpm at fluence of 0.7 mW/cm2. Growth was monitored by measuring light scattering (turbidity) at 600 nm.
Illumination
Cell cultures were illuminated by two 5-W white fluorescent light tubes mounted under a translucent screen (2.7 mW/cm2 at the level of the screen). Distance from the light source was selected such a way that a desired fluence was obtained. Light intensity was measured with a Laser Power Meter Orion/PD (Ophir-Spiricon, LLC, USA). No change of the temperature of the samples was detected during the illumination.
Determination of cellular viability by plating
The photodynamic killing efficiency of ZnPs was evaluated by colony formation. After illumination, the samples were diluted in sterile M9 salts and 50 μl portions of the resulting cell suspension were evenly spread on LB agar plates. The plates were incubated at 37 °C for 24 h in the dark and colonies were counted.
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide assay
Solutions of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT) and 10 % SDS were prepared as previously described (Thomas et al. 2015). Cell suspensions in M9CA medium (100-μl aliquots) were transferred into 96-well plates. ZnPs were added to 5.0 μM and plates were illuminated (19.5 J/cm2). Light and dark controls were run in parallel. At the end of the treatment, 10 μl of MTT reagent was added per well and plates were incubated for 30 min in the dark, followed by addition of 100 μl of SDS reagent. After 1-h incubation at room temperature, the absorbance was measured at 570 and 700 nm using a microplate reader (Tecan Sunrise, Tecan Instruments, Grodig, Austria).
Enzyme assays
Enzyme assays were performed essentially as previously described (Al-Mutairi et al. 2007a) using cell-free extracts. After illumination, the cells were centrifuged at 3000×g for 10 min at 4 °C and washed with 50 mM potassium phosphate buffer (pH 7.3). Cells were disrupted by sonication and debris was separated by centrifugation at 14,000×g for 10 min. The resulting supernatant was used for enzyme assays. Protein was determined by the Lowry assay (Lowry et al. 1951).
Glyceraldehyde-3-phosphate dehydrogenase
The protocol of Dagher (Dagher and Deal 1982) was followed to determine the activity of glyceraldehyde-3-phosphate dehydrogenase (GAPDH). The assay mixture contained 910 μl of 50 mM Tris buffer (pH 8.8), 25 μl of 10 mM NAD+, and 35 μl of cell-free extract. The reaction was started by the addition of 30 μl of (2:1) ratio of 20 mM GA3P and 10 mM sodium arsenate, and NADH formation was measured at 340 nm for 20 s. One unit was defined as the activity of GA3PD that is required to reduce 1 μmol of NAD+ to NADH per min at pH 8.8 at 25 °C.
Glucose-6-phosphate dehydrogenase
The activity of glucose-6-phosphate dehydrogenase (G6PD) was measured by following the increase in the absorbance at 340 nm due to the formation of NADPH as described by Lee (Lee 1982). One milliliter of a reaction mixture contained 0.1 M Tris-HCl buffer (pH 8.0), 10 mM glucose-6-phosphate, 10 mM NADP+, and 20–50 μl of cell-free extract. One unit of G6PD is defined as the activity of G6PD that reduces 1 μmol of NADP+ to NADPH per min at pH 8.0 at 25 °C.
Isocitrate dehydrogenase
Isocitrate dehydrogenase (ICD) activity was assayed as described by Fatania et al. (Fatania et al. 1993) by monitoring the production of NADPH at 340 nm. One-milliliter reaction mixture contained 33 mM Tris-EDTA buffer (pH 7.4), 1.33 mM MnCl2, 1.3 mM DL-isocitrate, 0.1 mM NADP+, and 20–50 μl of cell-free extract. One unit of the enzyme activity is defined as production of 1.0 μmol of NADPH per minute.
Cellular respiration
Ability of PSs to damage plasma membrane protein complexes was determined by measuring respiration after phototreatment. Experiments were performed essentially as previously described (Thomas et al. 2015). One-milliliter aliquots of cell suspension were transferred in each well of 8-well plates and ZnPs were added. Light and dark controls were run in parallel. The plates were illuminated for 15 min; cell suspensions were assayed by placement into a thermostatic chamber fitted with a Clark electrode. Oxygen consumption was recorded and respiration rates were calculated as nanomole O2 consumed per minute per milliliter cell suspension.
Assessment of ATP leakage
ATP leaking out of the cells was estimated by Sigma-Aldrich ATP Bioluminescent Assay Kit. Aliquots of M9CA E. coli culture were transferred into 96-well plates (200 μl/well), ZnPs were added, and plates were illuminated. Light and dark controls were run in parallel. Immediately after the illumination, the samples were centrifuged at 12,000×g for 3 min. Then, 25 μl of the supernatant was transferred to 25 μl of 2 % TCA. These steps were repeated at each time interval. Immediately before analysis, 20 μl of the TCA mixture was added to 180 μl of Tris buffer (100 μM, pH 8) for neutralization of the acidic medium. Aliquots were analyzed for ATP content.
DNA damage
M9CA E. coli cultures were transferred into 6-well plates (1 ml/well), 5 μM ZnPs were added, and plates were illuminated for 1 h (19.5 J/cm2). Light and dark controls were run in parallel. Genomic DNA was purified using a Wizard Genomic DNA Purification Kit (Promega) following protocols provided by the manufacturer.
Aliquots (1 ml) of cell suspension were centrifuged for 2 min at 12,000×g; 600 μl of lysis solution was added to the cells, and samples were incubated at 80 °C water bath for 5 min. RNase (3 μl) was added to degrade RNA, and the tubes were incubated for 15 min at 37 °C in a water bath; 200 μl of protein precipitation solution was then added and mixed well using a vortex mixer. The samples were kept for 5 min on ice. After centrifugation for 3 min at 12,000×g, the supernatant was transferred to a new tube containing 600 μl isopropanol. DNA was harvested by centrifugation and pellet washed with 70 % ethanol. Ethanol was aspirated and the pellet was air-dried. Rehydration solution was added at the end of the procedure. DNA was analyzed by electrophoresis on 1 % agarose gel and stained with ethidium bromide. A DNA ladder of 1 kb was used to calibrate the separated DNA.
A positive control demonstrating the sensitivity of the method to detect DNA fragments was obtained by 5–15-min microwave irradiation as described by Yang and Hang (2013).
All experiments were repeated at least three times with three to five replicates. Statistical analysis was performed using Student’s t test. P < 0.05 was considered statistically significant. Results are presented as mean ± SEM.
Results
Effect of photoactivated Zn porphyrins on bacterial growth
Initial experiments demonstrated that when E. coli liquid cultures were illuminated in the presence of 5 μM PS, the two ZnPs inhibited the growth completely. Such complete inhibition of cell division did not allow comparison of the efficacy of ZnPs. Therefore, lower concentrations of PSs were tested. As shown in Fig. 2a, at 0.5 μM, ZnTnHex-2-PyP completely inhibited bacterial growth while ZnTM-2-PyP had no substantial effect. Since optical and photochemical properties of the two PSs are very similar, the difference in photoefficiency should be attributed to differences in bacterial uptake, primarily resulting from differences in their lipophilicity (Thomas et al. 2015).

Effect of photoexcited ZnPs on bacterial proliferation (a) and viability (b). a Bacterial suspensions (1.5 × 107 cells/ml) were illuminated (0.7 mW/cm2) in the presence of 0.5 μM ZnPs. Dark controls and illuminated controls without sensitizer were run in parallel. Growth was monitored by light scattering (turbidity) using OD at 600 nm. Each point represents mean of three different readings in a single experiment. Qualitatively similar growth curves were obtained in three separate experiments. b Bacterial suspensions (1.5 × 107 cells/ml) were illuminated (2.7 mW/cm2) for 2 h in the presence of 5.0 μM ZnPs. Dark controls and controls without sensitizer were run in parallel. Viable cells were estimated by plating and counting colonies. *p < 0.05 compared to control
None of the PSs affected the growth of E. coli in the dark, and no effect was observed if cultures were illuminated in the absence of a PS (not shown).
Bactericidal effect of ZnPs
In order to determine if photoinduced E. coli killing efficacy of the two ZnPs paralleled growth inhibition effects, colony forming ability was assessed. At cell numbers corresponding to those in the growth experiments (~2 × 107 cells/ml), 0.5 μM of ZnTnHex-2-PyP killed about 95 % of the cells while ZnTM-2-PyP did not demonstrate statistically significant effect (not shown).
Increasing cell number to ~2 × 109 cells/ml while keeping PS concentration the same reduced the effect of ZnHex-2-PyP to insignificant. As previously reported, cells compete for the PS (Demidova and Hamblin 2005), and when the number of PS molecules per cell drops below a certain threshold, no effect can be observed. Increasing PS concentration to 5 μM resulted in >99 % killing by ZnHex-2-PyP, but the effect of ZnTM-2-PyP remained insignificant (Fig. 2b).
To make comparison possible, in all further experiments, the ratio of PS to cell number was strictly controlled.
Effect of photoactivated ZnPs on overall metabolic activity
Among the possible reasons for suppression of bacterial division is repression of metabolism. Enzymatic reduction of tetrazolium dyes to formazan, commonly used as surrogate assay for viability of mammalian (Berridge and Tan 1993; Sylvester 2011) and microbial (Tsukatani et al. 2009) cells, depends on NAD(P)H (Berridge et al. 2005; Berridge and Tan 1993) and can therefore serve as an integrative measure of metabolic activity. Figure 3 shows that illumination of E. coli suspension in the presence of ZnTnHex-2-PyP caused ~60 % inhibition of MTT reduction to formazan, but no effect of ZnTM-2-PyP was observed.

Effect of photoexcited ZnPs on overall metabolic activity. E. coli was grown in M9CA medium to mid-log phase. One-hundred-microliter aliquots were transferred to 96-well plate, and PSs were added to a final concentration of 5.0 μM. Plates were illuminated (19.44 J/cm2) and metabolic activity was assessed by the MTT test. Dark and illuminated controls were run in parallel. *p < 0.05 compared to control
No inhibition of MTT reduction occurred when the cells were incubated with PSs in the dark. Illumination of cells in the absence of PSs also had no effect (not shown).
Effect of phototreatment on metabolic enzymes and cellular respiration
Suppression of formazan formation by the phototreatment suggests that ZnTnHex-2-PyP inactivates enzymes catalyzing MTT reduction and/or involved in NAD(P)H production. To test this possibility, photoinactivation of cytosolic enzymes and respiratory complexes embedded in the inner membrane were studied.
The ability of the PSs to affect pathways maintaining a high NAD(P)H/NAD(P)+ ratio was assessed by measuring the activities of glyceraldehyde-3-phosphate dehydrogenase (glycolysis), glucose-6-phosphate dehydrogenase (pentose phosphate pathway), and isocitrate dehydrogenase (Krebs cycle) enzymes.
In order to minimize inhibition of enzymes by secondary reactive products, and changes of activity due to gene induction/repression, illumination time was reduced to 20 min (3.24 J/cm2). Figure 4 shows that under these conditions, all three tested enzymes were photoinactivated by ZnTnHex-2-PyP while the hydrophilic PS ZnTM-2-PyP did not suppress the activity of any of the enzymes.

Effect of photoactivated ZnPs on glyceraldehyde-3-phosphate dehydrogenase (GAPD), glucose-6-phosphate dehydrogenase (G6PD), and isocitrate dehydrogenase (ICD). Cell suspensions containing 5 μM ZnPs were illuminated for 20 min (2.7 mW/cm2); cells were collected by centrifugation, washed, and disrupted by sonication. Cell-free extracts were used for enzyme activity assay. *p < 0.05 compared to control
To assess the ability of photoactivated ZnPs to damage respiratory complexes, E. coli suspensions were illuminated in the presence of PSs, and oxygen consumption was determined 15 min after cessation of light exposure. Figure 5 shows that when cells were illuminated in the presence of ZnTnHex-2-PyP, almost complete loss of respiration occurred. For E. coli suspensions illuminated in the presence of ZnTM-2-PyP, suppression of respiration did not exceed 20 % (Fig. 5). No effect was observed when cells were illuminated in the absence of PS.

Effect of photoactivated ZnPs on cellular respiration. One-milliliter aliquots of E. coli grown to mid-log phase were transferred to 8-well plates and 5 μM of ZnPs were added. Cells were illuminated (19.5 J/cm2) and then transferred into thermostatic chamber fitted with a Clark electrode. Oxygen consumption was recorded and calculated as nanomole per minute per milliliter of cell suspension. *p < 0.05 compared to control
ATP leakage
Damage of the plasma membrane leads to inactivation of membrane-bound proteins and loss of membrane barrier function. To test the ability of ZnPs to target E. coli plasma membrane, leakage of ATP out of the cells was monitored by measurement of appearance of ATP in the extracellular medium during the phototreatment. Figure 6 shows a rapid increase of ATP in the medium when E. coli suspension was illuminated in the presence of ZnTnHex-2-PyP. This indicates that membrane damage and loss of membrane barrier functions are among the first events triggered by the photoactivated amphiphilic PS. In contrast, no significant increase of ATP concentration in the medium within the first 30 min was observed when the cells were illuminated in the presence of ZnTM-2-PyP. Neither of the PSs caused ATP leakage when cells were incubated with PSs in the dark or when illuminated in the absence of a PS (not shown).

Effect of photoactivated ZnPs on ATP leakage from E. coli. Cell suspensions were illuminated in presence of 5 μM of ZnTM-2-PyP or ZnTnHex-2-PyP; the cells were separated by centrifugation, and the supernatant was assayed to estimate ATP released in the medium. *p < 0.05 compared to control
DNA damage by photoactivated ZnPs
There is an ongoing debate regarding whether DNA is a primary PDI target leading to loss of microbial viability (summarized in Alves et al. 2014a). Analyses of genomic DNA modifications by agarose gel electrophoresis usually show smears indicating strand breaks (Alves et al. 2013). As seen in Fig. 7, panel 1, when cells were illuminated in the presence of 5 μM ZnPs, none of the PSs produced smears or measurable DNA structural alterations leading to fragmentation and aggregation. Similarly to what has been previously reported for its metal-free para analog (Alves et al. 2013), a weaker genomic DNA band was observed when E. coli was illuminated in the presence of ZnTnHex-2-PyP, which can be interpreted as loss of DNA (Fig. 7, panel 1) and which may represent a consistent experimental artifact caused either by changes to the efficiency of DNA release on treatment of cells with lysis medium or by co-precipitation with photomodified proteins. Illumination of purified E. coli genomic DNA in the presence of 5 μM ortho, meta, or para methyl or ortho hexyl ZnPs also did not result in DNA cleavage (Fig. 7, panel 2). No detectable DNA modification was observed even when ZnP concentration was increased to 10 μM (not shown).

Effect of photoactivated ZnPs on genomic E. coli DNA. panel 1 Cells were illuminated (19.5 J/cm2) in the presence of 5 μM ZnPs, and DNA was extracted and analyzed by 1 % agarose gel electrophoresis. Gels were stained with ethidium bromide. a DNA ladder, b control, c ZnTM-2-PyP, and d ZnTnHex-2-PyP. panel 2 Genomic DNA was isolated from intact cells and then illuminated in the presence of 5 μM of photosensitizers. a Non-treated, b ZnTM-2-PyP, c ZnTM-3-PyP, d ZnTM-4-PyP, e ZnTnHex-2-PyP, and f DNA fragmentation by microwave irradiation for 5 min (positive control). Experiments were repeated three times with similar results
Discussion
Singlet oxygen (1O2) is considered the main cell-damaging species generated by photoexcited ZnPs (Benov et al. 2012). Its lifetime in water is so short that 1O2 will not diffuse more than 150 nm from the site where it is generated (Redmond and Kochevar 2006). That distance is much smaller than the size of E. coli (1 × 2 μm). In biological environment, migration is further shortened by presence of chemical and physical 1O2 quenchers. Therefore, cellular structures/molecules will be damaged only when the PS is close enough to allow an adequate flux of 1O2 to reach the target.
Oxidative modification of two essential cellular components has been proposed to be responsible for killing of bacteria by PDI, DNA, and the cytoplasmic membrane (Hamblin and Hasan 2004).
Bacterial DNA damage including breaks in both single- and double-stranded DNA has been detected in Gram-positive and Gram-negative bacteria illuminated in the presence of PSs (Bertoloni et al. 2000; Menezes et al. 1990), but in general, it may represent a secondary event observed when cells have already been rendered non-viable (for details, see Alves et al. 2014a). PDI-induced DNA modification can occur through action of three different pathways: (i) via direct one-electron oxidation, (ii) by radicals generated via type I reactions, and (iii) by singlet oxygen (Cadet et al. 2006, 2012; Epe 2012). The first mechanism involves electron abstraction from a DNA base by the excited PS, while the other two are mediated by reactive oxygen species generated either by type I (mainly hydroxyl radical) or type II (singlet oxygen) reactions (Benov 2015). Irrespective of the mechanism, localization of the PS on, or very close to, DNA is an essential prerequisite for DNA damage; therefore, PSs that intercalate into double-stranded DNA are more efficient in causing damage (Hass and Webb 1981).
In our experiments, none of the tested PS produced detectable fragmentation or aggregation of DNA. At least two reasons can be listed for this negative result.
First, a PS needs to build up sufficiently high intracellular concentration to produce fragmentation and aggregation directly from photogenerated species or should generate substantial amounts of peroxidation products that can cause secondary DNA damage (Cadet et al. 2010). Such damage has been observed at relatively long irradiation times, when survival was drastically decreased (Bertoloni et al. 2000; Dosselli et al. 2012; Jori et al. 2006). For our experiments, we intentionally selected mild PDI treatment protocols to avoid secondary damage and to identify the most sensitive primary targets.
Second, singlet oxygen is highly selective in reacting with DNA, targeting mainly guanine (rate constant for reaction with guanine is 5 × 106 M−1 s−1 (Sies and Menck 1992; Cadet et al. 2010; Epe 1991)). Singlet oxygen does not react with deoxyribose and cannot directly break the DNA phosphodiester backbone structure; therefore, no major structural alteration should be expected to result from short exposures to PSs that generate predominantly 1O2. A specific 1O2 DNA modification product, 8-oxo-7,8-dihydro-2′-deoxyguanosine, has been detected in mammalian cells illuminated in the presence of ZnTM-3-PyP (Al-Mutairi et al. 2007b).
Previous studies demonstrated that brief illumination (up to 5 min) in the presence of the para metal-free porphyrin ligand TMPyP (H2TM-4-PyP) killed practically the entire bacterial population without damaging genomic DNA (Nitzan and Ashkenazi 2001). A possible explanation is that the short time of illumination did not allow PS to be released from the photodamaged cellular structures and to relocate to DNA. Experiments with Streptococcus mitis, however, demonstrated that no cleavage of genomic DNA occurred even after 2 h of illumination (Spesia and Durantini 2013). In our experiments, no genomic DNA cleavage was observed even when ZnPs were directly added to isolated E. coli DNA and illuminated. This finding indicates that the nature of the PS molecule and its interaction with DNA, as well as its mechanism of photophysical action and the reactive species it generates, determine if substantial DNA damage can occur. It also supports the view that DNA damage is not needed for efficient PDI-mediated photoeradication of bacteria.
Cell membranes are another potential PDI target whose photoinduced alterations could cause cell death. Our results show that leakage of metabolites out of the cells is an early event when cells were illuminated in the presence of the amphiphilic ZnTnHex-2-PyP. No such an effect was observed during phototreatment with the hydrophilic ZnTM-2-PyP, which accumulates to about 10-fold lower level in E. coli than ZnTnHex-2-PyP (Thomas et al. 2015). Due to its higher lipophilicity (Thomas et al. 2015), the hexyl derivative would accumulate to much higher levels in membranes than the hydrophilic ZnTM-2-PyP (Kos et al. 2009), which explains its high efficiency in causing membrane damage upon illumination.
In addition to impairment of membrane barrier functions that can be attributed to disruption of the lipid bilayer, membrane-bound proteins were also oxidatively damaged. A consequence is inactivation of respiratory complexes, which in turn leads to inability to aerobically generate ATP. Combined with leakage of metabolites out of the cells, and inactivation of cytosolic enzymes, this leads to a drastic depletion of ATP. Our data show that cytosolic enzymes are among the primary PDI targets. Their photoinactivation, which occurs at the early stages of illumination, deprives bacteria of energy and reducing equivalents (NADH, NADPH), which in turn compromises cellular defenses against oxidants. Since proteins are abundant, and some amino acid residues react with high rate constants with 1O2 (Benov 2015; Benov et al. 2012), structural and catalytic proteins should be considered as key PDI targets, whose oxidative damage is among the main reasons for PDI-induced microbial inactivation.
A comparison of the two structural analogs used in this study indicates that in order to be photoefficient at low concentrations, a PS must penetrate bacterial cells. Using a metal-free para analog of ZnTM-2-PyP, 5,10,15,20-tetrakis(1-methylpyridinium-4-yl)porphyrin (TMPyP), Preuß and coworkers determined that E. coli can be photoinactivated without intracellular uptake of the PS (Preuß et al. 2012). A reason is probably the ability of 1O2 to cross E. coli cell wall (Ragas et al. 2013) and penetration of the PS inside the cell as a result of photoinduced membrane disruption. Singlet oxygen generated outside the cell, however, has much greater chances to be quenched by organic molecules or water in the environment, which decreases the efficiency of the treatment. This can eventually be compensated by increasing the concentration of the PS, which initially bound to the outer surface of the cell, can subsequently enter as a result of cellular photodamage. Amphiphilic cationic PSs, in contrast to their hydrophilic analogs, disperse in the plasma membrane and penetrate into the cell. As a consequence, 1O2 is generated in close proximity to critical targets (membrane lipid and protein complexes, cytosolic enzymes, etc.) whose damage compromises cell integrity and metabolism, ultimately causing cell death.
In conclusion, this study shows that a PS which induces loss of plasma membrane integrity and disruption of cell metabolism can act as a highly efficient bactericidal agent when illuminated by conventional and widely available light sources (fluorescent tube lights), which makes it suitable for disinfection of surfaces and waste treatment. Such a PS would be also suitable for treatment of localized infections. Application of low concentrations of a PS that is rapidly taken up by microbes, followed shortly by illumination, is a promising strategy for preventing host tissue damage and increasing the selectivity of the photodynamic treatment (Sharma et al. 2011). The action of such PSs is not limited to a single target as is in the case of most antibiotics (Hogan and Kolter 2002; Liu and Imlay 2013), which makes cell repair and survival unlikely, and the probability for development of resistance, extremely low.
References
Almeida J, Tome JPC, Neves MGPMS, Tome AC, Cavaleiro JAS, Cunha A, Costa L, Faustino MAF, Almeida A (2014) Photodynamic inactivation of multidrug-resistant bacteria in hospital wastewaters: influence of residual antibiotics. Photochem Photobiol Sci 13(4):626–633. doi:10.1039/C3pp50195g
Al-Mutairi DA, Craik JD, Batinic-Haberle I, Benov LT (2007a) Inactivation of metabolic enzymes by photo-treatment with zinc meta N-methylpyridylporphyrin. Biochim Biophys Acta 1770(11):1520–1527
Al-Mutairi DA, Craik JD, Batinic-Haberle I, Benov LT (2007b) Induction of oxidative cell damage by photo-treatment with zinc N-methylpyridylporphyrin. Free Radic Res 41(1):89–96
Alves E, Faustino MAF, Tomé JPC, Neves MGPMS, Tomé AC, Cavaleiro JAS, Cunha A, Gomes NCM, Almeida A (2013) Nucleic acid changes during photodynamic inactivation of bacteria by cationic porphyrins. Bioorg Med Chem 21(14):4311–4318. doi:10.1016/j.bmc.2013.04.065
Alves E, Faustino MAF, Neves MGPMS, Cunha A, Tome J, Almeida A (2014a) An insight on bacterial cellular targets of photodynamic inactivation. Future Med Chem 6(2):141–164. doi:10.4155/fmc.13.211
Alves E, Faustino MAF, Neves MGPMS, Cunha T, Nadais H, Almeida A (2014b) Potential applications of porphyrins in photodynamic inactivation beyond the medical scope. J Photochem Photobiol C: Photochem Rev 22:34–57. doi:10.1016/j.jphotochemrev.2014.09.003
Ballatore MB, Durantini J, Gsponer NS, Suarez MB, Gervaldo M, Otero L, Spesia MB, Milanesio ME, Durantini EN (2015) Photodynamic inactivation of bacteria using novel electrogenerated porphyrin-fullerene C60 polymeric films. Environ Sci Technol 49(12):7456–7463. doi:10.1021/acs.est.5b01407
Benov L (2015) Photodynamic therapy: current status and future directions. Med Princ Pract 24:14–28
Benov L, Batinic-Haberle I, Spasojevic I, Fridovich I (2002) Isomeric N-alkylpyridylporphyrins and their Zn(II) complexes: inactive as SOD mimics but powerful photosensitizers. Arch Biochem Biophys 402(2):159–165
Benov L, Craik J, Batinic-Haberle I (2012) Protein damage by photo-activated Zn(II) N-alkylpyridylporphyrins. Amino Acids 42(1):117–128
Berridge MV, Tan AS (1993) Characterization of the cellular reduction of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT): subcellular localization, substrate dependence, and involvement of mitochondrial electron transport in MTT reduction. Arch Biochem Biophys 303(2):474–482
Berridge MV, Herst PM, Tan AS (2005) Tetrazolium dyes as tools in cell biology: new insights into their cellular reduction. Biotechnol Annu Rev 11:127–152
Bertoloni G, Lauro FM, Cortella G, Merchat M (2000) Photosensitizing activity of hematoporphyrin on Staphylococcus aureus cells. Biochim Biophys Acta Gen Subj 1475(2):169–174. doi:10.1016/S0304-4165(00)00071-4
Cadet J, Douki T, Ravanat JL (2006) One-electron oxidation of DNA and inflammation processes. Nat Chem Biol 2(7):348–349
Cadet J, Douki T, Ravanat JL (2010) Oxidatively generated base damage to cellular DNA. Free Radic Biol Med 49(1):9–21
Cadet J, Ravanat JL, TavernaPorro M, Menoni H, Angelov D (2012) Oxidatively generated complex DNA damage: tandem and clustered lesions. Cancer Lett 327(1–2):5–15. doi:10.1016/j.canlet.2012.04.005
Craig RA, McCoy CP, Gorman SP, Jones DS (2015) Photosensitisers-the progression from photodynamic therapy to anti-infective surfaces. Expert Opin Drug Deliv 12(1):85–101. doi:10.1517/17425247.2015.962512
Dagher SM, Deal WC Jr (1982) [54] Glyceraldehyde-3-phosphate dehydrogenase from pig liver. Methods Enzymol 89(p):310–316
Demidova TN, Hamblin MR (2005) Effect of cell-photo sensitizer binding and cell density on microbial photoinactivation. Antimicrob Agents Chemother 49(6):2329–2335. doi:10.1128/Aac.49.6.2329-2335.2005
Denis TGS, Dai TH, Izikson L, Astrakas C, Anderson RR, Hamblin MR, Tegos GP (2011) All you need is light antimicrobial photoinactivation as an evolving and emerging discovery strategy against infectious disease. Virulence 2(6):509–520. doi:10.4161/viru.2.6.17889
Dosselli R, Millioni R, Puricelli L, Tessari P, Arrigoni G, Franchin C, Segalla A, Teardo E, Reddi E (2012) Molecular targets of antimicrobial photodynamic therapy identified by a proteomic approach. J Proteome 77:329–343. doi:10.1016/j.jprot.2012.09.007
Epe B (1991) Genotoxicity of singlet oxygen. Chem Biol Interact 80(3):239–260
Epe B (2012) DNA damage spectra induced by photosensitization. Photochem Photobiol Sci 11(1):98–106. doi:10.1039/C1pp05190c
Ezzeddine R, Al-Banaw A, Tovmasyan A, Craik JD, Batinic-Haberle I, Benov LT (2013) Effect of molecular characteristics on cellular uptake, subcellular localization, and phototoxicity of Zn(II) N-alkylpyridylporphyrins. J Biol Chem 288(51):36579–36588. doi:10.1074/jbc.M113.511642
Fatania H, Al-Nassar KE, Sidhan V (1993) Purification and partial characterisation of NADP+-linked isocitrate dehydrogenase from rat liver cytosol. FEBS Lett 320(1):57–60. doi:10.1016/0014-5793(93)81657-L
Hamblin MR, Hasan T (2004) Photodynamic therapy: a new antimicrobial approach to infectious disease? Photochem Photobiol Sci 3(5):436–450
Hass BS, Webb RB (1981) Photodynamic effects of dyes on bacteria. IV. Lethal effects of acridine orange and 460- or 500-nm monochromatic light in strain of Escherichia coli that differ in repair capability. Mutat Res Fundam Mol Mech Mutagen 81(3):277–285. doi:10.1016/0027-5107(81)90116-0
Hogan D, Kolter R (2002) Why are bacteria refractory to antimicrobials? Curr Opin Microbiol 5(5):472–477. doi:10.1016/S1369-5274(02)00357-0
Jori G, Brown SB (2004) Photosensitized inactivation of microorganisms. Photochem Photobiol Sci 3(5):403–405. doi:10.1039/b311904c
Jori G, Fabris C, Soncin M, Ferro S, Coppellotti O, Dei D, Fantetti L, Chiti G, Roncucci G (2006) Photodynamic therapy in the treatment of microbial infections: basic principles and perspective applications. Lasers Surg Med 38(5):468–481
Jori G, Magaraggia M, Fabris C, Soncin M, Camerin M, Tallandini L, Coppellotti O, Guidolin L (2011) Photodynamic inactivation of microbial pathogens: disinfection of water and prevention of water-borne diseases. J Environ Pathol Toxicol 30(3):261–271
Kos I, Benov L, Spasojević I, Rebouças JS, Batinić-Haberle I (2009) High lipophilicity of meta Mn(III) N-alkylpyridylporphyrin-based superoxide dismutase mimics compensates for their lower antioxidant potency and makes them as effective as ortho analogues in protecting superoxide dismutase-deficient Escherichia coli. J Med Chem 52(23):7868–7872
Lee CY (1982) [43] Glucose-6-phosphate dehydrogenase from mouse. Methods Enzymol 89(p):252–257
Leung E, Weil DE, Raviglione M, Nakatani H (2011) The WHO policy package to combat antimicrobial resistance. Bull World Health Organ 89(5):390–392. doi:10.2471/BLT.11.088435
Levy SB, Marshall B (2004) Antibacterial resistance worldwide: causes, challenges and responses. Nat Med
Liu Y, Imlay JA (2013) Cell death from antibiotics without the involvement of reactive oxygen species. Science 339(6124):1210–1213. doi:10.1126/science.1232751
Lowry OH, Rosebrough NJ, Farr AL, Randall RJ (1951) Protein measurement with the Folin phenol reagent. J Biol Chem 193(1):265–275
Maisch T (2015) Photodynamic inactivation of multi-resistant bacteria. Photonics Lasers Med 4(4):332–334
Menezes S, Capella MAM, Caldas LR (1990) Photodynamic action of methylene blue: repair and mutation in Escherichia coli. J Photochem Photobiol B Biol 5(3–4):505–517. doi:10.1016/1011-1344(90)85062-2
Nitzan Y, Ashkenazi H (2001) Photoinactivation of Acinetobacter baumannii and Escherichia coli B by a cationic hydrophilic porphyrin at various light wavelengths. Curr Microbiol 42(6):408–414. doi:10.1007/s002840010238
Ogilby PR (2010) Singlet oxygen: there is indeed something new under the sun. Chem Soc Rev 39(8):3181–3209
Preuß A, Zeugner L, Hackbarth S, Faustino MAF, Neves MGPMS, Cavaleiro JAS, Roeder B (2012) Photoinactivation of Escherichia coli (SURE2) without intracellular uptake of the photosensitizer. J Appl Microbiol
Ragas X, He X, Agut M, Roxo-Rosa M, Gonsalves AR, Serra AC, Nonell S (2013) Singlet oxygen in antimicrobial photodynamic therapy: photosensitizer-dependent production and decay in E. coli. Molecules 18(3):2712–2725. doi:10.3390/molecules18032712
Redmond RW, Kochevar IE (2006) Spatially resolved cellular responses to singlet oxygen. Photochem Photobiol 82(5):1178–1186
Sharma SK, Dai T, Kharkwal GB, Huang YY, Huang L, de Arce VJB, Tegos GP, Hamblin MR (2011) Drug discovery of antimicrobial photosensitizers using animal models. Curr Pharm Des 17(13):1303–1319
Sharma SK, Dai TH, Hamblin MR (2012) Antimicrobial photosensitizers: harnessing the power of light to treat infections. Adv M C M (22):310–322 doi:10.1079/9781845939434.0000
Sies H, Menck CFM (1992) Singlet oxygen induced DNA damage. Mutat Res 275(3–6):367–375
Spesia MB, Durantini EN (2013) Photodynamic inactivation mechanism of Streptococcus mitis sensitized by zinc(II) 2,9,16,23-tetrakis[2-(N, N, N-trimethylamino) ethoxy]phthalocyanine. J Photochem Photobiol B Biol 125:179–187. doi:10.1016/j.jphotobiol.2013.06.007
Sylvester PW (2011) Optimization of the tetrazolium dye (MTT) colorimetric assay for cellular growth and viability. Methods Mol Biol 716:157–168. doi:10.1007/978-1-61779-012-6_9
Thomas M, Craik JD, Tovmasyan A, Batinic-Haberle I, Benov LT (2015) Amphiphilic cationic Zn-porphyrins with high photodynamic antimicrobial activity. Future Microbiol 10(5):709–724. doi:10.2217/Fmb.14.148
Tovmasyan A, Reboucas JS, Benov L (2014) Simple biological systems for assessing the activity of superoxide dismutase mimics. Antioxid Redox Signal 20(15):2416–2436
Tsukatani T, Higuchi T, Suenaga H, Akao T, Ishiyama M, Ezoe T, Matsumoto K (2009) Colorimetric microbial viability assay based on reduction of water-soluble tetrazolium salts for antimicrobial susceptibility testing and screening of antimicrobial substances. Anal Biochem 393(1):117–125. doi:10.1016/j.ab.2009.06.026
Vatansever F, de Melo WCMA, Avci P, Vecchio D, Sadasivam M, Gupta A, Chandran R, Karimi M, Parizotto NA, Yin R, Tegos GP, Hamblin MR (2013) Antimicrobial strategies centered around reactive oxygen species—bactericidal antibiotics, photodynamic therapy, and beyond. FEMS Microbiol Rev 37(6):955–989
Wainwright M (2014) In defence of “dye therapy”. Int J Antimicrob Agents
WHO (2014) Antimicrobial resistance: global report on surveillance 2014. April ISBN: 978 92 4 156474 8
Yang Y, Hang J (2013) Fragmentation of genomic DNA using microwave irradiation. J Biomol Tech 24(2):98–103. doi:10.7171/jbt.13-2402-005
Yin R, Hamblin MR (2015) Antimicrobial photosensitizers: drug discovery under the spotlight. Curr Med Chem 22(18):2159–2185
Acknowledgments
The authors are grateful to Milini Thomas and Amna Al-Shamali for their excellent technical assistance. We thank Kuwait University Research Sector (grant YM18/09) and College of Graduate Studies, Kuwait University, for their financial support. The help of the OMICS Research Unit (grant SRUL02/13) is highly appreciated. IBH acknowledges her General Research Funds.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This study was funded by Kuwait University (grant YM18/09) and by the College of Graduate Studies.
Conflict of interest
Mariam M. Awad declares that she has no conflict of interest.
Artak Tovmasyan declares that he has no conflict of interest.
James D. Craik declares that he has no conflict of interest.
Ines Batinic-Haberle declares that she has no conflict of interest.
Ludmil T. Benov declares that he has no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Results are derived from a M. Sc. Thesis submitted by Marim Mubarak Awad to the College of Graduate Studies, Kuwait University (supervisor L. Benov, co-supervisor J. Craik).
Rights and permissions
About this article

Cite this article
Awad, M.M., Tovmasyan, A., Craik, J.D. et al. Important cellular targets for antimicrobial photodynamic therapy. Appl Microbiol Biotechnol 100, 7679–7688 (2016). https://doi.org/10.1007/s00253-016-7632-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-016-7632-3