
Abstract
The main aim of the current study is to explore the bioactive potential of Streptomyces sp. VITJS8 isolated from the marine saltern. The cultural, biochemical, and morphological studies were performed to acquire the characteristic features of the potent isolate VITJS8. The 16Sr DNA sequencing was performed to investigate the phylogenetic relationship between the Streptomyces genera. The structure of the compound was elucidated by gas chromatography-mass spectrometry (GC-MS), infra-red (IR), and ultra-violet (UV) spectroscopic data analysis. The GC-MS showed the retention time at 22.39 with a single peak indicating the purity of the active compound, and the molecular formula was established as C14H9ONCl2 based on the peak at m/z 277 [M]+. Furthermore, separated by high-performance liquid chromatography (HPLC), their retention time (t r) 2.761 was observed with the absorption maxima at 310 nm. The active compound showed effective inhibitory potential against four clinical pathogens at 500 μg/mL. The antioxidant activity was found effective at the IC50 value of 500 μg/mL with 90 % inhibition. The 3-(4,5-dimethylthiazol-2-yl)-2,5-ditetrazolium bromide (MTT) assay revealed the cytotoxicity against HepG2 cells at IC50 of 250 μg/mL. The progression of apoptosis was evidenced by morphological changes by nuclear staining. The DNA fragmentation pattern was observed at 250 μg/mL concentration. Based on flow cytometric analysis, it was evident that the compound was effective in inhibiting the sub-G0/G1 phase of cell cycle. The in vitro findings were also supported by the binding mode molecular docking studies. The active compound revealed minimum binding energy of −7.84 and showed good affinity towards the active region of topoisomerase-2α that could be considered as a suitable inhibitor. Lastly, we performed 30 ns molecular dynamic simulation analysis using GROMACS to aid in better designing of anticancer drugs. Simulation result of root mean square deviation (RMSD) analysis showed that protein-ligand complex reaches equilibration state around 10 ns that illustrates the docked complex is stable. We propose the possible mechanism of sesquiterpenes to play a significant role in antitumor cascade. Hence, our studies open up a new facet for a potent drug as an anticancer agent.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Microbial products have a central role in the area of drug discovery and used extensively to cure numerous diseases (Xiang et al. 2009). Recent studies showed the disease burden of cancer remains as one of the world’s most serious health problem (ACS 2013). A survey conducted by the International Association of Pain stated that 6.6 million people die from cancer each year (IASP 2013). The resistance of cancer cells towards the treatment of anticancer drugs is due to various factors including expression of more energy-dependent transporters that detect and eject anticancer drugs from cells, somatic cell genetic differences, insensitivity to drug-induced apoptosis, and induction of drug-detoxifying mechanisms. The mechanism of cancer drugs has much importance to improve cancer chemotherapy (Michael and Gottesman 2002). Chemotherapy is currently the standard treatment for most cancers. However, many side effects and adverse reactions of chemotherapeutics are mainly due to insufficient selectivity for tumor cells, which represents a major limitation to this type of therapy (Covington 1988). The recent expansion of antimicrobial and anticancer drug research has occurred since there is a critical need for drug designing as to understand drug-receptor interaction for treating life-threatening invasive diseases. Therefore, to develop new anticancer agents, it is essential to accelerate the progress of clinical cancer research and search for new substances, with minimal side effects (Mutai et al. 2009). Recently, much attention was directed towards the biologically active compounds isolated from marine bacteria. Actinomycetes are typically useful in the pharmaceutical industry for their unique compounds with diverse chemical structures and biological activities (Taylor and Stark 2009; Arasu et al. 2008). Among the actinomycetes, with particular reference to the genus Streptomyces are prolific and can produce various antibiotics that are biologically active secondary metabolites. They cover 80 % of total antibiotic products, with other genera trailing numerically. They are non-fastidious in their growth requirements and well adapted to life under fluctuating environmental conditions (Kutzner 1986). Streptomyces genera produce variety of bioactive compounds, which have an enormous capacity for the production of secondary metabolites with broad spectrum of biological activities (Demain 1999). Hence, the bioactive molecules from Streptomyces are considered to be a rich biotechnological resource with the range of biological activities such as antifungal, antitumor, antibacterial, immunosuppressive, insecticidal, and enzyme inhibitors. The most structurally diverse pool of secondary metabolites was constituted by terpenes (Wendt and Schulz 1998). Novobiocin an antibiotic with a terpenoid side chain was reported from the marine Streptomyces niveus (Hoeksema and Smith 1961). Azamerone is a meroterpenoid produced by new marine Streptomyces with cytotoxicity potential (Cho et al. 2006). Cyclomarin, a cyclic peptide isolated from Streptomyces sp., exhibits anti-inflammatory and antiviral activities (Fenical et al. 1999). Caprolactones obtained from marine Streptomyces sp. showed phytotoxicity and cytotoxicity against cancer cells (Stritzke et al. 2004). The exploitation of Streptomyces for novel secondary metabolite production holds considerable promise to combat various diseases.
Induction of apoptosis is recognized as an aggressive strategy to arrest the proliferation of cancer cells (Vermeulen et al. 2005). Molecular docking provides useful information about drug-receptor binding and predicts the binding orientation to find the affinity and activity of a small molecule. The bioactive principle studied by in vitro analysis could be a significant breakthrough in cancer therapy. The parallel study using molecular docking and dynamics simulation analysis showed precise understanding of ligand specificity. Henceforth, the proposed study is towards the discovery and development of novel selective chemotherapeutic drugs devoid of unpleasant side effects.
Materials and methods
We confirm that the proposed field study did not involve endangered or protected species. Based on this, we state clearly that no specific permissions were required for these locations for sample collection.
Sample collection, isolation, characterization, and screening
Marine soil samples were collected from southeast coast of India, Ramanathapuram-SethuKarai (latitude 9° 50″ N and 78° 10′ E) at the depth of 10–100 cm at the littoral zone. The isolation of actinomycetes was performed on starch casein agar along with 25 % marine water and 25 % marine soil extract for effective isolation (Balagurunathan and Subramanian 2001). All the plates were incubated for 7 days at 30 °C (Ravikumar et al. 2008). The isolate VITJS8 was cross-streaked on modified nutrient glucose agar against a broad range of gram-positive and gram-negative bacteria at the right angle and incubated for 3 days (Alexander 1977). The zone of inhibition was measured after 2 days of incubation. The morphological, biochemical, and cultural characteristics were determined on various International streptomyces project (ISP) media. The spore chain morphology and spore surface ornamentation were evaluated by scanning electron microscopy.
Molecular characterization and phylogenetic analysis
Genomic DNA was isolated, and PCR amplification of 16S rDNA was carried out (Rainey et al. 1996). The query sequence was subjected to similarity search using BLAST and submitted to the GenBank database under the accession number (KJ725090). The generic level was identified (Altschul et al. 1997). The phylogenetic tree was constructed via neighbor-joining method using the EvolView program. Bootstrap analysis was performed to validate the reproducibility of the branching pattern. The study strain Streptomyces sp. VITJS8 was assigned with culture collection NCIM No. 5575 under the National Collection of Industrial Microorganisms (NCIM), Pune, India.
Fermentative production of active compound
The potent isolate VITJS8 was inoculated on starch casein broth at a seed concentration of 100 mL in a 250-mL Erlenmeyer flask with pH 7.2 and incubated for 7 days at room temperature. The solvent extraction method was performed to obtain the principal bioactive compound from the fermented broth. After filtration, 100 mL of the fermented broth was taken in 250-mL separating funnels. Different organic solvents (100 mL) ranging from polar to non-polar were utilized for the product recovery (Remya and Vijayakumar 2007). The obtained concentrate was dried using a rotary evaporator and lyophilized.
Purification of active compound
The crude ethyl acetate extract (2.0 g) was subjected to column chromatography using silica gel (100–200 mesh) and eluted which was followed by a combination of methanol/chloroform ranging 95:5. The single fraction was collected in 200-mL conical flasks. An aliquot of purified compound was spotted onto the silica gel plate developed with the same mixture of solvent, and the spots were visualized using thin layer chromatography (TLC) (20 × 20 cm with 0.2 mm thickness, silica gel GF254, Merck, Darmstadt, Germany) (Duraipandiyan and Ignacimuthu 2007). The compound was subjected to spectroscopic analysis.
Characterization of pure compound
The purified active compound was subjected to chemical characterization by ultra-violet (UV), infra-red (IR), gas chromatography-mass spectrometry (GC-MS), and high-performance liquid chromatography (HPLC) analysis.
Determination of λ max
The purified compound was noticed with the absorption spectrum in methanol determined by Shimadzu UV-160A spectrometer. This mixture was scanned at 200–400 nm to measure the maximum absorption.
Infrared spectroscopy
Characterization and identification of a compound or chemical bonds were carried out using Fourier transform infrared (FT-IR). The FT-IR spectra of the purified compound were recorded using Thermo Nicolet Avatar 370 spectrometer over the 4,000–400-cm−1 range at the resolution of 4 cm−1, and a maximum source aperture and the infrared spectra of the compound were measured (as KBr discs).
Gas chromatography-mass spectrum analysis
GC-MS technique was incorporated to determine the presence of volatile compounds in the constitute. NIST08 and WILEY8 database were used for the identification of the separated peaks.
HPLC
The method based on HPLC separation of purified compound was carried out using LC-10 AT vp model HPLC using 250 × 4.60 mm Rheodyne column (C-18) and a mobile phase containing methanol/water (95:5) at a flow rate of 1 mL/min using UV detection at 310 nm. The retention time for the peak was identified.
Bioactivity
Test organisms
Pathogens, namely, Staphylococcus aureus (MTCC No. 7405), Salmonella typhi (MTCC No. 1167), Pseudomonas aeruginosa (MTCC No. 4676), and Escherichia coli (MTCC No. 1588) were obtained from Microbial Culture Collection, IMTECH, Chandigarh, India.
In vitro antibacterial activity
The in vitro antibacterial activity of the purified compound from the isolate VITJS8 was determined by agar well diffusion method (Pandey et al. 2004).
Free radical scavenging activity
2,2-Diphenyl-1-picrylhydrazyl (DPPH) scavenging assay was incorporated to determine the antioxidant activity (Khalaf et al. 2008). Various concentrations of compound (100, 150, 200, 250 μg/mL) of VITJS8 were analyzed with ascorbic acid as reference compound (100, 150, 200, 250 μg/mL).
HepG2 cell maintenance
The cells were obtained from NCSS, Pune and cultured in the RPMI 1640 medium on 10-cm tissue culture plates (Greiner Bio-one™, Germany) supplemented with 10 % heat-inactivated fetal bovine serum. Cells were incubated in a humidified incubator with 5 % CO2 at 37 °C and subcultured when confluence reached up to 80 % (Freshney et al. 1982).
MTT assay
MTT assay assessed the viability of the cells (Mosmann 1983). In the 96-well plates, per well, 5 × 103 HepG2 were added. One hundred microliters of the VITJS8 pure compound in triplicates with different concentrations ranging from 250, 500, 750, and 1,000 μg/mL was tested. Five micrograms per milliliter doxorubicin and 100 μL of Dulbecco's modified Eagle's medium (DMEM) were used as an internal positive and negative control, and wells without any cells were treated as blank.
Effect of pure compound on cell morphology
The HepG2 cells were incubated with purified compound at 250 μg/mL IC50 dose for 16 h and stained with 4',6-diamidino-2-phenylindole (DAPI). Fluorescence microscope (Zeiss, Germany) was used to observe the morphology of the cells.
Apoptotic DNA fragmentation
The cytotoxicity of the compound that leads to the DNA cleavage pattern was assessed by varying the concentrations, 250 and 500 μg/mL, and analyzed by agarose gel electrophoresis. The DNA was analyzed by electrophoresis at 50 V/cm (Sánchez-Alcázar et al. 1997).
Caspase assay
HepG2 cells grown to 70–80 % confluence in 12-well plates were incubated for 24 h with concentration ranging from 100, 150, 200, and 250 μg/mL with 10 % FBS-DMEM. The activity of caspase-3, caspase-8, and caspase-9 were determined by the cleavage of chromogenic substrates (Kohler et al. 2002). The caspase activity was quantified and calculated as fold increase over control.
Cell cycle analysis by flow cytometry
The cell cycle analysis was determined by flow cytometry (Pozarowski and Darzynkiewicz 2004). The cells were treated with IC50 value at 250 μg/mL compound in DMEM with 10 % FBS and processed. The control and the treated samples were subjected to FACS analysis utilizing Beckman Coulter flow cytometer (Beckman Coulter, Indianapolis, IN). Events (10,000) were defined as a minimum range. Each cell cycle phase was defined as the percentage of cells.
Molecular docking
Docking study was performed to study the drug activity on the protein. We obtained ligand structure from the SMILES String (pubchem.ncbi.nlm.nih.gov) and protein structure with (PDB ID: 1ZXM) from Protein Data Bank (http://www.rcsb.org/). The structures were minimized using Swiss PDB viewer. The binding regions were predicted using Metapocket server (Guex and Peitsch 1997). The minimized structures were docked using Auto dock 4.2 (Morris et al. 2009) with Lamarckian Genetic Algorithms (LGA) to get the best docking conformations. The grid size was set to 60, 60, and 60 along the X, Y, and Z axis, respectively, with 0.375 spacing.
Molecular dynamics
The protein-ligand complex with the best conformation and binding energy was taken for the molecular simulation (MD) analysis. MD simulation was performed with GROMACS 4.6.3 using the GROMOS96 43al force field (Van Gunsteren et al. 1996) in octahedron box of a simple point charge. Ligand topology was generated using PRODRG Server (Schüttelkopf and Van Aalten 2004). Two Na+ ions were replaced with solvent molecules to attain system neutrality. Energy minimization was carried out with the steepest descent of 50,000 steps. To extend the equilibrium, the system was subjected to position-restrained dynamic simulation (NVT and NPT) with 50,000 steps at 300 K temperature. Finally, MD was performed at 300 K temperature for 30 ns.
GROMACS output analysis
The GROMACS output can be achieved in the form of trajectory files. These trajectory files were further investigated using root mean square deviation (g_rmsd), radius of gyration (g_gyrate), and hydrogen bond (g_hbond) graphs were plotted using xmgrace.
Results
Description of Streptomyces sp. VITJS8
VITJS8 was identified based on the morphological characteristic by the presence of spiral spores with the smooth-spore surface. The aerial and substrate mycelium was found to be white and pinkish purple in color, respectively (Fig. S1). The various patterns of growth morphology were observed on different ISP media. Cells were identified as Gram-positive, oxidase-negative, and catalase-negative. Growth occurred at 15–60 °C (optimum, 25 °C) and pH 6.0–8.0 (optimum, pH 7.2). Effect of NaCl is 0.5–5 % (optimum, 1.5 %). Several carbon and nitrogen substrates could distinguish the studied strain VITJS8 from all other strains by assimilating d-glucose, d-lactose, d-galactose, d-mannose, d-sucrose, d-arabinose, l-rhamnose, d-mannitol, and nitrogen sources including arginine and phenylalanine. No growth occurred when cysteine, threonine, alanine, aspartic acid, glycine, histidine, and lysine were supplied. The phenotypic characteristics, such as assimilation of carbon, nitrogen, and growth, were useful in the discrimination of the strain VITJS8 from the closely associated species of Streptomyces.
Phylogenetic affiliation of the strain VITJS8 was determined using 16S rRNA gene sequence analysis and submitted to GenBank under the accession number KJ725090. The top 30 related sequences obtained from BLAST along with the most similar Streptomyces type strains were used for further phylogenetic analysis. Based on 16S rRNA gene sequence matches, the studied strain VITJS8 belongs to the family Streptomycetaceae and genus Streptomyces. A maximum of 98 % similarity was observed between the Streptomyces rameus strain NBRC 3782 and 97 % with Streptomyces cellostaticus strain CSSP 188 (Fig. 1a, b). The strain VITJS8 allowed the parallel relatedness to its closest genotypic neighbors and exhibited a characteristic that represents distinguishable taxa phenotypically and phylogenetically for which the name Streptomyces sp. VITJS8 was proposed.

a Multiple sequence alignment of Streptomyces sp. VITJS8 (ACC: KJ725090) 16S rDNA gene sequences from NCBI database. b Phylogenetic tree based on 16S rRNA gene sequences from strains of related recognized Streptomyces species, generated using neighbor-joining algorithm within the Evolview software package. Bootstrap percentages based on 1,000 resamplings are shown
Characterization of the compound
In the present study, the bioactive guided fractionation of the crude fraction. The fraction showed single spot on TLC and fraction was eluted. The chromatographic profile of the compound was visualized under the visible light that showed the presence of a single band on the TLC plate. The intense orange yellow color band at \( Rf \) = 0.52 using methanol/chloroform (95:5). The yellowish precipitate afforded 250 mg. The spectral analysis of the principal compound UV spectra showed λ max at 310 nm (Fig. 2a). HPLC chromatogram indicated the presence of purified compound with the retention time at 2.761 min (Fig. 2b). In subsequent experiment, the characteristic spectrum of the purified compound was recorded by FT-IR spectroscopy which determines the characteristics of the functional groups present in the compound. An infrared spectrum was recorded on a FT-IR spectrophotometer using KBr dispersion method. All the samples were recorded in a range of 4,000–400 cm. Characteristic peaks at 3,469.94 cm−1 (N–H stretching), 1,573.5 cm−1 (C=O bending), 1,400.32 cm−1 (C–H bending), 1,382.96 cm−1 (CH2 and CH3 deformation), 1,242.16 cm−1 (C–H bending), 1,153.43 cm−1 (C–H bending), 856.32 cm−1 (C–Cl stretching), 1,062.07, 1,633.71 cm−1 (C–stretching), 3,140.11 cm−1 (H-bonded), 3,130.47 cm−1 (H-bonded), 3,008.96 cm−1 (C–H stretching), and 2,929.89 cm−1 (O–H stretching) were observed. FT-IR data supports that the isolated compound tends to prove with biological activity due to the presence of important functional groups (Fig. 2c). GC-MS showed peaks with retention time of 22.39 min having relative abundance of 98.586 %. The molecular formula of compound was established as C14H9ONCl2 based on the peak at m/z 277 [M]+ (Fig. 3a, b). The structures of compounds were determined by interpretation of their spectroscopic data. The identified compound was tested towards antimicrobial, antioxidant, and cytotoxic effects with significant activities.

a IR spectra of compound (2R-acetoxymethyl-1,3,3-trimethyl-4t-(3-methyl-2-buten-1-yl)-1T-cyclohexanol). b UV–Vis of compound (2R-acetoxymethyl-1,3,3-trimethyl-4t-(3-methyl-2-buten-1-yl)-1T-cyclohexanol). c GC spectra of the active principal (2R-acetoxymethyl-1,3,3-trimethyl-4t-(3-methyl-2-buten-1-yl)-1T-cyclohexanol)

a Mass spectrum of the active compound (2R-acetoxymethyl-1,3,3-trimethyl-4t-(3-methyl-2-buten-1-yl)-1T-cyclohexanol). b Gradient HPLC chromatogram of compound (2R-acetoxymethyl-1,3,3-trimethyl-4t-(3-methyl-2-buten-1-yl)-1T-cyclohexanol)
Antibacterial and antioxidant activity
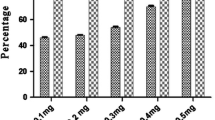
Screening of the compound for in vitro antibacterial activity was performed against pathogens. Among the four pathogens tested, the compound showed good inhibitory activity at 250 μg/mL against S. aureus (MTCC No. 7405) (25 mm), S. typhi (MTCC No. 1167) (12 mm), E. coli (MTCC No. 1588) (15 mm), and P. aeruginosa (MTCC No. 4676) (29 mm) (Fig. S2). The antiradical activity of the compound was studied by screening its possibility to bleach the stable DPPH radical. The scavenging activity was found to be higher with increasing the compound concentration. This compound was shown to exert potent antioxidant activity of 90 % with IC50 of 500 μg/mL against DPPH radicals (Fig. S3).
Microscopy and cytotoxicity
The inhibitory effect of the compound on hepatocellular cell survival was examined. Following treatment, cells grown for subsequent days were photographed under the light microscope to evaluate the cell morphology. The compound promotes cell death at 250 μg/mL, and the morphological changes during apoptosis were observed with cell shrinkage and chromatin condensation, which indicates cell death. DAPI staining was performed to analyze DNA morphology. The nuclear compartment with chromatin condensation and apoptotic bodies were observed. The cells treated with the compound had reduced cell density and incomplete nuclear membrane. Hence, the compound triggered the process of apoptosis (Fig. 4). The MTT assay revealed the cytotoxic effects of the compound treated with IC50 concentration at 250 μg/mL for 8 and 16 h (Fig. 5).

a Light microscopy images showing the morphological response of HepG2 cells to compound (i) control cells (no treatment), (ii) cells treated with (150 μg), (iii) cells treated with (200 μg), and (iv) cells treated with (250 μg). The images were taken 20 h after the addition of the compound. Scale bar, 10 mm. b Fluorescence microscopic images of (i) control cells (no treatment), (ii) cells treated with (150 μg), (iii) cells treated with (200 μg), and (iv) cells treated with (250 μg). The control cells found were intact nuclei, whereas treated cells showed intense fragments of nucleus as signs of apoptosis by DAPI staining

Cytotoxicity of (2R-acetoxymethyl-1,3,3-trimethyl-4t-(3-methyl-2-buten-1-yl)-1T-cyclohexanol) against HepG2 cells
DNA fragmentation
The classical hallmark of apoptosis was measured by DNA fragmentation assay. DNA fragmentation study indicated that the cytotoxic effect of the compound is mediated through induction of apoptosis. The compound produced fragmented DNA in HepG2 cells when treated at 150 and 250 μg/mL concentrations, respectively, as compared to the control cells. Agarose gel electrophoresis demonstrated the presence of the ladder pattern of DNA fragmentation (Fig. 6).

DNA laddering was visualized in HepG2 cell lines after treatment with IC50 dose of the compound for 24 h. L1 150 μg/mL treated DNA, L2 200 μg/mL treated DNA, L3 250 μg/mL treated DNA, L4 control (untreated), L5 1 Kb DNA ladder
Compound regulates cell cycle progression in hepatocellular carcinoma cells
The identified compound was found to induce cell growth inhibition that occurred through the cell cycle arrest. The cell cycle distribution was analyzed by flow cytometric analysis. The exposure of the compound (250 μg) resulted in 30 % increase in S phase cells. The compound induced an increase in G2/M fraction that was accompanied by a decrease in G0/G1 phase cells. These findings indicated that the cytotoxic effects produced by the compound are exerted to cell cycle arrest in HepG2 cell lines (Fig. 7).

a Flow cytometric analysis of compound-induced apoptotic HepG2 cancer cells. b Cell cycle analysis by flow cytometry. After being treated with compound (zero, 250 μg) for 24 h, cells were fixed in ethanol and stained with propidium iodide. DNA content was determined. c HepG2 cells were treated with compound (zero, 150, 250 μg) for 24 h and were analyzed with Annexin-V/PI staining by flow cytometry. Note that the compound-treated cells (bottom panel) have a higher percentage of apoptotic cells (indicated by A) than the basal level of apoptosis seen in the control cells (top panel). L = live cells, D = dead cells
Caspase pathway activation under the regulation of Bcl-2 family proteins
The induction of apoptosis-associated proteins on compound treatment induced cleavage of caspase-9 and caspase-3 in HepG2 cells after 24 h of treatment. The activity of caspases 3, 8, and 9 was assessed to identify the mechanism involved. The results revealed a cleavage in caspase-9, caspase-8, and caspase-3 in a dose-dependent manner (Fig. 8). Hence, the apoptosis was found in the regulation of Bcl-2 family proteins indicating caspases were activated during the process and suggesting a caspase-dependent pathway.

Analysis of the apoptosis-associated caspase activities in HepG2 cells upon treatment with (2R-acetoxymethyl-1,3,3-trimethyl-4t-(3-methyl-2-buten-1-yl)-1T-cyclohexanol) for 24 h
Analysis of molecular binding and dynamic simulation studies
Considering the well-obtained in vitro results, the molecular docking studies were performed. The energy-minimized ligand was docked in the binding site of PDB ID: 1ZXM Human TopoIIa ATPase/AMP-PNP. The potential ligand binding sites were predicted by Metapocket server, namely, ARG162, LYS378, ASN163, GLN376, GLY 161, TYR 165, GLY164, ASN150, SER148, GLY166, ASN91, ALA166, ALA167, SER149, LYS168, ASN95, PHE142, ILE141, ASN120, ARG98, and THR215, respectively. The in silico conformational studies showed the comparative and automated docking abilities. The docking of the receptor with ligand exhibited well-established bonds with the active receptor pocket (Fig. 9a–c) and obtained a good binding energy of −7.84 kJ mol–1 towards the target protein (Table 1). To study the protein-ligand interaction, the docked configuration was visualized using Ligplot (Fig 9a). PyMOL was used to visualize the amino acids that are closely interacting (4 Å) with the ligand (Fig 9c).

a Active site for the ligand molecule. b 3D view of ligand molecule. c Schematic representation of docking revealing the compound (2R-acetoxymethyl-1,3,3-trimethyl-4t-(3-methyl-2-buten-1-yl)-1T-cyclohexanol) interactions with the residues in topoisomerase-2α in 4 Å using PyMOL
Furthermore, we used molecular dynamic simulation analysis by GROMACS to compute the stability, integrity, and hydrogen bonds between the ligand [2R-acetoxymethyl-1,3,3-trimethyl-4t-(3-methyl-2-buten-1-yl)-1T-cyclohexanol)] and protein [topoisomerase-2α]. We analyzed root mean square deviation (RMSD), the radius of gyration, and hydrogen bond formation from 30 ns trajectory files to understand better binding interaction between the protein and protein-ligand complex. To measure the stability of the protein and protein-ligand complex, we analyzed RMSD of protein backbone atoms. From Fig. 10a simulation graph of 30 ns, it is clearly revealed that protein and protein-ligand complex reached equilibration after ∼6 ns and the deviation ranging from 0.25 to 0.45 nm in protein and 0.3 to 0.4 nm in the protein-ligand complex. However, in between the time periods of 10 to 30 ns in protein-ligand complex, it was observed that the amount of deviation has turned minimal which indicates the binding was stable. This proved that the simulation achieved was stable (Fig. 10a). To find out the compactness of the protein, the radius of gyration graph was plotted (Fig. 10b). It was observed that the fluctuation in a radius of gyration of protein and protein-ligand complex ranged from 2.17 to 2.25 nm and 2.0–2.15 nm. This indicates protein-ligand complex is more compact than the protein. Based on RMSD and radius of gyration analysis, we conclude that the binding of protein-ligand complex is much more stable than protein. Furthermore, we performed hydrogen bond analysis to measure the stability, and binding affinity exists between protein-ligand complexes (Fig. 10c). An average number of hydrogen bonds formed in protein and protein-ligand complex is ∼250 to ∼280 and ∼260 to ∼310, respectively. Notably, the number of hydrogen bond formation is slightly high in protein-ligand complex as compared to protein leading to the conclusion that protein-ligand complex had a good binding affinity. Lastly, to measure the binding affinity, we calculated the surrounding amino acids within 4 Å using PyMOL (Fig. 9c). The above results suggest that protein-ligand interaction is more stable over the entire simulation period of 30 ns.

a RMSD backbone of protein (green) and protein-ligand complex (magenta) at 30 ns. b Radius of gyration graph representing protein (green) and protein-ligand complex (magenta) at 30 ns. c Hydrogen bond analysis graph representing protein (green) and protein-ligand complex (magenta) at 30 ns
Discussion
Marine actinomycetes are considered as one of the most significant resources for the new isolation of compounds (Fenical and Jensen 2006). Despite the standard treatment for advanced diseases including chemotherapy and antimicrobial therapy, the mortality rate remains high. P. aeruginosa, S. aureus, E. coli, and Salmonella sp. are the most common pathogens that cause bacteremia, gastrointestinal, skin infections, and toxic shock syndrome (Silva et al. 2012; Kerr and Snelling 2009; Lodise et al. 2007). Some of these pathogens are commonly implicated in serious nosocomial infections. Simultaneously, therapies with cytotoxic compounds are the primary way to cure cancer at the disseminated stage. Drug resistance and their side effects are the major obstacles for effective treatment (Miller and Larkin 2009). Developing anticancer targets with specificity and higher potency against cancer cells has become the thrust area of research, and there is a high demand for safer alternative and effective chemotherapeutic agents.
During the last two decades, sesquiterpenes and their biological activities have been the focus of numerous phytochemical, pharmacological, and synthetic studies. To date, only very few sesquiterpenes have been reported from the marine microorganisms (Denning 2003). Marine actinomycete-derived terpenoids, marinone (Pathirana et al. 1992), neomarinone (Hardt et al. 2000), amorphane sesquiterpenes (Wu et al. 2006), new T-muurolol sesquiterpenes (Ding et al. 2009), (5b,6a)-6,11-dihydroxyeudesmane from Streptomyces sp. LR4612 (Zhao et al. 2006), oxaloterpins A, B, C, D, and E, diterpenes from Streptomyces sp. KO-3988 (Motohashi et al. 2007), and napyradiomycins from Streptomyces antimycoticus NT17 (Motohashi et al. 2008) were reported. These terpenoids showed interesting biological properties such as antitumor, antibacterial, or antioxidative activities. Previous reports have demonstrated the antibacterial activity of four-streptothricin-group antibiotics from Streptomyces qinlingensis (Ji et al. 2007). Previous studies also support the present study. Echinoside A, a marine-derived anticancer drug, targets the topoisomerase-2α by unique interference with its DNA binding and catalytic cycle (Li et al. 2010). Bivittoside A, holothurin A, holothurinoside A, holotoxin A, and cucumarioside A were effective against human DNA topoisomerase-2α inhibitor (PDB: 1ZXM) (Patil and Thakare 2012). Anticancer agents, 4-cycloalkylidineamino 1,2-naphthoquinone semicarbazones (Shukla et al. 2012), and xanthone derivatives have also inhibited the activity of DNA topoisomerase-2α. The present study showed significant data with expanded knowledge of terpenes and considered to be first report on sesquiterpenes (2R-acetoxymethyl-1,3,3-trimethyl-4t-(3-methyl-2-buten-1-yl)-1T-cyclohexanol) from Streptomyces sp. VITJS8 expected to be a promising candidate for drug discovery. The inhibitory potential of the pure compound and the effect on HepG2 cell cycle prompted us to carry out a mechanistic investigation. This research provides a basis for understanding the biological insights into the compound activation mechanism. The apparent anticancer activities against human tumors were collectively reflected by docking analysis that competitively binds to the ligand. Molecular dynamic simulation analysis (30 ns) by GROMACS was used to calculate the stability of the compound. RMSD, the radius of gyration, and hydrogen bond analysis of the complex illustrated the stability, flexibility, and microenvironment of the protein and complex. RMSD analysis showed that protein-ligand complex attained equilibration state around 10 ns which illustrates that the docked complex is stable. The current study highlights the possibility of using this new marine-derived compound as a potential source of anticancer drugs which clearly infers the special mode of action and provides an ideal design for a novel type of topoisomerase-2α inhibitor. The close parallelism between the docking and experimental results in this study refined the inhibitory activity towards topoisomerase. The direction of the work implemented computational studies provided valuable insights into the binding pattern of the compound at molecular level, and the sesquiterpenes are proven to play a significant role in antitumor cascade. Hence, adverse reactions and high-cost chemotherapeutics clearly underscore the necessity to extend new anticancer agents.
References
Alexander M (1977) Soil microbiology, 2nd edn. Wiley, New York, p 207
Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSLBLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402
American Cancer Society. Atlanta: cancer facts and figures, 2013. Available from URL:http://www.cancer.org
Arasu MV, Duraipandiyan V, Agastian P, Ignacimuthu S (2008) Antimicrobial activity of Streptomyces sp. ERI-26 recovered from Western Ghats of Tamil Nadu. J Mycol MeD 18:147–153
Balagurunathan R, Subramanian A (2001) Antagonistic Streptomycetes from marine sediments. Adv Bio Sc 20:71–6
Cho JY, Kwon HC, Williams PG, Jensen PR, Fenical W (2006) Azamerone, a terpenoidphthalazinone from a marine derived bacterium related to the genus Streptomyces (Actinomycetales). Org Lett 8:2471–2474
Covington RT (1988) Management of diarrhea. Drug News Lett 13:1–2
Demain AL (1999) Pharmaceutically active secondary metabolites of microorganisms. Appl Microbiol Biotechnol 52:455–463
Denning DW (2003) Echinocandin antifungal drugs. Lancet 362:1142–1151
Ding L, Pfoh R, Ruhl S, Qin S, Laatsch H (2009) T-muurololsesquiterpenes from the marine Streptomyces sp. M491 and revision of the configuration of previously reported amorphanes. J Nat Prod 72(1):99–101
Duraipandiyan V, Ignacimuthu S (2007) Antibacterial and antifungal activity of Cassia fistula L.: an ethnomedicinal plant. J Ethnopharmacol 112:590–594
Fenical W, Jensen PR (2006) Developing a new resource for drug discovery: marine actinomycete bacteria. Nat Chem Biol 2:666–673
Fenical W, Lobkovsky E, Cladry J (1999) Cyclomarins A-C, new antiinflammatory cyclic peptides produced by a marine bacterium (Streptomyces sp). J Am Chem Soc 21:11273–11276
Freshney RI, Hart E, Russell JM (1982) Isolation and purification of cell cultures from human tumors. In: Reid E, Cook GMW, Moore DJ (eds) (B): Biochemistry, vol 2. Horwood, Chicester
Guex N, Peitsch MC (1997) SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis 18:2714–2723
Hardt IH, Jensen PR, Fenical (2000) Neomarinone, and new cytotoxic marinone derivatives, produced by a marine filamentous bacterium (Actinomycetales). Tetrahedron Lett 41:2073–2076
Hoeksema H, Smith CG (1961) Novobiocin. Prog Ind Microbiol 3:91–139
International Association for the Study of Pain, Epidemiology of Cancer Pain [Accessed March 25, 2013]. Available from URL: http://www.iasppain.org
Ji Z, Wang M, Zhang J, Wei S, Wu W (2007) Two new members of streptothricin class antibiotics from Streptomyces qinlingensis sp. nov. J Antibiot 60:739–744
Kerr KG, Snelling AM (2009) Pseudomonas aeruginosa: a formidable and ever present adversary. J Hosp Infect 73:338–344
Khalaf NA, Shakya AK, Al-Othman A, El-Agbar Z, Farah (2008) Antioxidant activity of some common plants. Turk J Biol 32:51–55
Kohler C, Orrenius S, Zhivotovsky B (2002) Evaluation of caspase activity in apoptotic cells. J Immunol Methods 265:97–110
Kutzner KJ (1986) The family Streptomycetaceae. In: Starr MP, Stolp H, Trper HG, Balows A, Schlegel HG (eds) The prokaryotes. A handbook on habitats, isolation, and identification of bacteria, vol 2. Springer, New York, pp 2028–2090
Li M, Miao ZH, Chen Z, Chen Q, Gui M, Lin LP, Sun P, Yi YH, Ding J (2010) Echinoside A, a new marine-derived anticancer saponin, targets topoisomerase 2 alpha by unique interference with its DNA binding and catalytic cycle. Ann Oncol 21(3):597–607
LodiseJr PTP, Kwa N, Graves A, Furuno J, Graffunder E, Lomaestro B, McGregor JC (2007) Predictors of 30-day mortality among patients with Pseudomonas aeruginosa blood stream infection: impact of delayed appropriate antibiotic selection. Antimicrob Agents Chemother 51:3510–3515
Michael M, Gottesman (2002) Mechanisms of cancer drug resistance. Annu Rev Med 53:615–27
Miller RE, Larkin JM (2009) Combination systemic therapy for advanced renal cell carcinoma. Oncology 4(12):1218–1224
Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ (2009) Autodock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem 16:2785–2791
Mosmann T (1983) Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods 65:55–63
Motohashi K, Ueno R, Sue M, Furihata K, Matsumoto T, Dairi T, Omura S, Seto H (2007) Studies on terpenoids produced by actinomycetes: oxaloterpins A, B, C, D, and E, diterpenes from Streptomyces sp. KO-3988. J Nat Prod 70:1712–1717
Motohashi K, Sue M, Furihata K, Ito S, Seto H (2008) Terpenoids produced by actinomycetes: napyradiomycins from Streptomyces antimycoticus NT17. J Nat Prod 71(4):595–601
Mutai C, Bii C, Vagias C, Abatis D, Roussis V (2009) Antimicrobial activity of Acacia mellifera extracts and lupanetriterpenes. J Ethnopharmacol 123:143–148
Pandey B, Ghimire P, Agrawal VP (2004) Studies on the antimicrobial activity of actinomycetes isolated from Khumbu region of Nepal. Tribhuvan University, Kathmandu
Pathirana C, Jensen PR, Fenical W (1992) Marinone and debromomarinone: antibiotic sesquiterpenoidnaphthoquinones of a new structure class from a marine bacterium. Tetrahedron Lett 33:7663–7666
Patil TD, Thakare SV (2012) In silico evaluation of selected triterpene glycosides as a human DNA topoisomerase II alpha (α) inhibitor. Int J Pharm Pharm Sci 4(4):201–204
Pozarowski P, Darzynkiewicz Z (2004) Analysis of cell cycle by flow cytometry. Methods Mol Biol 281:301–311
Rainey FA, Ward-Rainey NL, Kroppenstedt RM, Tackebrandt E (1996) The genus Norcardiopsis represents a phylogenetically coherent taxon and a distinct actinomycetes lineage: proposal of Nocaridiopsaceae fam. nov. Int J Syst Bacteriol 46:1088–1092
Ravikumar YS, Mahadevan KM, Kumaraswamy MN, Vaidya VP, Manjunatha H, Kumar V, Satyanarayana ND (2008) Antioxidant, cytotoxic and genotoxic evaluation of alcoholic extract of Polyalthia cerasoides (Roxb.) Bedd. Environ Toxicol Pharmacol 26(2):142–146
Remya M, Vijayakumar R (2007) Isolation and characterization of marine antagonistic actinomycetes from West Coast of India. Med Biol 5:13–19
Sánchez-Alcázar JA, Ruíz-Cabello J, Hernández-Muñoz I, Pobre PS, de la Torre P, Siles-Rivas E, García I, Kaplan O, Muñoz-Yagüe MT, Solís-Herruzo JA (1997) Tumor necrosis factor-α increases ATP content in metabolically inhibited L929 cells preceding cell death. J Biol Chem 272(48):30167–30177
Schüttelkopf AW, Van Aalten DMF (2004) PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr Sect D: Biol Crystallogr 60(8):1355–1363
Shukla S, Srivastava RS, Shrivastava SK, Sodhi A, Kumar P (2012) Synthesis, molecular docking and biological evaluation of 4-cycloalkylidineamino 1,2-naphthoquinone semicarbazones as anticancer agents. Asian Pac J Tro Biomed 2(2):S1040–S1046
Silva JC, Rodrigues S, Feas X, Estevinho LM (2012) Antimicrobial activity, phenolic profile and role in the inflammation of propolis. Food ChemToxicol 50:1790–1795
Stritzke K, Schulz S, Laatsch H, Helmke E, Beil W (2004) Novel caprolactones from a marine Streptomycete. J Nat Prod 67:395–401
Taylor WR, Stark GR (2009) Regulation of the G2/M transition by p53. Oncogene 20:1803–1815
Van Gunsteren WF, Billeter SR, Eising AA, Hunenberger PH and Kruger P (1996) Biomolecular simulation: the GROMOS96 manual and user guide, vdf Hochschulverlag AG an der ETH Zurich and BIOMOS b.v, Zurich, Switzerland, pp 1–1042
Vermeulen K, VanBockstaele DR, Berneman ZN (2005) Apoptosis: mechanisms and relevance in cancer. Ann Hematol 84:627–639
Wendt KU, Schulz GE (1998) Isoprenoid biosynthesis: manifold chemistry catalyzed by similar enzymes. Structure 6:127–133
Wu SJ, Fotso S, Li F, Qin S, Kelter G, Fiebig HH, Laatsch H (2006) N-carboxamido-staurosporine and selina-4(14),7(11)-diene-8,9-diol, new metabolites from a marine Streptomyces sp. J Antibiot (Tokyo) 59(6):331–337
Xiang WS, Wang JD, Wang XL, Zhang J (2009) A novel macrolide compound from Streptomyces bingchenggensis: fermentation, isolation, structure elucidation and biological properties. J Antibiot 62:229–231
Zhao PJ, Li GH, Shen YM (2006) New chemical constituents from the endophyte Streptomyces species LR4612 cultivated on Maytenus hookeri. Chem Biodivers 3:337–342
Acknowledgments
The authors take this opportunity to thank the management of VIT University and CSIR for providing the facilities and encouragement to carry out this work.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Ethical approval
This article does not contain any studies with human participants performed by any of the authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 166 kb)
Rights and permissions
About this article

Cite this article
Naine, S.J., Devi, C.S., Mohanasrinivasan, V. et al. Binding and molecular dynamic studies of sesquiterpenes (2R-acetoxymethyl-1,3,3-trimethyl-4t-(3-methyl-2-buten-1-yl)-1t-cyclohexanol) derived from marine Streptomyces sp. VITJS8 as potential anticancer agent. Appl Microbiol Biotechnol 100, 2869–2882 (2016). https://doi.org/10.1007/s00253-015-7156-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-015-7156-2