
Abstract
Antimicrobial dendrimeric peptides (AMDP) are a relatively new class of agents displaying repetitive functional groups on a branched core. Previously, we have investigated the length requirement for antimicrobial activity of peptides consisting of repeated arginine (R) and tryptophan (W) side chains and found that even short linear RW repeats are active, providing a starting point for a de novo design of multivalent structures. In this study, we synthesized and tested a new synthetic dendrimer, 2D-24, for its antimicrobial activity against Pseudomonas aeruginosa, including the wild-type PAO1 and its mucoid mutant PDO300. This synthetic AMDP was found to kill planktonic cells of both PAO1 and PDO300 in a dose-dependent manner, with nearly complete killing of both strains observed when treated with 50 μM of this agent. In addition to planktonic cells, 2D-24 was also found to kill biofilm cells of both strains in a dose-dependent manner. For example, treatment with 30 μM 2D-24 led to 94.4 ± 1.4 and 93.9 ± 4.2 % killing of PAO1 and PDO300 biofilm cells, respectively. Furthermore, 2D-24 was effective in killing multidrug-tolerant persister cells of PAO1 and PDO300. While higher concentrations of 2D-24 were required to kill persister cells, combinations of 2D-24 with ciprofloxacin, tobramycin, or carbenicillin showed synergistic effects on killing persister cells of both strains. Based on hemolysis assays using sheep erythrocytes and a coculture model of PAO1 and human epithelial cells, 2D-24 was found to kill P. aeruginosa cells at concentrations that are not toxic to mammalian cells.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Bacteria have evolved diverse mechanisms to survive from the attack of antibiotics. One such mechanism involves persister cell formation, by which a small population in a bacterial culture enters an inactive stage and exhibits high tolerance to antibiotics and other forms of stress (Lewis 2013). Consistently, persister cell formation has been implicated in chronic infections by Mycobacterium tuberculosis and Pseudomonas aeruginosa (Mulcahy et al. 2010; Ojha et al. 2008).
In addition to persister formation, bacteria can also evade antibiotics by forming multicellular structures, known as biofilms, in which bacterial cells adhere to a surface and secrete an extracellular matrix that protects them from environmental stresses (Flemming and Wingender 2010). Due to limited mass transfer (Gilbert et al. 1997), biofilm cells grow slowly with reduced metabolism and exhibit high tolerance to antimicrobial agents (Keren et al. 2004; Lewis 2007). It is estimated that 80 % of bacterial infections in humans involve biofilms (Romero et al. 2008). Thus, biofilms present a serious complication associated with chronic infections especially in patients with compromised immune systems (Costerton et al. 1987, 1999; Hall-Stoodley et al. 2004; McDougald et al. 2012).
To address the challenge of drug-resistant infections, it is important to develop new antimicrobials that are effective against biofilms and persister cells. Antimicrobial peptides (AMP) are promising alternative antibiotics due to their rapid killing effects, low frequency of resistance development, and broad spectrum of target microbes (Bahar and Ren 2013). Thousands of AMPs have been identified to date (Wang et al. 2009), and many synthetic analogs have been developed and tested against infectious microorganisms including protozoa, fungi, bacteria, and viruses (Alhoot et al. 2013; Arrighi et al. 2002; He et al. 2007; Lee et al. 2004; Powell et al. 1995). However, most AMPs are not appropriate for therapeutic use due to cytotoxicity, high MIC (minimum inhibitory concentration) values, in vivo inactivation by proteases, and/or high production cost (Hancock and Sahl 2006). Therefore, it is important to develop new AMPs to overcome these challenges. One such strategy is to modify the AMP molecules by amidation of carboxyl ends (Kim et al. 2011), cyclization, covalent modification with disulfide bonds (Uteng et al. 2003), and/or alteration of amino acid content such as adding prolines (Noto et al. 2008). Modifications of this kind can affect critical physiochemical properties of natural AMPs and thus the target spectrum, cytotoxicity to eukaryotic cells, antimicrobial activity, and stability against proteases (Bahar and Ren 2013).
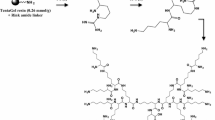
Antimicrobial dendrimeric peptides (AMDP) are a relatively new class of AMPs with repeating functional groups linked to a multivalent scaffold. The Kallenbach group has investigated the length requirement for antimicrobial activity of peptides only consisting of amino acid arginine (R) and tryptophan (W) repeats. Even short repeats of RW—trimers, for example—are active, providing a starting point for a de novo design of minimal structures (Tam et al. 2002). Further studies found that dendrimeric displays of di- or tripeptides of arginine and tryptophan can render the AMPs more potent and less cytotoxic than natural AMPs (Liu et al. 2007). Recently, we showed that some synthetic AMPs with RW repeats are effective against planktonic and biofilm cells of Escherichia coli (Hou et al. 2009, 2010) including persister cells (Chen et al. 2011). To further evaluate the potential of AMDPs as more effective antibacterial agents, the agent 2D-24, containing RWR and RTtbR(2) tripeptide branches (Fig. 1), was synthesized for this study and tested for its antimicrobial activities against planktonic, biofilm, and persister cells of the wild-type P. aeruginosa PAO1 and its mucoid mutant PDO300.

Structure of 2D-24
P. aeruginosa is a gram-negative bacterium with exceptional capabilities to adapt to different living environments (Winsor et al. 2011). It is a well-known opportunistic human pathogen (Bodey et al. 1983), causing serious infections in patients with comprised immunity or with cystic fibrosis (de Bentzmann and Plesiat 2011; Schurr et al. 1996). P. aeruginosa can acquire antibiotic tolerance through biofilm and persister formation (Lewis 2010; Mah and O'Toole 2001). In addition, during chronic colonization in the lungs of cystic fibrosis patients, P. aeruginosa commonly acquires mutations that lead to a mucoid phenotype overproducing the polysaccharide alginate and therefore acquires a higher-level antibiotic tolerance (Lowbury et al. 1969; Ma et al. 2009; Mathee et al. 1999). With its strong clinical relevance, P. aeruginosa is a good model bacterium for studying antibiotic resistance and for testing new antimicrobials.
Materials and methods
Synthesis of 2D-24
The synthesis of 2D-24 (50-μmol scale) was performed manually by stepwise solid phase reactions on rink amide resin. Briefly, the resin (200 mg, 0.62 mmol/g) was swelled in dichloromethane for 1 h, and 20 % piperidine/DMF (2 × 10 min) was added to remove the Fmoc-protecting group. A mixture of Fmoc-β-Ala-OH (50 μmol), HBTU/HOBt (50 μmol), and DIEA (100 μmol) was added to the resin and shaken overnight. After DMF and DCM washes, the unreacted amines were capped with an Ac2O/DIEA/DMF (1:2:2) mixture to give a final loading of 0.25 mmol/g (estimated by UV absorption of Fmoc at 290 nm in 20 % piperidine DMF). Next, Fmoc-Lys(Fmoc)-OH (4 eq.), Fmoc-Arg(Pbf)-OH (8 eq.), Fmoc-Tbt-OH (8 eq.), and Fmoc-Arg(Pbf)-OH (8 eq.) were coupled to the resin using HBTU/HOBt as activating agent, DIEA as base, and 20 % piperidine/DMF as Fmoc-deprotecting solution. Upon final Fmoc-deprotection, the product was cleaved from the resin using a TFA/Water/triisopropylsilane (95:2.5:2.5) mixture, precipitated with cold ether, and dried in vacuum. The resulting crude product was purified by reverse-phase high-performance liquid chromatography (RP-HPLC) in a preparative C18-column (VYDAC C18 cat# 218TP101522, 22 mm × 250 mm, 10–15 μm) using water + 0.1 % TFA with acetonitrile + 0.1 % TFA as eluents. The pure product was characterized by 1H-NMR and MALDI-TOF.
H-Tbt-OH was synthesized as described earlier (Haug et al. 2008). A mixture of H-Trp-OH (10.0 g, 49 mmol) and t-Butanol (38.67 g, 0.52 mol) in trifluoroacetic acid (150 mL) was stirred at room temperature for 69 h. The resulting dark solution was evaporated to give a dark oily material which was resuspended in water, and KHCO3 was added until the pH became neutral. Constant trituration of the mixture during addition of the base resulted in the formation of a pinkish gum-like solid. The aqueous layer was decanted off, and the product recrystallized twice from 50 % ethanol in water to afford the title compound as colorless crystals (6.2 g, 33.9 % yield).
For the synthesis of Fmoc-Tbt-OH, a solution of H-Ttb-OH (2.0 g, 5.37 mmol) in water/dioxane 1:1 (80 mL) was added to Fmoc-OSu (5.37 mmol) and NaHCO3 (1.35 g, 16.11 mmol); the resulting mixture was stirred at room temperature for 18 h. The solvents were evaporated, and the residual solid separated between ethyl acetate and 5 % KHCO3 in water. The aqueous phase was extracted twice more with ethyl acetate, and the combined organic phases were washed with 5 % KHCO3, saturated NaCl, and dried under Na2SO4. Evaporation of the solvent under vacuum gave the title compound as a yellow foam which was further purified by flash chromatography (Fig. S1, Fig. S2).
Bacterial strains and growth media
P. aeruginosa PAO1 (ATCC BAA-47, henceforth PAO1) and P. aeruginosa PDO300 (mucA22 mutant of P. aeruginosa PAO1, constructed based on a clinical isolate of P. aerugionsa FRD1 from cystic fibrosis patient (DeVries and Ohman 1994); henceforth, PDO300 (Mathee et al. 1999)) were cultured in Lysogeny broth (LB) medium (Sambrook and Russell 2001) containing 10 g/L tryptone, 5 g/L yeast extract, and 10 g/L NaCl at 37 °C with shaking at 200 rpm. To minimize the difference in persistence between overnight cultures, all overnight cultures were inoculated with single-use glycerol stocks prepared from the same overnight culture.
Effects on planktonic cells
Exponential cultures were prepared by inoculating 3 mL LB medium with an overnight culture to an optical density at 600 nm (OD600) of 0.01 and incubated at 37 °C with 200 rpm shaking until OD600 reached 0.5. Then, different concentrations of 2D-24 were added. The control and treated samples were incubated for 3 h at 37 °C with 200 rpm shaking. Then, the cells were washed three times with phosphate buffered saline (PBS, pH 7.4). The viability of each sample was determined by counting colony-forming units (CFUs) using a drop-plate method as described previously (Chen et al. 2011). Briefly, each sample was diluted in a 10× series with 0.85 % NaCl solution. The CFU was determined by loading 10 μL of each diluted sample on a LB agar plate and counting the CFUs after incubation at 37 °C overnight.
Biofilm-killing experiments
For each treatment, three separate polished (using 1000 grit sand paper, 3 M, St. Paul, MN, USA) sterile 2 cm × 1 cm 316-L stainless steel coupons were placed in a petri dish containing 20 mL LB medium. To initiate biofilm formation, the medium was inoculated with an overnight culture to OD600 of 0.01. These coupons were incubated for 24 h at 37 °C without shaking. The coupons were then washed gently with PBS three times and placed in a 12-well plate (one coupon in each well) with 2 mL PBS in each well and 2D-24 supplemented at concentrations of 0.1 to 30 μM. After incubation for 3.5 h, biofilm coupons were washed with PBS and transferred to 15-mL sterile conical tubes with 3 mL PBS buffer in each. The samples were gently sonicated for 4 min in a sonication bath (Branson B200 Ultrasonic, Danbury, CT, USA) and vortexed for 15 s (this condition was confirmed not to kill the cells; data not shown). The cells, thus detached from coupons, were plated using the drop-plate method (Chen et al. 2011) to determine the CFUs.
Persister isolation and killing
Persister cells of PAO1 and PDO300 were isolated by killing normal cells in overnight cultures with 200 μg/mL ciprofloxacin (Cip) for 3.5 h at 37 °C, as described previously (Niepa et al. 2012; Pan et al. 2012). The treated cells were then washed twice with PBS buffer to remove any remaining antibiotic. The collected cells were resuspended in 3 mL PBS supplemented with 2D-24 at different concentrations and incubated for 3.5 h at 37 °C with 200 rpm shaking. After treatment, the cells were washed with PBS and plated to determine the viability by counting CFU as described above.
Fluorescence microscopy
To corroborate the CFU results, the LIVE/DEAD BacLight™ bacterial viability kit (Life Technologies Inc., Carlsbad, CA, USA) was used to determine the viability of biofilm cells of PAO1 and PDO300 on 316-L stainless steel coupons. The 24-h P. aeruginosa biofilms grown on these surfaces were washed gently with 0.85 % NaCl (w/v) solution three times to remove planktonic cells. Then, 1 μL of 20 mM propidium iodide and 1 μL of 3.34 mM SYTO 9 stains were added in 1 mL PBS to stain each biofilm sample for 10 min in the dark. The stained biofilm samples were examined with an Axio Imager M1 fluorescence microscope (Carl Zeiss Inc., Berlin, Germany). At least five randomly selected spots were examined for each sample.
Synergistic effects between 2D-24 and antibiotics
Three different antibiotics were tested including Cip (a fluoroquinolone targeting DNA gyrase) (Drlica and Zhao 1997), tobramycin (Tob, an aminoglycoside targeting translation by binding to ribosome subunits) (Le Goffic et al. 1979), and carbenicillin (Car, a carboxypenicillin targeting bacterial cell wall synthesis) (Butler et al. 1970). Normal and persister cells of PAO1 and PDO300 were treated with antibiotic alone, 2D-24 alone, or a combination of an antibiotic and 2D-24 to assess any synergistic effects in bacterial killing. The viability of bacterial cells was determined using the drop-plate method as described above.
Hemolysis assays
The hemolytic activity of 2D-24 was assessed on fresh sheep erythrocytes (Fitzgerald Inc., Concord, MA). Peptide concentrations yielding 50 % hemolysis were used as the hemolytic dose (HD50), determined from dose-response curves. The red blood cell suspension was incubated in PBS buffer (pH 7.2) with various volumes of peptide stocks at 37 °C for 30 min and then spun down at 3000 rpm for 10 min. The resulting supernatant was diluted by a factor of 40 in distilled water. The absorbance levels of the supernatants at λ that was 540 nm (OD540) were measured with a UV spectrophotometer. Zero-hemolysis and 100 %-hemolysis controls were obtained by incubating the cells with buffer and 1 % Triton X, respectively.
Cytotoxicity and effects in cocultures
The cytotoxicity of 2D-24 on eukaryotic cells was evaluated using IB3-1 cells and LIVE/DEAD® viability/cytotoxicity kit (Invitrogen, NY, USA). IB3-1 cells were grown in LHC-8 basal medium (Invitrogen, NY, USA) supplemented with 10 % (v/v) fetal bovine serum (FBS) in flasks pre-coated with 100 μg/mL bovine serum albumin (BSA) and 30 μg/mL collagen at 37 °C with 5 % CO2. All treatments were performed in antibiotic-free medium. To test the cytotoxicity, IB3-1 (Andersson et al. 2008) (a compound heterozygote bronchial epithelial cell line from a CF patient) cells were seeded into black-sided clear-bottom 96-well plates (5 × 104 cells per well) and incubated until the cell density reached 1 × 106 cells per well. Then, 2D-24 was added at concentrations of 1 to 32 μM. After treatment for 24 h, EthD-1 and calcein AM were used to assess the viability of IB3-1 cells. The optimal concentrations of EthD-1 (1 μM) and calcein AM (2 μM) were determined prior to the treatments to label live and dead IB3-1 cells distinctively. After staining for 30 min at 37 °C, samples were washed with PBS and analyzed using a fluorescence microplate reader (model FLx800, Bio-Tek Instruments, Winooski, VT, USA). To test the effects of 2D-24 on PAO1 in cocultures, PAO1 cells (4 × 106 cells/mL) were added either with 2D-24 together or at 4 h prior to 2D-24 treatment (20 h after the inoculation of IB3-1 cells). The viability of IB3-1 cells was determined as described above. To compare the effects on bacteria, PAO1 cells were treated in the same medium (LHC-8 basal medium) but in the absence of IB3-1 cells. The viability of PAO1 cells was determined by counting CFU using the drop-plate method as described above. All treatments were tested in triplicate.
Statistical analysis
The data from CFU experiments were analyzed with one-way ANOVA followed by Tukey test using SAS version 9.3 (SAS Institute, Cary, NC, USA). Differences with p < 0.05 were considered to be statistically significant.
Results
Synthesis of 2D-24
Synthesis of the dendrimer (Fig. 1) was confirmed by 1H-NMR (Fig. S1) and MALDI-TOF mass spectrometry (Fig. S2). To overcome several limitations of linear peptides as antibacterial agents, the Kallenbach group has developed a series of small dendrimeric peptide analogs, such as (RW)4D (Liu et al. 2007), which mimic the core sequence pattern in natural AMPs yet offer advantages in economy of synthesis, protease resistance, reduced cytotoxicity, and chemical stability. A second-generation dendrimeric AMP mimetic, 2D-24, has been confirmed to show broad-spectrum activities against drug-resistant clinical isolates (data not shown). The indole side chain of tryptophan plays an important role in the pharmacology of antimicrobial activity (Liu et al. 2007). Since inactivation of AMPs by serum is a major problem limiting their potential in clinical applications, we have modified the indole side chain of tryptophan to include bulky substituents that afford greater likelihood of in vivo stability and activity.
Antimicrobial effects of 2D-24 on planktonic cells
First, the effects of 2D-24 on normal planktonic cells were evaluated by treating cells in exponential phase cultures with different concentrations of 2D-24. As shown in Fig. 2, 2D-24 was effective in killing both PAO1 and PDO300 planktonic cells in a dose-dependent manner. For example, 5 μM 2D-24 was found to kill 53.3 ± 3.3 % PAO1 (p < 0.001) and 40.3 ± 6.2 % PDO300 (p < 0.005) cells, respectively. The killing efficiency increased with the concentration of 2D-24. At 20 μM, 79.9 ± 4.7 % (p < 0.001) and 84.6 ± 7.1 % (p < 0.001) killing was obtained for PAO1 and PDO300, respectively. No viable cells of either strain were found after treatment with 50 μM 2D-24, suggesting that complete killing is possible. Interestingly, while the mucoid strain PDO300 is more tolerant to antibiotics than the wild-type PAO1 (Mathee et al. 1999), 2D-24 exhibited similar activities in killing PAO1 and PDO300.

Effects of 2D-24 on planktonic cells of PAO1 (a) and PDO300 (b). The dendrimer 2D-24 was added at different concentrations to exponential cultures, and the viability was determined by counting CFUs
Antimicrobial effects of 2D-24 on biofilm cells
In addition to planktonic cells, 2D-24 was also found to kill PAO1 and PDO300 biofilm cells effectively (Fig. 3). At 5 μM, 2D-24 is slightly more effective against PAO1 than PDO300. For example, 2D-24 exhibited 55.8 ± 6.5 % killing (p < 0.001) of PAO1 biofilm cells, while the killing of PDO300 biofilm cells with the same concentration of 2D-24 was 31.6 ± 8.8 % (p < 0.05). The activity increased with 2D-24 concentration for both strains, e.g., 2D-24 killed 87.8 ± 3.1 % (p < 0.001) and 94.4 ± 1.4 % (p < 0.001) of PAO1 biofilm cells when treated at 20 and 30 μM, respectively. Similar activities against PDO300 biofilms were observed, e.g., 2D-24 exhibited 81.7 ± 3.5 and 93.9 ± 4.2 % killing of PDO300 biofilm cells when added at 20 and 30 μM, respectively (p < 0.001 for both). Since the mucoid strain PDO300 is tolerant to multiple antibiotics (Hengzhuang et al. 2011; Hentzer et al. 2001), the finding that 2D-24 is equally effective in killing PAO1 and PDO300 biofilms is encouraging. The effective killing of biofilm cells, especially those of PDO300, suggests that 2D-24 is able to penetrate the biofilm matrix and the alginate layer of the mucoid strain.

Effects of 2D-24 on PAO1 (a) and PDO300 (b) biofilm cells. The biofilms were cultured for 24 h in LB medium before treatment with 2D-24 for 3.5 h in PBS
Live-dead staining of biofilms
To corroborate the CFU results of biofilm killing, the control and 2D-24-treated biofilm samples were analyzed using LIVE/DEAD staining (Fig. 4). Consistent with CFU data, the LIVE/DEAD images also showed dose-dependent killing of biofilms cells by 2D-24. While the cells in untreated control were healthy (green), killing was observed (red) with addition of 2D-24 and the effects were dose dependent (Fig. 4).

LIVE/DEAD images of PAO1 and PDO300 biofilms. A1–A3: PAO1 biofilms treated with 0, 10, and 30 μM 2D-24, respectively. B1–B3: PDO300 biofilms treated with 0, 10, and 30 μM 2D-24, respectively. Bar = 10 μm
Antimicrobial effect of 2D-24 on persister cells
Because persister cells are highly tolerant to antibiotics (Kint et al. 2012), we were motivated to also test 2D-24 against persister cells of PAO1 and PDO300 (Fig. 5). It was found that 2D-24 is effective in killing persister cells of both strains, although higher concentrations of 2D-24 than those required to kill normal cells are needed. For example, 200 μM of 2D-24 killed 68.7 ± 6.7 % (p < 0.001) of PAO1 persisters and 89.0 ± 3.2 % (p < 0.001) of PDO300 persisters. At concentrations below 50 μM, 2D-24 exhibited a stronger killing effect on PAO1 persister cells compared to PDO300. For example, at 30 μM, 2D-24 showed 30.3 ± 6.1 % (p < 0.05) killing of PAO1 persister cells, while the same condition did not show any significant killing (p = 0.08) of PDO300 persister cells. Interestingly, there was a sharp increase in the susceptibility of PDO300 persister cells between 30 and 50 μM of 2D-24; the killing increased from 10.71 ± 6.2 to 87.6 ± 3.3 % (p < 0.05) when 2D-24 was added at 30 and 50 μM, respectively. These results suggest that a threshold may exist for the activity of 2D-24 to kill PDO300 persister cells effectively.

Effects of 2D-24 on persister cells of PAO1 (a) and PDO300 (b). Persister cells were isolated by killing normal cells in overnight cultures with 200 μg/mL Cip for 3.5 h
Synergistic effects between 2D-24 and antibiotics
Since some membrane-active AMPs can synergize with traditional antibiotics in bacterial killing (Naghmouchi et al. 2012), we also tested if the activity of 2D-24 can be enhanced through synergy with antibiotics. To achieve this, 1 μg/mL Cip, 1.5 μg/mL Tob, and 0.75 μg/mL Car (which can kill ~0.5 log of normal planktonic cells) were tested in the presence and absence of 2D-24. As shown in Fig. 6, treatment with 1.5 μg/mL Tob or 5 μM 2D-24 alone killed 0.4 log (59.2 ± 0.7 %) and 0.3 log (54.1 ± 1.7 %) of normal planktonic PAO1 cells, respectively. In comparison, co-treatment with both agents caused 2.3 logs of killing (99.6 ± 0.6 %). Similar results were obtained for Cip and Car. Thus, there is indeed a synergy between 2D-24 and these three antibiotics. Co-treatments of normal planktonic cells of PDO300 with antibiotics and 2D-24 gave similar results. Thus, 2D-24 and the tested antibiotics show synergy in killing both strains.

Co-treatment of planktonic PAO1 and PDO300 cells with 2D-24 and antibiotics. Cip, Tob, and Car antibiotics were tested on exponential cultures (OD600 = 0.5). Antibiotics or 2D-24 alone were used as controls
Previously, we reported that some membrane-active AMPs, containing tryptophan and arginine repeats, synergize with ampicillin in killing E. coli persister cells (Chen et al. 2011). Thus, we tested if there is also a synergy in killing persister cells with 2D-24 and antibiotics. The results showed that there are synergies in killing persisters, especially the co-treatment with 2D-24 and Tob (Fig. 7). For example, Tob (100 μg/mL) or 2D-24 (30 μM) alone killed <1.5 % (data now shown) and 0.1 log (20.7 ± 1.4 %) of PAO1 persister cells, respectively. In comparison, co-treatment with these two agents led to 0.58 log (73.8 ± 3.8 %) killing of persister cells. Similar results were obtained for PDO300 persister cells.

Co-treatment of PAO1 and PDO300 persister cells with 2D-24 and antibiotics. Cip, Tob, and Car antibiotics were tested in the presence and absence of 30 μM 2D-24. The persister cells were isolated from overnight cultures by killing normal cells with 200 μg/mL Cip for 3.5 h. Antibiotics were tested at different concentrations (1–1000 μg/mL tested), but only the results of 100 μg/mL are shown. The CFU data of untreated controls in each run were normalized as 100 % for the convenience of data comparison across the samples
Hemolysis assays
To understand if 2D-24 is selective against bacteria, the cytotoxicity on fresh sheep erythrocytes was tested. The results showed that 2D-24 is not toxic to red blood cells even at high concentrations with HD50 > 1 mg/mL (870 μM). Since 2D-24 killed P. aeruginosa cells including biofilm and persister cells significantly at 50 μM or lower, this dendrimer is more selective against bacteria.
Cytotoxicity and effects on PAO1 in a coculture model with IB3-1 lung epithelial cell line
It is encouraging to find that 2D-24 is effective against the biofilm and persister cells of P. aeruginosa. To determine if 2D-24 is safe to mammalian cells at the effective concentrations, we further tested the cytotoxicity of 2D-24 on the IB3-1 lung epithelial cell line. The results showed that 2D-24 is not toxic to IB3-1 cells at concentrations up to 32 μM after treatment for 24 h (Fig. 8), e.g., no significant change in viability was found compared to the 2D-24-free control (p = 0.28, one-way ANOVA). In comparison, when PAO1 cells were treated by 2D-24 at the same concentration in the same medium, significant killing was observed (Fig. 8). For example, 84.9 ± 9.5 % of PAO1 cells were killed by 16 μM 2D-24. Consistently, 2D-24 was found to protect IB3-1 epithelial cells from the infection of PAO1 in a coculture model. In the absence of 2D-24, only 45.7 ± 2.6 % of IB3-1 cells remained viable after 24 h incubation with PAO1. In comparison, the presence of 2D-24 increased the viability of IB3-1 cells in a concentration-dependent manner. For example, 94.7 ± 2.7 and 82.6 ± 5.2 % of IB3-1 cells remained viable when 16 μM 2D-24 was added along with the inoculation of PAO1 and at 4 h after inoculation, respectively.

Cytotoxicity of 2D-24 on IB3-1 cells and the effects in a coculture model. Different 2D-24 concentrations, from 1 to 32 μM, were tested on IB3-1 cells alone (circles), added together with PAO1 inoculation (squares), or 4 h after PAO1 inoculation (triangles). Besides, PAO1 cells alone in LHC-8 medium with no IB3-1 cells (crossings) were treated with the same concentrations of 2D-24 to evaluate bacterial killing. The viability of IB3-1 cells was determined using the LIVE/DEAD staining kit by following the manufacturer’s protocol. The viability of PAO1 was determined by counting CFUs
Discussion
Although more than 5000 AMPs have been identified to date (Zhao et al. 2013), most previous studies of their activities have focused on activities against normal planktonic bacterial cells (Bahar and Ren 2013). In comparison, the effects of AMPs on biofilm cells have been less studied, and few reports exist regarding their effects on persister cells. In this study, a new synthetic AMDP, 2D-24, was prepared and tested against planktonic and biofilm cells, including persister cells of P. aeruginosa PAO1 and PDO300. Significant killing effects were observed for all the cell types tested.
Most synthetic AMPs are designed based on the characteristics of natural AMPs. In general, they are positively charged, amphiphilic molecules (Martin et al. 1995; Zasloff 2002) and act on negatively charged cell walls and membranes, impairing the membrane potential and leading to cell death (Bahar and Ren 2013). AMPs have a wide target spectrum of microbes. It has been postulated that the effectiveness of AMPs on bacteria depends on the peptide/lipid ratio during treatment (Huang 2006). In this study, the killing of PDO300 persister cells increased drastically when the concentration of 2D-24 reached 30 μM, which indicates that the killing may be dependent on peptide/lipid ratio as well. However, the biofilm and normal planktonic cells of PDO300 and all types of PAO1 cells tested here did not show a threshold concentration of 2D-24 for killing. It will be helpful to further investigate if persister formation affects the interaction between 2D-24 and the cell membrane of this mucoid strain.
Biofilms are composed of bacterial populations that include cells at different stages embedded in a complex extracellular matrix (Lam et al. 1980); cells in biofilms can be up to 1000 times more tolerant to antibiotics than their planktonic counterparts (Olson et al. 2002). Intriguingly, 2D-24 appeared to be equally effective against normal planktonic cells and biofilm cells of PAO1 and PDO300. This finding suggests that 2D-24 can penetrate biofilm matrix and is effective in killing biofilm cells. In comparison, persister cells tolerated higher concentrations of 2D-24. Overall, concentrations higher than 20 μM of 2D-24 were found sufficient to kill more than 80 % of both planktonic and biofilm cells of PAO1 and PDO300, while concentrations higher than 50 μM are required to achieve a similar level of killing of persister cells of these strains. Interestingly, when treated with higher concentrations of 2D-24, persister cells of PDO300 were slightly more susceptible than PAO1 persister cells. For example, 50 and 200 μM 2D-24 exhibited 19.8 and 57.3 % more killing of PDO300 persister cells compared to PAO1, respectively.
Mucoid conversion with overproduction of alginate is thought to play an important role in chronic infections by P. aeruginosa (Pritt et al. 2007) since mucoid strains are generally more tolerant to antibiotics than the wild-type strain (Hengzhuang et al. 2011; Hentzer et al. 2001). It is interesting that the killing effects of 2D-24 on biofilm cells of PDO300 and PAO1 were similar. This suggests that 2D-24 is able to penetrate alginate and interact with PDO300 cellular targets.
Besides the killing of P. aeruginosa cells by 2D-24 alone, intriguing synergistic effects were observed when the cells were treated with 2D-24 and Cip, Tob, or Car, which are antibiotics targeting DNA, protein, and cell wall synthesis, respectively. Similar synergistic effects were observed for killing both PAO1 and PDO300 normal planktonic and persister cells (Figs. 6 and 7). Among these antibiotics, the strongest synergy was observed for Tob, while Cip exhibited lower synergy. This is probably because the persister cells had been exposed to Cip during persister isolation. The mechanism of synergistic killing of bacteria by 2D-24 and antibiotics is unknown. Most AMPs target cell membrane and thus alter the membrane potential (Bahar and Ren 2013). We speculate that membrane disruption, depolarization, or permeabilization by 2D-24 can lead to reduced membrane potential along with lower energy production. This may favor the penetration of cell membrane by some antibiotics, which is consistent with a recent study by Schmidt et al. (2014) showing a peptide-tobramycin conjugate -Pentobra- can effectively enter the cells of P. aeruginosa, E. coli, and Staphylococcus aureus. The peptide domain of this molecule is rich in arginine and tryptophan residues (RQIKIWFQNRRW), similar to the amino acid content of 2D-24.
We speculate that the increase in intracellular antibiotic concentration along with membrane damage by 2D-24 may render the persister cells unable to wake up, leading to cell death. Alternatively, it may also be possible that the stress response to 2D-24 treatment could activate certain cellular activities targeted by antibiotics and thus increase the antibiotic susceptibility of persister cells. It will be interesting to test the effects of 2D-24 on the integrity of treated cells and compare the penetration of antibiotics through cell membrane in the presence and absence of 2D-24.
It is generally appreciated that AMPs and analogs can have more than one inactivating target in bacteria. Even though persister cells are known to be latent and different from normal active cells, they still need to maintain intact cell membrane structures. While the cell surface and membrane are generally implicated in the antibacterial action by AMPs, we still lack a complete understanding of the detailed mechanism(s) involved. Similar to bactericidal antibiotics, we recently reported that a different AMDP analog, (RW)4D, generates hydroxide radicals in target bacterial cells via a Fenton reaction (Marchand et al. 2006). It will be interesting to study the interaction of 2D-24 with cellular targets among regular planktonic, persister, and biofilm cells of P. aeruginosa.
In addition, we obtained data showing that 2D-24 is effective in killing P. aeruginosa at concentrations not toxic to IB3-1 epithelial cells. Similar to the uninfected control, the viability of IB3-1 cells increased to near 90 % when 2D-24 is added into the culture medium together with PAO1 at inoculation. This finding is intriguing since cytotoxicity is a major challenge in bacterial control with AMPs. Further in vivo tests will help evaluate the clinical potential of this new dendrimer.
In summary, a new antimicrobial dendrimer 2D-24 was synthesized in this study and tested for its effects on the biofilm and planktonic cells (including persister cells) of PAO1 and its mucoid mutant PDO300. Similar killing activities were observed for regular planktonic and biofilm cells of both strains, and this agent was also found to kill persister cells of both strains. Synergy between 2D-24 and antibiotics in killing P. aeruginosa was also observed. Further testing using sheep erythrocytes and cocultures of PAO1 and human IB3-1 cells demonstrated that 2D-24 is effective against bacteria at concentrations nontoxic to mammalian cells. Thus, our in vitro data provide encouraging evidence for potential application of 2D-24 in treatment of chronic infections caused by P. aeruginosa. In addition to the investigation of mechanistic aspects of 2D-24 action, animal studies will be needed to demonstrate its efficacy in vivo.
References
Alhoot MA, Rathinam AK, Wang SM, Manikam R, Sekaran SD (2013) Inhibition of dengue virus entry into target cells using synthetic antiviral peptides. Int J Med Sci 10:719–729
Andersson C, Al-Turkmani MR, Savaille JE, Alturkmani R, Katrangi W, Cluette-Brown JE, Zaman MM, Laposata M, Freedman SD (2008) Cell culture models demonstrate that CFTR dysfunction leads to defective fatty acid composition and metabolism. J Lipid Res 49:1692–1700
Arrighi RB, Nakamura C, Miyake J, Hurd H, Burgess JG (2002) Design and activity of antimicrobial peptides against sporogonic-stage parasites causing murine malarias. Antimicrob Agents Chemother 46:2104–2110
Bahar AA, Ren D (2013) Antimicrobial peptides. Pharmaceuticals 6:1543–1575
Bodey GP, Bolivar R, Fainstein V, Jadeja L (1983) Infections caused by Pseudomonas aeruginosa. Rev Infect Dis 5:279–313
Butler K, English AR, Ray VA, Timreck AE (1970) Carbenicillin: chemistry and mode of action. J Infect Dis 122(Suppl):S1–S8
Chen X, Zhang M, Zhou CH, Kallenbach NR, Ren DC (2011) Control of bacterial persister cells by Trp/Arg-containing antimicrobial peptides. Appl Environ Microbiol 77:4878–4885
Costerton JW, Cheng KJ, Geesey GG, Ladd TI, Nickel JC, Dasgupta M, Marrie TJ (1987) Bacterial biofilms in nature and disease. Annu Rev Microbiol 41:435–464
Costerton JW, Stewart PS, Greenberg EP (1999) Bacterial biofilms: a common cause of persistent infections. Science 284:1318–1322
de Bentzmann S, Plesiat P (2011) The Pseudomonas aeruginosa opportunistic pathogen and human infections. Environ Microbiol 13:1655–1665
DeVries CA, Ohman DE (1994) Mucoid-to-nonmucoid conversion in alginate-producing Pseudomonas aeruginosa often results from spontaneous mutations in algT, encoding a putative alternate sigma factor, and shows evidence for autoregulation. J Bacteriol 176:6677–6687
Drlica K, Zhao X (1997) DNA gyrase, topoisomerase IV, and the 4-quinolones. Microbiol Mol Biol Rev 61:377–392
Flemming HC, Wingender J (2010) The biofilm matrix. Nat Rev Microbiol 8:623–633
Gilbert P, Das J, Foley I (1997) Biofilm susceptibility to antimicrobials. Adv Dent Res 11:160–167
Hall-Stoodley L, Costerton JW, Stoodley P (2004) Bacterial biofilms: from the natural environment to infectious diseases. Nat Rev Microbiol 2:95–108
Hancock REW, Sahl HG (2006) Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat Biotechnol 24:1551–1557
Haug BE, Stensen W, Kalaaji M, Rekdal O, Svendsen JS (2008) Synthetic antimicrobial peptidomimetics with therapeutic potential. J Med Chem 51:4306–4314
He J, Eckert R, Pharm T, Simanian MD, Hu CH, Yarbrough DK, Qi FX, Anderson MH, Shi WY (2007) Novel synthetic antimicrobial peptides against Streptococcus mutans. Antimicrob Agents Chemother 51:1351–1358
Hengzhuang W, Wu H, Ciofu O, Song Z, Hoiby N (2011) Pharmacokinetics/pharmacodynamics of colistin and imipenem on mucoid and nonmucoid Pseudomonas aeruginosa biofilms. Antimicrob Agents Chemother 55:4469–4474
Hentzer M, Teitzel GM, Balzer GJ, Heydorn A, Molin S, Givskov M, Parsek MR (2001) Alginate overproduction affects Pseudomonas aeruginosa biofilm structure and function. J Bacteriol 183:5395–5401
Hou SY, Zhou CH, Liu ZG, Young AW, Shi ZH, Ren DC, Kallenbach NR (2009) Antimicrobial dendrimer active against Escherichia coli biofilms. Bioorg Med Chem Lett 19:5478–5481
Hou SY, Liu ZG, Young AW, Mark SL, Kallenbach NR, Ren DC (2010) Effects of Trp- and Arg-containing antimicrobial-peptide structure on inhibition of Escherichia coli planktonic growth and biofilm formation. Appl Environ Microbiol 76:1967–1974
Huang HW (2006) Molecular mechanism of antimicrobial peptides: the origin of cooperativity. Biochim Biophys Acta 1758:1292–1302
Keren I, Shah D, Spoering A, Kaldalu N, Lewis K (2004) Specialized persister cells and the mechanism of multidrug tolerance in Escherichia coli. J Bacteriol 186:8172–8180
Kim JY, Park SC, Yoon MY, Hahm KS, Park Y (2011) C-terminal amidation of PMAP-23: translocation to the inner membrane of Gram-negative bacteria. Amino Acids 40:183–195
Kint CI, Verstraeten N, Fauvart M, Michiels J (2012) New found fundamentals of bacterial persistence. Trends Microbiol 20:577–585
Lam J, Chan R, Lam K, Costerton JW (1980) Production of mucoid microcolonies by Pseudomonas aeruginosa within infected lungs in cystic fibrosis. Infect Immun 28:546–556
Le Goffic F, Capmau ML, Tangy F, Baillarge M (1979) Mechanism of action of aminoglycoside antibiotics. Binding studies of tobramycin and its 6′-N-acetyl derivative to the bacterial ribosome and its subunits. Eur J Biochem 102:73–81
Lee DG, Hahm KS, Shin SY (2004) Structure and fungicidal activity of a synthetic antimicrobial peptide, P18, and its truncated peptides. Biotechnol Lett 26:337–341
Lewis K (2007) Persister cells, dormancy and infectious disease. Nat Rev Microbiol 5:48–56
Lewis K (2010) Persister cells. Annu Rev Microbiol 64:357–372
Lewis K (2013) Platforms for antibiotic discovery. Nat Rev Drug Discov 12:371–387
Liu ZG, Young AW, Hu P, Rice AJ, Zhou CH, Zhan YK, Kallenbach NR (2007) Tuning the membrane selectivity of antimicrobial peptides by using multivalent design. Chembiochem 8:2063–2065
Lowbury EJL, Kidson A, Lilly HA, Ayliffe GAJ (1969) Sensitivity of Pseudomonas aeruginosa to antibiotics—emergence of strains highly resistant to carbenicillin. Lancet 2:448–452
Ma L, Conover M, Lu H, Parsek MR, Bayles K, Wozniak DJ (2009) Assembly and development of the Pseudomonas aeruginosa biofilm matrix. PLoS Pathog 5:e1000354
Mah TF, O'Toole GA (2001) Mechanisms of biofilm resistance to antimicrobial agents. Trends Microbiol 9:34–39
Marchand C, Krajewski K, Lee HF, Antony S, Johnson AA, Amin R, Roller P, Kvaratskhelia M, Pommier Y (2006) Covalent binding of the natural antimicrobial peptide indolicidin to DNA abasic sites. Nucleic Acids Res 34:5157–5165
Martin E, Ganz T, Lehrer RI (1995) Defensins and other endogenous peptide antibiotics of vertebrates. J Leukoc Biol 58:128–136
Mathee K, Ciofu O, Sternberg C, Lindum PW, Campbell JI, Jensen P, Johnsen AH, Givskov M, Ohman DE, Molin S, Hoiby N, Kharazmi A (1999) Mucoid conversion of Pseudomonas aeruginosa by hydrogen peroxide: a mechanism for virulence activation in the cystic fibrosis lung. Microbiology 145:1349–1357
McDougald D, Rice SA, Barraud N, Steinberg PD, Kjelleberg S (2012) Should we stay or should we go: mechanisms and ecological consequences for biofilm dispersal. Nat Rev Microbiol 10:39–50
Mulcahy LR, Burns JL, Lory S, Lewis K (2010) Emergence of Pseudomonas aeruginosa strains producing high levels of persister cells in patients with cystic fibrosis. J Bacteriol 192:6191–6199
Naghmouchi K, Le Lay C, Baah J, Drider D (2012) Antibiotic and antimicrobial peptide combinations: synergistic inhibition of Pseudomonas fluorescens and antibiotic-resistant variants. Res Microbiol 163:101–108
Niepa TH, Gilbert JL, Ren D (2012) Controlling Pseudomonas aeruginosa persister cells by weak electrochemical currents and synergistic effects with tobramycin. Biomaterials 33:7356–7365
Noto PB, Abbadessa G, Cassone M, Mateo GD, Agelan A, Wade JD, Szabo D, Kocsis B, Nagy K, Rozgonyi F, Otvos L Jr (2008) Alternative stabilities of a proline-rich antibacterial peptide in vitro and in vivo. Protein Sci 17:1249–1255
Ojha AK, Baughn AD, Sambandan D, Hsu T, Trivelli X, Guerardel Y, Alahari A, Kremer L, Jacobs WR Jr, Hatfull GF (2008) Growth of Mycobacterium tuberculosis biofilms containing free mycolic acids and harbouring drug-tolerant bacteria. Mol Microbiol 69:164–174
Olson ME, Ceri H, Morck DW, Buret AG, Read RR (2002) Biofilm bacteria: formation and comparative susceptibility to antibiotics. Can J Vet Res 66:86–92
Pan J, Bahar AA, Syed H, Ren D (2012) Reverting antibiotic tolerance of Pseudomonas aeruginosa PAO1 persister cells by (Z)-4-bromo-5-(bromomethylene)-3-methylfuran-2(5H)-one. PLoS One 7:e45778
Powell WA, Catranis CM, Maynard CA (1995) Synthetic antimicrobial peptide design. Mol Plant Microbe Interact 8:792–794
Pritt B, O'Brien L, Winn W (2007) Mucoid Pseudomonas in cystic fibrosis. Am J Clin Pathol 128:32–34
Romero R, Schaudinn C, Kusanovic JP, Gorur A, Gotsch F, Webster P, Nhan-Chang CL, Erez O, Kim CJ, Espinoza J, Goncalves LF, Vaisbuch E, Mazaki-Tovi S, Hassan SS, Costerton JW (2008) Detection of a microbial biofilm in intraamniotic infection. Am J Obstet Gynecol 198(135):e131–e135
Sambrook J, Russell DW (2001) Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor
Schmidt NW, Deshayes S, Hawker S, Blacker A, Kasko AM, Wong GC (2014) Engineering persister-specific antibiotics with synergistic antimicrobial functions. ACS Nano 8:8786–8793
Schurr MJ, Yu H, Martinez-Salazar JM, Boucher JC, Deretic V (1996) Control of AlgU, a member of the sigma E-like family of stress sigma factors, by the negative regulators MucA and MucB and Pseudomonas aeruginosa conversion to mucoidy in cystic fibrosis. J Bacteriol 178:4997–5004
Tam JP, Lu YA, Yang JL (2002) Antimicrobial dendrimeric peptides. Eur J Biochem 269:923–932
Uteng M, Hauge HH, Markwick PRL, Fimland G, Mantzilas D, Nissen-Meyer J, Muhle-Goll C (2003) Three-dimensional structure in lipid micelles of the pediocin-like antimicrobial peptide sakacin P and a sakacin P variant that is structurally stabilized by an inserted C-terminal disulfide bridges. Biochemistry 42:11417–11426
Wang G, Li X, Wang Z (2009) APD2: the updated antimicrobial peptide database and its application in peptide design. Nucleic Acids Res 37:D933–D937
Winsor GL, Lam DK, Fleming L, Lo R, Whiteside MD, Yu NY, Hancock RE, Brinkman FS (2011) Pseudomonas genome database: improved comparative analysis and population genomics capability for Pseudomonas genomes. Nucleic Acids Res 39:D596–D600
Zasloff M (2002) Antimicrobial peptides of multicellular organisms. Nature 415:389–395
Zhao X, Wu H, Lu H, Li G, Huang Q (2013) LAMP: a database linking antimicrobial peptides. PLoS One 8:e66557
Acknowledgments
We thank the US National Science Foundation (CAREER-1055644) for supporting this work. We are grateful to Prof. Matthew R. Parsek at the University of Washington for sharing P. aeruginosa PAO1 and PDO300 and Prof. Rebecca Bader at Syracuse University for sharing the IB3-1 cells.
Conflict of interest
The authors declare no conflict of interest.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Ali Adem Bahar and Zhigang Liu contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 414 kb)
Rights and permissions
About this article

Cite this article
Bahar, A.A., Liu, Z., Totsingan, F. et al. Synthetic dendrimeric peptide active against biofilm and persister cells of Pseudomonas aeruginosa . Appl Microbiol Biotechnol 99, 8125–8135 (2015). https://doi.org/10.1007/s00253-015-6645-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-015-6645-7