
Abstract
A recombinant putative lipoxygenase from Burkholderia thailandensis with a specific activity of 26.4 U mg−1 was purified using HisTrap affinity chromatography. The native enzyme was a 75-kDa dimer with a molecular mass of 150 kDa. The enzyme activity and catalytic efficiency (k cat/K m) were the highest for linoleic acid (k cat of 93.7 s−1 and K m of 41.5 μM), followed by arachidonic acid, α-linolenic acid, and γ-linolenic acid. The enzyme was identified as an omega-6 linoleate lipoxygenase (or a linoleate 13S-lipoxygenase) based on genetic and HPLC analyses as well as substrate specificity. The reaction conditions for the enzymatic production of 13-hydroxy-9,11(Z,E)-octadecadienoic acid (13-HODE) were optimal at pH 7.5, 25 °C, 20 g l−1 linoleic acid, 2.5 g l−1 enzyme, 0.1 mM Cu2+, and 6 % (v/v) methanol. Under these conditions, linoleate 13-lipoxygenase from B. thailandensis produced 20.8 g l−1 13-HODE (70.2 mM) from 20 g l−1 linoleic acid (71.3 mM) for 120 min, with a molar conversion yield of 98.5 % and productivity of 10.4 g l−1 h−1. The molar conversion yield and productivity of 13-HODE obtained using B. thailandensis lipoxygenase were 151 and 158 % higher, respectively, than those obtained using commercial soybean lipoxygenase under the optimum conditions for each enzyme at the same concentrations of substrate and enzyme.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Lipoxygenases (EC 1.13.11) are a family of dioxygenases that catalyze regio- and stereo-specific dioxygenation of polyunsaturated fatty acids with one or several cis, cis-1,4-pentadiene units to form hydroperoxy fatty acids, which reduce to hydroxy fatty acids (Joo and Oh 2012). Linoleate 13-lipoxygenase (EC 1.13.11.12), a non-heme-iron-containing enzyme with 896–941 amino acids, can convert linoleic acid to 13-hydroperoxy-9,11(Z,E)-octadecadienoic acid (13-HPOD) (Bannenberg et al. 2009; Chohany et al. 2011), which can be easily reduced to 13-hydroxy-9,11(Z,E)-octadecadienoic acid (13-HODE) by reducing agents. Linoleate 13-lipoxygenases exist in a wide variety of plants, and they have been reported in only two microorganisms such as Gaeumannomyces graminis tritici (Villaverde et al. 2013b) and Pseudomonas aeruginosa (Villaverde et al. 2013a).
13-HPOD, produced mostly by soybean linoleate 13-lipoxygenase (Iacazio et al. 1990), has been used for the production of green note aroma compounds such as hexanal and hexanol by hydroxyperoxide lyase and alcohol dehydrogenase (Buchhaupt et al. 2012). 13-HODE, a reduced form of 13-HPOD, can be used as a preservative emulsifier in the cosmetics industry because hydroxy unsaturated fatty acids have antifungal activities against Aspergillus niger, a typical contaminant fungus in cosmetic products (Black et al. 2013), and reduce surface tension (Parra et al. 1990). Therefore, the increased production of 13-HODE should be achieved. However, 13-HODE (or 13-HPOD) production by soybean linoleate 13-lipoxygenase, thus far, has proven inefficient in terms of specific activity. To produce 13-HODE with greater efficiency, a linoleate 13-lipoxygenase with a higher catalytic efficiency is required.
In the present study, we identified a putative lipoxygenase from Burkholderia thailandensis that has high catalytic efficiency for linoleic acid. The gene encoding this putative lipoxygenase was cloned and expressed in Escherichia coli. The expressed enzyme was identified as a linoleate 13S-lipoxygenase based on genetic and HPLC analyses as well as substrate specificity. The reaction conditions such as pH, temperature, metal ions, solvent, and concentrations of enzyme and substrate were optimized for the production of 13-HODE from linoleic acid. Under the optimized conditions, 13-HODE production using the enzyme was performed.
Materials and methods
Materials
Commercial soybean lipoxygenase as well as the fatty acid standards linoleic acid, arachidonic acid, α-linolenic acid, and γ-linolenic acid were purchased from Sigma (St. Louis, MO, USA). 9-HODE, 13S-HODE, and 13R-HODE standards were purchased from Cayman Chemical (Ann Arbor, MI, USA).
Bacterial strains, plasmid, and culture conditions
B. thailandensis KCTC 23190 (Korean Collection for Type Cultures, Deajeon, Republic of Korea), which was the same strain as B. thailandensis E264 (Haraga et al. 2008), was used as the source of genomic DNA encoding the putative lipoxygenase. E. coli ER2566 and pET-28a plasmid were host cells and expression vector, respectively. Recombinant E. coli cells were cultivated in a 2000-ml flask containing 500 ml of Luria-Bertani (LB) medium and 20 μg ml−1 kanamycin at 37 °C with shaking at 200 rpm. When the optical density of the bacterial culture at 600 nm reached 0.6, isopropyl-β-d-thiogalactopyranoside was added to a final concentration of 0.1 mM to induce enzyme expression, and the culture was then incubated with shaking at 150 rpm at 16 °C for 16 h.
Gene cloning
The gene encoding the putative lipoxygenase was amplified by PCR using B. thailandensis genomic DNA as a template. The primers used for gene cloning were designed based on the DNA sequence of a putative lipoxygenase from B. thailandensis (GenBank accession number, YP_442874). Forward (5′-GCTAGCATGGTCAATCACAAAACCGG-3′) and reverse primers (5′-AAGCTTTCAAATGTTCGTGCTTGCCG-3′) were designed to introduce the NheI and HindIII restriction sites (underlined), respectively, and were synthesized by Bioneer (Daejon, Korea). The DNA fragment obtained by PCR amplification using pfu polymerase (Solgent, Daejon, Korea) was extracted using a gel extraction kit (Promega, Madison, WI, USA) and was cloned into pGEM-T Easy vector (Promega). The NheI-HindIII fragment harboring the gene encoding the putative lipoxygenase was subcloned from the pGEM-T Easy vector into the same sites of pET-28a. The resulting plasmid was transformed into E. coli ER2566, which was plated on LB agar containing 20 μg ml−1 kanamycin. A kanamycin-resistant colony was selected, and plasmid DNA from the transformant was isolated using a plasmid purification kit (Promega). DNA sequencing was performed at the Macrogen facility (Seoul, Korea).
Homology modeling
Homology modeling of lipoxygenase from B. thailandensis was performed using Build Homology Models module in the MODELER application of Discoversy Studio 4.1 (Accelrys Software, San Diego, CA, USA) using the crystal structure of linoleate 13-lipoxygenase from P. aeruginosa (Protein Data Bank (PDB) entry 4G32) as a template. Comparative modeling was used to generate the most probable structure of the query protein by aligning it with the template sequence, simultaneously considering spatial restraints, and local molecular geometry. The generated structure was improved by subsequent refinement of the loop conformations by assessing the compatibility of amino acid sequences with known PDB structures (Protein Health module, DS 4.1). The geometry of loop region was corrected using the Refine Loop/MODELER, and the best model was chosen. To confirm consistency of the model, the quality of the model was analyzed by PROCHECK software as a different validation tool. Hydrogen atoms were added to the model; these atoms were minimized to have stable energy conformation and to relax the conformation from close contacts.
Enzyme purification
Washed recombinant cells were resuspended in 50 mM phosphate buffer (pH 8.0) containing 300 mM NaCl, 10 mM imidazole, and 0.1 mM phenylmethylsulfonyl fluoride as a protease inhibitor. Resuspended cells were disrupted using a sonicator at 18 W on ice for 2 min. The cell debris was removed by centrifugation at 13,000×g for 20 min at 4 °C, and the supernatant was filtered through a 0.45-μm pore size filter. The filtrate was applied to a HisTrap HP chromatography column (Amersham Biosciences, Uppsala, Sweden) equilibrated with 50 mM phosphate buffer (pH 8.0) containing 300 mM NaCl. The column was washed extensively with the same buffer, and the bound protein was eluted with a linear gradient of 10–250 mM imidazole at a flow rate of 1 ml min−1. The active fractions were collected and dialyzed against 50 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) buffer (pH 7.5). After dialysis, the resulting solution was used as the purified enzyme. The purification step using the column was carried out in a cold room at 4 °C using a fast protein liquid chromatography system (Bio-Rad Laboratories, Hercules, CA, USA).
Circular dichroism
Circular dichroism (CD) spectra of the enzymes without the addition of metal ions and with 0.1 mM Cu2+ were recorded with a spectrophotometer (Jasco J-815, Tokyo, Japan) using a 0.5-cm path length quartz cell. Acquisition of far-UV (190–250 nm) CD spectra was performed in distilled water containing 3 mg ml−1 enzyme at 25 °C with a rate of 100 nm min−1. CD spectra were corrected for base line, and ellipticity for each sample was expressed in millidegrees.
Molecular mass determination
The subunit molecular mass of the putative lipoxygenase was examined using SDS-PAGE. The molecular mass of the native enzyme was determined by gel filtration chromatography using a Sephacryl S-300 HR 16/60 preparative-grade column (Amersham Biosciences). The enzyme solution was applied to the column and eluted with 50 mM Tris-HCl buffer (pH 7.5) containing 150 mM NaCl at a flow rate of 1 ml min−1. The column was calibrated with ferritin (440 kDa), catalase (232 kDa), aldolase (158 kDa), conalbumin (75 kDa), ovalbumin (44 kDa), and carbonic anhydrase (29 kDa) as reference proteins (Amersham Biosciences).
Determination of specific activity and kinetic parameters
The specific activity and kinetic parameters were determined using a Beckman Coulter DU-700 spectrophotometer (Brea, CA, USA). The lipoxygenase activity was assayed at 25 °C by the increase in absorbance at 234 nm. An extinction coefficient of 25,000 M−1 cm−1 was used to calculate the activity of the enzyme for 13-HPOD (Gibian and Vandenberg 1987; Graff et al. 1990). Unless otherwise stated, the reaction was performed in 50 mM HEPES (pH 7.5) buffer containing 0.1 mM substrate, 0.05 μg ml−1 enzyme, 0.1 mM Cu2+, and 6 % (v/v) methanol at 25 °C for 2 min. One unit of activity was defined as the amount of enzyme required to produce 1 μmol min−1 13-HPOD at 25 °C and pH 7.5. The specific activity was defined as the amount of product per amount of protein per unit reaction time. Various amounts of unsaturated fatty acid (10–200 μM) were used to determine the kinetic parameters of the enzyme; the reactions were performed in 50 mM HEPES (pH 7.5) at 25 °C for 1 min. The K m (μM) and k cat (s−1) were calculated using the enzyme concentration and Lineweaver-Burk plot of the Michaelis-Menten equation.
Effects of pH, temperature, metal ions, and solvent on lipoxygenase activity
To examine the effects of pH and temperature on the activity of B. thailandensis lipoxygenase, the pH was varied from 6.5 to 8.5 using 50 mM HEPES buffer (pH 6.5–7.5) and 50 mM Tris-HCl buffer (pH 7.0–8.5) at a constant temperature of 25 °C, and the temperature was varied from 10 to 40 °C at a constant pH of 7.5. The reactions were performed in the buffer solution containing 0.1 mM substrate and 0.05 μg ml−1 enzyme for 2 min. The effect of temperature on enzyme stability was monitored as a function of incubation time by incubating the enzyme reaction solutions at different temperatures ranging from 20 to 40 °C in 50 mM HEPES buffer solution (pH 7.5). The experimental data for enzyme inactivation were fitted to a first-order curve, and the half-life of the enzyme was calculated using SigmaPlot 9.0 (Systat Software, San Jose, CA, USA).
To investigate the effect of metal ions on the activity of lipoxygenase from B. thailandensis, the reactions were performed in 50 mM HEPES (pH 7.5) buffer containing 0.1 mM linoleic acid, 0.05 μg ml−1 enzyme, and 0.1 mM or 1 mM metal ions (Fe2+, Fe3+, Ca2+, Zn2+, Co2+, Mg2+, Mn2+, Ni2+, or Cu2+) at 25 °C for 2 min. The effect of Cu2+ concentration was investigated in the range of 0–0.25 mM. The effect of solvent on the activity of lipoxygenase from B. thailandensis was evaluated using methanol, ethanol, pentane, ethyl acetate, iso-propanol, dimethyl sulfoxide, iso-octane, butanol, and toluene. The reactions were performed in 50 mM HEPES (pH 7.5) buffer containing 0.1 mM linoleic acid, 0.05 μg ml−1 enzyme, and 2 or 6 % (v/v) solvents at 25 °C. The effect of methanol concentration on lipoxygenase activity was investigated by varying its concentration from 0 to 10 % (v/v).
Measurement of the activities of linoleate 13-lipoxygenases from B. thainlandensis and soybean
The activity of linoleate 13-lipoxygenase from B. thainlandensis was measured in 50 mM HEPES (pH 7.5) buffer containing 0.1 mM linoleic acid, 0.1 μg ml−1 enzyme, 0.1 mM Cu2+, and 6 % (v/v) methanol at 25 °C for 3 min. The activity of soybean linoleate 13-lipoxygenase was determined in 50 mM 4-(2-hydroxyethyl)-1-piperazinepropanesulfonic acid (EPPS) (pH 8.5) buffer containing 0.1 mM linoleic acid, 0.1 μg ml−1 enzyme, and 0.01 mM Tween 40 at 25 °C for 3 min.
Optimization of enzyme and substrate concentrations for 13-HODE production
To determine the optimal concentration of enzyme, the enzyme concentration was varied from 0 to 3.0 g l−1. The reactions were performed at 25 °C for 1 h in 50 mM HEPES (pH 7.5) buffer containing 20 g l−1 linoleic acid, 0.1 mM Cu2+, and 6 % (v/v) methanol with shaking at 200 rpm in a 100 ml baffled flask containing a working volume of 10 ml. To decide the optimal concentration of substrate, the substrate concentration was varied from 5 to 30 g l−1. The reactions were performed in 50 mM HEPES (pH 7.5) buffer containing 2.5 g l−1 enzyme, 0.1 mM Cu2+, and 6 % (v/v) methanol at 25 °C for 1 h.
13-HODE production by linoleate 13-lipoxygenase from B. thailandensis and soybean under optimized conditions
13-HODE production by B. thailandensis linoleate 13-lipoxygenase was performed in 50 mM HEPES (pH 7.5) buffer containing 20 g l−1 linoleic acid, 2.5 g l−1 enzyme, 0.1 mM Cu2+, and 6 % (v/v) methanol at 25 °C for 2 h. 13-HODE production by soybean linoleate 13-lipoxygenase was performed in 50 mM EPPS (pH 8.5) buffer containing 20 g l−1 linoleic acid, 2.5 g l−1 enzyme, and 0.01 mM Tween 40 at 20 °C for 1 h. After the reactions, 13-HPOD in the reaction solutions was reduced to 13-HODE by adding one reaction volume of 5 % NaBH4 dissolved in 0.2 N NaOH solution and incubating for 10 min. The reaction mixture was extracted with an equal volume of ethyl acetate. The solvent was removed from the extract using a rotary evaporator.
Analytical methods
The concentrations of linoleic acid and 13-HODE were determined using an HPLC (Agilent 1100, Santa Clara, CA, USA) with a UV detector at a detection wavelength of 234 nm and a reverse phase Nucleosil C18 column (3.2 × 150 mm; Phenomenex, Torrance, CA, USA). The column was eluted at 40 °C using a gradient of solvent A (acetonitrile/water/acetic acid, 50/50/0.1, v/v/v) and solvent B (acetonitrile/acetic acid, 100/0.1, v/v) as follows: 100 % solvent A at a flow rate of 0.25 ml min−1 for 0–5 min; solvent B at 0.25 ml min−1 for 5–21 min; at 0.4 ml min−1 for 21–22 min; 100 % solvent B at 0.4 ml min−1 for 22–27 min; solvent A at 0.4 ml min−1 for 27–32 min; and 100 % solvent A at 0.25 ml min−1 for 32–35 min. The chirality of 13-HODE was determined using the HPLC with a chiral phase Chirlcel OD-H column (2.1 × 150 mm; Daicel, West Chester, PA, USA). The column was eluted at a flow rate of 0.2 ml min−1 with a solvent system of n-hexane/2-propanol/acetic acid (88/12/0.1, v/v/v) at 30 °C.
Results
Gene cloning and molecular mass of the putative lipoxygenase from B. thailandensis
The gene (2,088 bp) encoding a putative lipoxygenase from B. thailandensis, with the same sequence as that reported in GenBank (YP_442874), was cloned and expressed in E. coli. The enzyme was purified from crude extract obtained from harvested cells by HisTrap HP affinity chromatography. The expressed protein was almost expressed in soluble form, but inclusion body was little observed. The putative lipoxygenase from B. thailandensis was purified with a purification fold of 10, yield of 35 %, and specific activity of 26.4 U mg−1. The purified protein showed a single band in SDS-PAGE with a molecular mass of approximately 75 kDa (Fig. 1a), which is consistent with the value calculated based on the 695 amino acids and 6 histidine residues. The molecular mass of the native protein was determined to be 150 kDa by gel filtration chromatography (Fig. 1b), indicating that it is a dimer.

SDS-PAGE analysis and molecular mass determination of the putative lipoxygenase from B. thailandensis. a SDS-PAGE stained with Coomassie blue of the putative lipoxygenase from B. thailandensis. M, molecular mass marker proteins (180,135, 100, 75, 63, 48, 35, 25, 17, and 11 kDa); lane 1, crude extract; lane 2, pellet; and lane 3, purified linoleate 13-lipoxygenase. b Determination of molecular mass of the native enzyme purified using Sephacryl S-300 HR gel filtration chromatography. The reference proteins (filled circle) were ferritin (440 kDa), catalase (232 kDa), aldolase (158 kDa), conalbumin (75 kDa), ovalbumin (44 kDa), and carbonic anhydrase (29 kDa). The putative lipoxygenase eluted at a position corresponding to 150 kDa (empty circle)
The amino acid sequence of the putative lipoxygenase from B. thailandensis was aligned with those of the characterized linoleate 13-lipoxygenases from soybean and P. aeruginosa, the characterized linoleate 9-lipoxygenase from Acaryochloris marina, and the characterized linoleate 8-lipoxygenase from Plexaura homomalla (Fig. 2). The putative lipoxygenase from B. thailandensis exhibited 49.2, 20.3, and 19.6 % identity in amino acid sequence to the characterized linoleate 13-lipoxygenases from P. aeruginosa, soybean, and G. graminis tritici, respectively. Metal-binding residues (HHHNI) (Liavonchanka and Feussner 2006) of the putative lipoxygenase from B. thailandensis were completely conserved. A homology model of the new enzyme was constructed and compared with the determined structure of soybean lipoxygenase (Fig. S1). Nine residues of the active-site residues within a sphere of 7-Å radius centered on Fe2+ were conserved. However, Leu389, Thr693, and Asn694 of B. thailandensis lipoxygenase were corresponded to Try500, Ile837, and Ser838 of soybean lipoxygenase, respectively. Coffa site, a determinant site for isomer chirality, is Ala for S-lipoxygenase and Gly for R-lipoxygenase (Coffa and Brash 2004), and the positional isomer residue in linoleate 13-lipoxygenase is Phe (Sloane et al. 1991). Coffa site and positional isomer residue of lipoxygenase from B. thailandensis were Ala and Phe, respectively, indicating that the enzyme was linoleate 13S-lipoxygenase.

Amino acid sequence alignment of lipoxygenases. The GenBank accession numbers of the aligned lipoxygenase sequences are as follows: soybean 13-lipoxygenase (SB 13LOX), P08170; Pseudomonas aeruginosa 13-lipoxygenase (PA 13LOX), Q8RNT4; Acaryochloris marina 9-lipoxygenase (AM 9LOX), B0C6P0; Plexaura homomalla 8-lipoxygenase (PH 8LOX), O16025; and B. thailandensis 13-lipoxygenase (BT 13LOX), Q2SW25. The metal-binding residues are indicated in bold and boxed. The positional isomer and Coffa site residues are indicated in bold with black, white, and gray backgrounds, respectively. Asterisk, colon, and dot indicate identity, conserved substitutions, and semi-conserved substitution, respectively
Substrate specificity of the putative lipoxygenase from B. thailandensis
The substrate specificity of B. thailandensis lipoxygenase for fatty acid was investigated by measuring the increase in absorbance at 234 nm. The specific activity and k cat/K m were the highest for linoleic acid (C18:2Δ9Z,12Z), followed by arachidonic acid (20:4Δ5Z,8Z,11Z,14Z), α-linolenic acid (C18:3Δ9Z,12Z,15Z), and γ-linolenic acid (18:3Δ6Z,9Z,12Z) (Table 1). The k cat/K m for linoleic acid (2.26 s−1 μM−1) was approximately 2-fold higher than that for the second favored substrate, arachidonic acid (1.15 s−1 μM−1). However, the enzyme showed no activity for stearic acid (C18), oleic acid (C18:1Δ9Z), or conjugated linoleic acids (C18:2Δ9Z,11E, C18:2Δ9E,11E, and C18:2Δ10E,12Z). The enzyme showed activity only for unsaturated fatty acids containing omega-6 cis, cis-1,4-pentadiene double bonds.
Effects of pH, temperature, metal ions, and solvent on the activity of linoleate lipoxygenase from B. thailandensis
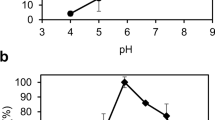
The effect of pH on the activity of linoleate 13-lipoxygenase from B. thailandensis for linoleic acid was investigated in a pH range from 6.5 to 8.5 at 25 °C. Maximum activity was recorded at pH 7.5 (Fig. 3a). The enzyme activity was examined over a temperature ranging from 10 to 40 °C at pH 7.5 (Fig. 3b), and maximum activity was observed at 25 °C. The thermal stability of linoleate 13-lipoxygenase from B. thailandensis was assessed by measuring the residual activity after incubation at different temperatures between 20 and 40 °C (Fig. 4). Thermal inactivation of the enzyme followed first-order kinetics, with half-lives for linoleic acid of 900, 122, 51, and 21 min at 20, 25, 30, 35, and 40 °C, respectively. Because of the instability of the enzyme above 30 °C, the optimum temperature of the enzyme was selected as 25 °C.

Effects of pH and temperature on the activity of linoleate lipoxygenase from B. thainlandensis. a pH effect. The used buffers in the pH experiments were 50 mM HEPES buffer (filled circle, pH 6.5–7.5) and 50 mM Tris-HCl buffer (empty circle, pH 7.0–8.5). The reactions were performed in enzyme solution containing 0.1 mM linoleic acid and 0.05 μg ml−1 enzyme at 25 °C for 2 min. b Temperature effect. The reactions were performed in 50 mM HEPES (pH 7.5) buffer containing 0.1 mM linoleic acid and 0.05 μg ml−1 enzyme for 2 min. Data represent the means of three experiments, and error bars represent the standard deviation

Thermal inactivation of the activity of linoleate lipoxygenase from B. thailandensis. The enzyme was incubated at 20 °C (filled triangle), 25 °C (open square), 30 °C (filled square), 35 °C (open circle), and 40 °C (filled circle) in 50 mM HEPES (pH 7.5) buffer. A sample was withdrawn at each time point and assayed in 50 mM HEPES (pH 7.5) buffer containing 0.1 mM linoleic acid and 0.05 μg ml−1 enzyme at 25 °C for 2 min. Data represent the means of three experiments, and error bars represent the standard deviation
The effect of metal ions on the activity of linoleate 13-lipoxygenase from B. thainlandensis was evaluated in the presence of metal ions at 0.1 or 1 mM (Fig. 5a). Cu2+ at 0.1 mM, Zn2+ at 1 mM, Ca2+, and Mg2+ ions enhanced lipoxygenase activity, whereas Ni2+, Co2+, Fe2+, Mn2+, Cu2+ at 1 mM, and Zn2+ ions at 0.1 mM inhibited activity. Enzyme activity was the highest when Cu2+ was used, and the optimal concentration of Cu2+ was 0.1 mM (Fig. 5b). The far-UV CD spectra of the enzymes without the addition of metal ions and with 0.1 mM Cu2+ were determined to analyze the change of the secondary structures. The two spectra were almost the same, indicating that the secondary structure was not changed by adding 0.1 mM Cu2+ (Fig. S2).

Effect of metal ions on the activity of linoleate lipoxygenase from B. thailandensis. a Effect of the type of metal ions. The reactions were performed in 50 mM HEPES (pH 7.5) buffer containing 0.1 mM linoleic acid, 0.05 μg ml−1 enzyme, and 0.1 mM (black bar) or 1 mM (gray bar) of various metal ions at 25 °C for 1 min. b Effect of Cu2+ concentration. The reactions were performed by varying the Cu2+ concentration from 0 to 0.25 mM in 50 mM HEPES (pH 7.5) buffer containing 0.1 mM linoleic acid and 0.05 μg ml−1 enzyme at 25 °C for 2 min. Data represent the means of three experiments, and error bars represent the standard deviation
The effect of different solvents (methanol, ethanol, pentane, ethyl acetate, iso-propanol, dimethyl sulfoxide, iso-octane, butanol, and toluene) on the activity of linoleate lipoxygenase from B. thailandensis was also investigated. Enzyme activity was the highest when methanol was used as the solvent (Fig. S3a), and its optimal concentration was 6 % (v/v) (Fig. S3b). Thus, 6 % (v/v) methanol was used for the enzymatic reaction.
Optimization of enzyme and substrate concentrations for 13-HODE production by linoleate 13-lipoxygenases from B. thailandensis
The product was obtained from linoleic acid catalyzed by B. thailandensis linoleate 13-lipoxygenase and reduced by adding NaBH4 as a reducing agent. The reduced reaction product was analyzed by HPLC using a reverse-phase column and identified as 13-HODE based on the retention times using 13-HODE and 10-HODE standards (Fig. S4a). The chirality of the 13-HODE product was identified as 13S-HODE as the same retention time of the 13S-HODE standard by HPLC using a chiral phase column with the 13S-HODE and 13R-HODE standards (Fig. S4b). Based on the reported analytical method using 2 % 2-propanol (Meruvu et al. 2005), the concentration of 2-propanol was adjusted to 12 % to separate the 13S-HODE and 13R-HODE standards (Fig. S5).
13-HODE production was investigated at enzyme concentrations ranging from 0.5 to 3.0 g l−1 enzyme with 20 g l−1 linoleic acid as the substrate after 60 min (Fig. 6a). 13-HODE production increased with increasing enzyme concentrations up to 2.5 g l−1 and reached a plateau thereafter. Therefore, the optimal enzyme concentration was 2.5 g l−1. 13-HODE production at linoleic acid concentrations ranging from 5 to 30 g l−1 with 2.5 g l−1 enzyme was tested after 60 min (Fig. 6b). Up to 20 g l−1 linoleic acid, 13-HODE production increased with increasing linoleic acid concentrations, while the molar conversion yield was almost constant as approximately 98 %. Above 20 g l−1 linoleic acid, the production and conversion yield decreased. Thus, the optimal substrate concentration was 20 g l−1.

Effect of the concentrations of enzyme and substrate on the production of 13-HODE from linoleic acid by linoleate 13-lipoxygenase from B. thailandensis. a Effect of enzyme concentration. The reactions were performed by varying the enzyme concentration from 0.5 to 3.0 g l−1 in 50 mM HEPES (pH 7.5) buffer containing 20 g l−1 linoleic acid, 0.1 mM Cu2+, and 6 % (v/v) methanol at 25 °C for 1 h. b Effect of substrate concentration. Conversion yield (filled square) and 13-HODE production (open circle). The reactions were performed by varying the substrate concentration from 5 to 30 g l−1 in 50 mM HEPES (pH 7.5) buffer containing 2.5 g l−1 enzyme, 0.1 mM Cu2+, and 6 % (v/v) methanol at 25 °C for 1 h. Data represent the means of three separate experiments, and error bars represent the standard deviation
Production of 13-HODE from linoleic acid by linoleate 13-lipoxygenases from B. thailandensis
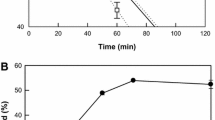
The optimal reaction conditions for 13-HODE production from linoleic acid by linoleate 13-lipoxygenase from B. thailandensis were pH 7.5, 25 °C, 20 g l−1 linoleic acid, 2.5 g l−1 enzyme, 0.1 mM Cu2+, and 6 % (v/v) methanol. Under these optimized conditions, time-course reactions of 13-HODE production were performed for 120 min (Fig. 7a). B. thailandensis linoleate 13-lipoxygenase produced 20.8 g l−1 13-HODE (70.2 mM) from 20 g l−1 linoleic acid (71.3 mM) for 120 min, with a molar conversion yield of 98.5 % and productivity of 10.4 g l−1 h−1.

Time-course reactions of the conversion of linoleic acid to 13-HPOD by linoleate 13-lipoxygenases from B. thailandensis and soybean using the same concentrations of enzyme and substrate under the optimum conditions for each enzyme. a HPLC detection at a high substrate concentration. The reaction using B. thailandensis 13-lipoxygenase was performed in 50 mM HEPES (pH 7.5) buffer containing 20 g l−1 linoleic acid, 2.5 g l−1 enzyme, 0.1 mM Cu2+, and 6 % (v/v) methanol at 25 °C for 120 min. The reaction using soybean 13-lipoxygenase was performed in 50 mM EPPS (pH 8.5) buffer containing 20 g l−1 linoleic acid, 2.5 g l−1 enzyme, and 0.01 mM Tween 40 at 20 °C for 120 min. The produced 13-HPOD was reduced to 13-HODE by adding one reaction volume of 5 % NaBH4 dissolved in 0.2 N NaOH solution. Data represent the means of three separate experiments, and error bars represent the standard deviation. b Spectrophotometric detection at a low substrate concentration. The activity of B. thailandensis 13-lipoxygenase was evaluated in 50 mM HEPES (pH 7.5) buffer containing 0.1 mM linoleic acid, 0.1 μg ml−1 enzyme, 0.1 mM Cu2+, and 6 % (v/v) methanol at 25 °C for 3 min. The activity of soybean 13-lipoxygenase was evaluated 50 mM EPPS (pH 8.5) buffer containing 0.1 mM linoleic acid, 0.1 μg ml−1 enzyme, and 0.01 mM Tween 40 at 25 °C for 3 min. The activities of linoleate 13-lipoxygenases from B. thailandensis (filled circle) and soybean (empty circle) were measured based on the increase of absorbance at 234 nm
Discussion
B. thailandensis lacks polyunsaturated fatty acids (Inglis et al. 2003), and lipoxygenase from this bacterium convert arachidonic acid into 15-hydroxyeicosatetraenoic acid (15-HETE), which can be metabolized to animal bioactive lipid mediators such as leukotrienes and lipoxins (Joo and Oh 2012). Thus, lipoxygenase from B. thailandensis as a pathogen may modulate host defense and inflammation via alteration of the biosynthesis of local chemical mediators (Vance et al. 2004).
Metal-binding residues, coffa site, and positional isomer residue in the amino acid sequence of the putative lipoxygenase from B. thailandensis suggested that the enzyme was 13S-lipoxygenase (Fig. 2). The product obtained from linoleic acid by 13S-lipoxygenase from B. thailandensis was confirmed as 13S-HODE (Fig. S1). The activity of B. thailandensis lipoxygenase was observed for only polyunsaturated fatty acids containing omega-6 cis, cis-1,4-pentadiene double bonds such as linoleic acid, arachidonic acid, α-linolenic acid, and γ-linolenic acid (Table 1). The specific activity and k cat/K m of the enzyme were the highest for linoleic acid. Therefore, the putative lipoxygenase from B. thailandensis was an omega-6 linoleate lipoxygenase (or a linoleate 13S-lipoxygenase).
The activity of linoleate 13-lipoxygenase from B. thailandensis for linoleic acid was compared with that of soybean linoleate 13-lipoxygenase using the same concentrations of enzyme and substrate. As shown in Fig. 6b, the initial formation rate of 13-HPOD by B. thainlandensis linoleate 13-lipoxygenase was higher than that by soybean linoleate 13-lipoxygenase, and after 1 min, the 13-HPOD concentration produced by B. thailandensis linoleate 13-lipoxygenase was 4-fold higher. The specific activity of B. thailandensis linoleate 13-lipoxygenase for linoleic acid was 26.4 U mg−1, which was 2.2-fold higher than the reported specific activity of soybean linoleate 13-lipoxygenase (Kermasha et al. 2001). The reaction optimum conditions of soybean linoleate 13-lipoxygenase were pH 8.5, 20 °C, and 0.01 mM Tween 40. 13-HODE production by soybean 13-lipoxygenase was performed under the optimum conditions with 20 g l−1 linoleic acid and 2.5 g l−1 enzyme. Soybean linoleate 13-lipoxygenase produced 13.8 g l−1 13-HODE (46.6 mM) for 120 min, with a molar conversion yield of 65.3 % and a productivity of 6.9 g l−1 h−1. The molar conversion yield and productivity of 13-HODE obtained by B. thailandensis linoleate 13-lipoxygenase under the optimum conditions were 151 and 158 % higher, respectively, than those obtained using soybean linoleate 13-lipoxygenase. These results indicate that B. thailandensis linoleate 13-lipoxygenase is a more efficient enzyme than soybean linoleate 13-lipoxygenase for use in commercial applications.
The specific activity and kinetic parameters of microbial lipoxygenases for unsaturated fatty acids are summarized in Table 1. The specific activity and k cat/K m of B. thailandensis lipoxygenase for linoleic acid were the highest among microbial lipoxygenases. The k cat/K m (0.53 s−1 μM−1) of potato lipoxygenase for linoleic acid was highest among those of plant lipoxygenases (Hughes et al. 2001). The k cat/K m of B. thailandensis lipoxygenase was 5-fold higher than that of potato lipoxygenase. To the best of our knowledge, the k cat/K m of B. thailandensis lipoxygenase is the highest of all lipoxygenases reported. The production of 13-HPOD from linoleic acid by B. thailandensis linoleate 13-lipoxygenase was compared with those by other linoleate 13-lipoxygenases (Table 2). G. graminis tritici produced 134 g l−1 13-HPOD from 300 g l−1 linoleic acid for 37 h, with a molar conversion yield of 75 % and a volumetric productivity of 3.6 g l−1 h−1 (Villaverde et al. 2013b). This is the highest reported concentration of 13-HPOD. The production of 13-HPOD from linoleic acid using soybean lipoxygenase showed the previous highest volumetric and specific productivities as well as molar conversion yield (22, 11 g l−1 h−1, and 80 % respectively) (Iacazio et al. 1990). The molar conversion yield of B. thailandensis lipoxygenase was 1.2-fold higher than that obtained using soybean lipoxygenase. However, the concentration, volumetric productivity, and specific productivity of 13-HPOD in the present study were 6.1-, 1.5-, and 1.9-fold lower, respectively, than the highest results reported previously.
In conclusion, the putative lipoxygenase from B. thailandensis was identified as linoleate 13S-lipoxygenase based on genetic and HPLC analyses as well as substrate specificity. The specific activity and k cat/K m of B. thailandensis 13-lipoxygenase for linoleic acid are the highest among the reported lipoxygenases, and B. thainlandensis linoleate 13-lipoxygenase is a more efficient enzyme than soybean linoleate 13-lipoxygenase as a commercial enzyme. These results indicate that this enzyme is an effective producer of 13-HODE. These results will contribute to the industrial production of 13-HODE.
References
Andreou AZ, Vanko M, Bezakova L, Feussner I (2008) Properties of a mini 9R-lipoxygenase from Nostoc sp PCC 7120 and its mutant forms. Phytochemistry 69:1832–1837
Andreou A, Gobel C, Hamberg M, Feussner I (2010) A bisallylic mini-lipoxygenase from cyanobacterium Cyanothece sp. that has an iron as cofactor. J Biol Chem 285:14178–14186
Bannenberg G, Martinez M, Hamberg M, Castresana C (2009) Diversity of the enzymatic activity in the lipoxygenase gene family of Arabidopsis thaliana. Lipids 44:85–95
Black BA, Zannini E, Curtis JM, Ganzle MG (2013) Antifungal hydroxy fatty acids produced during sourdough fermentation: microbial and enzymatic pathways, and antifungal activity in bread. Appl Environ Microbiol 79:1866–1873
Buchhaupt M, Guder JC, Etschmann MMW, Schrader J (2012) Synthesis of green note aroma compounds by biotransformation of fatty acids using yeast cells coexpressing lipoxygenase and hydroperoxide lyase. Appl Microbiol Biotechnol 93:159–168
Chohany LE, Bishop KA, Camic H, Sup SJ, Findeis PM, Clapp CH (2011) Cationic substrates of soybean lipoxygenase-1. Bioorg Chem 39:94–100
Coffa G, Brash AR (2004) A single active site residue directs oxygenation stereospecificity in lipoxygenases: stereocontrol is linked to the position of oxygenation. Proc Natl Acad Sci U S A 101:15579–15584
Gao BL, Boeglin WE, Brash AR (2010) Omega-3 fatty acids are oxygenated at the n-7 carbon by the lipoxygenase domain of a fusion protein in the cyanobacterium Acaryochloris marina. Biochim Biophys Acta 1801:58–63
Gibian MJ, Vandenberg P (1987) Product yield in oxygenation of linoleate by soybean lipoxygenase: the value of the molar extinction coefficient in the spectrophotometric assay. Anal Biochem 163:343–349
Graff G, Anderson LA, Jaques LW (1990) Preparation and purification of soybean lipoxygenase-derived unsaturated hydroperoxy and hydroxy fatty acids and determination of molar absorptivities of hydroxy fatty acids. Anal Biochem 188:38–47
Haraga A, West TE, Brittnacher MJ, Skerrett SJ, Miller SI (2008) Burkholderia thailandensis as a model system for the study of the virulence-associated type III secretion system of Burkholderia pseudomallei. Infect Immun 76:5402–5411
Hughes RK, West SI, Hornostaj AR, Lawson DM, Fairhurst SA, Sanchez RO, Hough P, Robinson BH, Casey R (2001) Probing a novel potato lipoxygenase with dual positional specificity reveals primary determinants of substrate binding and requirements for a surface hydrophobic loop and has implications for the role of lipoxygenases in tubers. Biochem J 353:345–355
Iacazio G, Langrand G, Baratti J, Buono G, Triantaphylides C (1990) Preparative, enzymic synthesis of linoleic acid (13S)-hydroperoxide using soybean lipoxygenase-1. J Org Chem 55:1690–1691
Inglis TJ, Aravena-Roman M, Ching S, Croft K, Wuthiekanun V, Mee BJ (2003) Cellular fatty acid profile distinguishes Burkholderia pseudomallei from avirulent Burkholderia thailandensis. J Clin Microbiol 41:4812–4814
Joo YC, Oh DK (2012) Lipoxygenases: potential starting biocatalysts for the synthesis of signaling compounds. Biotechnol Adv 30:1524–1532
Kermasha S, Dioum N, Bisakowski B (2001) Biocatalysis of lipoxygenase in selected organic solvent media. J Mol Catal B Enzym 11:909–919
Kuribayashi T, Kaise H, Uno C, Hara T, Hayakawa T, Joh T (2002) Purification and characterization of lipoxygenase from Pleurotus ostreatus. J Agric Food Chem 50:1247–1253
Liavonchanka A, Feussner I (2006) Lipoxygenases: occurrence, functions and catalysis. J Plant Physiol 163:348–357
Lu XY, Zhang J, Liu S, Zhang DX, Xu Z, Wu J, Li JH, Du GC, Chen J (2013) Overproduction, purification, and characterization of extracellular lipoxygenase of Pseudomonas aeruginosa in Escherichia coli. Appl Microbiol Biotechnol 97:5793–5800
Meruvu S, Walther M, Ivanov I, Hammarstrom S, Furstenberger G, Krieg P, Reddanna P, Kuhn H (2005) Sequence determinants for the reaction specificity of murine (12R)-lipoxygenase: targeted substrate modification and site-directed mutagenesis. J Biol Chem 280:36633–36641
Parra JL, Pastor J, Comelles F, Manresa MA, Bosch MP (1990) Studies of biosurfactants obtained from olive oil. Tenside Surf Det 27:302–306
Sloane DL, Leung R, Craik CS, Sigal E (1991) A primary determinant for lipoxygenase positional specificity. Nature 354:149–152
Vance RE, Hong S, Gronert K, Serhan CN, Mekalanos JJ (2004) The opportunistic pathogen Pseudomonas aeruginosa carries a secretable arachidonate 15-lipoxygenase. Proc Natl Acad Sci U S A 101:2135–2139
Villaverde JJ, Santos SAO, Haarmann T, Neto CP, Simoes MMQ, Domingues MRM, Silvestre AJD (2013a) Cloned Pseudomonas aeruginosa lipoxygenase as efficient approach for the clean conversion of linoleic acid into valuable hydroperoxides. Chem Eng J 231:519–525
Villaverde JJ, van der Vlist V, Santos SAO, Haarmann T, Langfelder K, Pirttimaa M, Nyyssola A, Jylha S, Tamminen T, Kruus K, de Graaff L, Neto CP, Simoes MMQ, Domingues MRM, Silvestre AJD, Eidner J, Buchert J (2013b) Hydroperoxide production from linoleic acid by heterologous Gaeumannomyces graminis tritici lipoxygenase: optimization and scale-up. Chem Eng J 217:82–90
Acknowledgments
This study was supported by grants from the Bio-industry Technology Development Program, Ministry for Agriculture, Food and Rural Affairs (No. 112002-3) and the Korea Healthcare Technology R&D Project, Ministry for Health & Welfare, Republic of Korea (No. 2012-009).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 502 kb)
Rights and permissions
About this article

Cite this article
An, JU., Kim, BJ., Hong, SH. et al. Characterization of an omega-6 linoleate lipoxygenase from Burkholderia thailandensis and its application in the production of 13-hydroxyoctadecadienoic acid. Appl Microbiol Biotechnol 99, 5487–5497 (2015). https://doi.org/10.1007/s00253-014-6353-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-014-6353-8