
Abstract
Isobutanol is attracting attention as a potential biofuel because it has higher energy density and lower hygroscopicity than ethanol. To date, several effective methods for microbial production of isobutanol have been developed, but they require expensive reagents to maintain expression plasmids and induce expression, which is not suitable for practical production. Here, we describe a simple and efficient method for isobutanol production in Escherichia coli. It is noteworthy that no expression plasmids or inducers were used during the production. Instead, heterologous genes necessary for isobutanol production were all knocked into the genome, and the expression of those genes was induced by xylose, which is present in most biomass feedstocks. The constructed strain (mlcXT7-LAFC-AAKCD) contains Bacillus subtilis alsS, E. coli ilvCD, Lactococcus lactis adhA, and L. lactis kivd genes in its genome and efficiently produced isobutanol from glucose and xylose in flask batch cultures. Under conditions in which the temperature and pH of the medium and the aeration in the culture were all optimized, the final isobutanol concentration reached 8.4 g L−1 after 48 h. Isobutanol was also produced using hydrolysate from Japanese cedar as the carbon source without supplemented glucose, xylose, or yeast extract. Under those conditions, isobutanol (3.7 g L−1) was produced in 96 h. Taken together, these results indicate that the developed strain is potentially useful for industrial isobutanol production.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Global energy demand continues to increase as the world population and its prosperity rise. To reduce dependence on petroleum and establish an environment-friendly society, microbiologically produced biofuels are being studied intensively. But, although biofuels have been produced using various approaches (Zinoviev et al. 2010), their application for industrial use has been very limited. The main reason is the higher cost of microbiological methods as compared to petrochemical ones.
Because isobutanol has high energy density, close to that of gasoline, and lower hygroscopicity than ethanol, it is considered a potential alternative to gasoline (Karabektas and Hosoz 2009). Recently, for example, the suitability of isobutanol-diesel fuel blends was investigated experimentally in diesel engines (Karabektas and Hosoz 2009). In addition, isobutanol and its derivatives are used in a variety of industrial applications, including solvents, paint additives, ink ingredients, and raw materials for organic compounds. As a result, isobutanol is currently being produced on a scale of about 500,000 t year−1 worldwide (Wang et al. 2012).
The current method used for industrial isobutanol production is carbonylation of propylene (Hahn et al. 2013). This method is based on petrochemical reactions and arouses fears of environmental destruction. As potential alternatives, several methods for microbial production of isobutanol have been developed and studied on a lab scale (Table 1). Using glucose as the carbon source, microbial methods often show high yield and high specificity but always require expensive reagents necessary for expressing heterologous genes and supporting cell growth, which elevates the production cost. Also reported has been the production of isobutanol from cheap carbon dioxide using Ralstonia eutropha (Li et al. 2012) and Synechococcus elongatus (Atsumi et al. 2009); however, the yields were considerably lower (Table 1), and the availability of these methods is limited by the necessity for special expertise and expensive equipment. Thus, a simple and economical method for microbial production of isobutanol has yet to be reported and would be highly desirable.
We recently developed a simple and efficient method for producing valuable chemicals from biomass in Escherichia coli (Nakashima et al. 2014). With our biomass-inducible chromosome-based expression system (BICES) method, foreign genes are expressed without the use of plasmids or expensive inducers. The BICES starter strain, mlc-XT7, harbors the “xylose-inducible promoter (PxylF)-T7 RNA polymerase gene (T7RNAP)” gene cassette in its genome, enabling expression of T7RNAP in the presence of xylose, which is the second most abundant component after glucose in lignocellulose biomass. In addition, the mlc gene (encoding a negative regulator of sugar permease genes) is mutated in this strain (mlc*), so that PxylF works even in the co-presence of xylose and glucose; in other words, the carbon catabolite repression (CCR) mechanism is released by the mlc* mutation. Consequently, when a gene cassette containing a “T7 promoter (Pt7) target gene” is knocked into the genome of this strain, strong expression of the target gene is driven in the presence of lignocellulose biomass.
Here, we report production of isobutanol using BICES. After five heterologous genes were introduced into the E. coli genome, optimal culture conditions for isobutanol production were determined, after which isobutanol was produced from a promising biomass, hydrolysate from Japanese cedar.
Materials and methods
Construction of plasmids for knock-in of alsS, adhA, and kivd
To enable isobutanol production, five heterologous genes were introduced into the E. coli genome (Fig. 1). A codon-optimized alsS gene (from Bacillus subtilis; encoding α-acetolactate dehydrogenase; accession number: LC002203) was synthesized by Operon Biotechnologies Inc. (Tokyo, Japan). A DNA fragment containing the sequence was PCR-amplified using primers sSN1727 and sSN1728 (Supplementary Table S1). The amplified fragment was then cut with NcoI and BamHI and cloned into the NcoI-BamHI site of pHN1948 (Nakashima et al. 2014), yielding pHN1953. pHN1953 contains alsS under Pt7 as well as the pACYC replication origin (pACYC ori) and the chloramphenicol resistance gene (chl r).

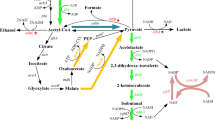
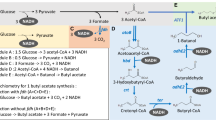
Pathway for isobutanol production. AlsS acetolactate synthase, IlvC acetohydroxy acid isomeroreductase, IlvD dihydroxy acid dehydratase, Kivd ketoisovalerate decarboxylase, Adh alcohol dehydrogenase
To knock-in the above-mentioned “Pt7-alsS” gene cassette into the yghX pseudogene locus, plasmid pHN2143 was constructed from pHN1975. pHN1975 contains a temperature-sensitive version of plasmid replication origin (pSC101ts ori), a counterselective gene (for sucrose; sacB), and the chl r and yghX flanking regions (Nakashima et al. 2014). To create pHN2143, the Pt7-alsS gene cassette was excised from pHN1953 using NsiI and BamHI, after which the excised fragment was cloned into the PstI-BamHI site of pHN1975.
To knock-in the “Pt7-adhA” gene cassette into the ybeM pseudogene locus, plasmid pHN2158 was constructed from pHN1997. Codon-optimized adhA (from Lactococcus lactis; encoding alcohol dehydrogenase; accession number: LC002204) was synthesized by Operon Biotechnologies Inc. pHN1997 contains pSC101ts ori, sacB, and the chl r and ybeM flanking regions, and a “Pt7-mCherry” gene cassette (Nakashima et al. 2014). A fragment containing adhA was PCR-amplified using primers sSN1956 and sSN1957. To replace mCherry in pHN1997 with adhA, the fragment was cut with NcoI and XhoI and cloned into the NcoI-XhoI site of pHN1997 (Nakashima et al. 2014), yielding pHN2158.
Plasmid pHN1936 (Nakashima et al. 2014) contains pSC101ts ori, sacB, and the chl r and lacY flanking regions. A fragment containing the Pt7 sequence was excised from pHN1948 using NsiI and NcoI. In addition, a fragment containing the codon-optimized kivd gene (from L. lactis; encoding 2-keto acid decarboxylase; accession number: AJ746364) was excised from pUC57-kivd (Nakashima and Tamura 2012) using SalI and NcoI. These two fragments were cloned together into the XhoI-PstI site of pHN1936, yielding pHN2157.
Construction of plasmids for knock-in of ilvCD
Plasmid pHN1377 (Nakashima and Tamura 2013) contains the RK2 replication ori (RK2 ori) and apramycin resistance gene (apr r). A DNA fragment containing the pN25 promoter sequence (Deuschle et al. 1986) was synthesized using primers sSN1801, sSN1802, sSN1803, and sSN1804 as described previously (Di Donato et al. 1993). This fragment was cut with PstI and NcoI and cloned into the PstI-NcoI site of pHN1377, yielding pHN1418. A DNA fragment containing Pt7 was PCR-amplified from pHN1948 using primers sSN1902 and sSN1903. The amplified fragment was digested with PstI and KpnI and ligated into the PstI-KpnI site of pHN1418, yielding pHN2129. A DNA fragment containing ilvCD (from E. coli; encoding ketol acid reductoisomerase and dihydroxy acid dehydratase; accession numbers are P05793 and P05791, respectively) was PCR-amplified from pHN1451 (Nakashima and Tamura 2013) using primers sSN1937 and sSN1330. The amplified fragment was digested with PciI and SpeI and ligated into the NcoI-SpeI site of pHN2129, yielding pHN2181. A DNA fragment containing attP was PCR-amplified from λ DNA-Hind III Digest (New England Biolabs, MA, USA) using primers sSN1986 and sSN1987. The amplified fragment was digested with NsiI and ligated into the NsiI-SnaBI site of pHN2181, yielding pHN2182. pHN2182 contains RK2 ori, apr r, the “Pt7-ilvCD” gene cassette, and the attP sequence. With the aid of a helper plasmid, apr r and the “Pt7-ilvCD” gene cassette from pHN2182 were knocked into the attB site of the E. coli genome.
After pHN1532 was constructed by PCR-amplifying pHN1234 (Nakashima and Tamura 2012) using primers sSN1417 and sSN1173, the amplified fragment was self-ligated. A DNA fragment containing the λ cI857 repressor and λ PL/PR promoter was PCR-amplified from pND707 (National BioResource Project E. coli, National Institute of Genetics, Japan) using primers sSN1988 and sSN1989. The amplified fragment was digested with SpeI and PciI and ligated into the XbaI-NcoI site of pHN1532, yielding pHN2183. A DNA fragment containing λ integrase was PCR-amplified from λ DNA-Hind III Digest using primers sSN1990 and sSN1991. The amplified fragment was digested with NdeI and XhoI and ligated into the NdeI-XhoI site of pHN2183, yielding pHN2184. pHN2184 contains the pSC101ts ori, chl r, and λ cI857 genes as well as a “λ PL/PR promoter-λ integrase” gene cassette and was used as a helper plasmid for site-specific recombination. The sequences of all primers from sSN1330 to sSN1991 are shown in Supplementary Table S1.
Knock-in of genes into the genome
The mlc-XT7 strain was constructed from the MG1655 strain (The Coli Genetic Stock Center: CGSG6300) earlier (Nakashima et al. 2014), and its derivatives were used as host strains for isobutanol production. The Pt7-alsS, Pt7-adhA, and “Pt7-kivd” gene cassettes were knocked into the yghX, ybeM, and lacY loci using pHN2143, pHN2158, and pHN2157, respectively, as described previously (Emmerson et al. 2006; Nakashima and Miyazaki 2014). The Pt7-ilvCD gene cassette was knocked-in using pHN2182 and pHN2184 according to the procedure established by Diederich et al. (1992).
Standard culture conditions for isobutanol production
E. coli strains were pregrown overnight and then diluted 1:100 with fresh M9Y medium (pH 7.0) containing 17 g L−1 Na2HPO4·12H2O, 3 g L−1 KH2PO4, 0.5 g L−1 NaCl, 1 g L−1 NH4Cl, 0.24 g L−1 MgSO4·7H2O, 0.011 g L−1 CaCl2·2H2O, and 5 g L−1 of Bacto yeast extract and a carbon source (glucose and xylose or cedar hydrolysate) and cultured at 37 °C in a conical flask with a screw cap. The volume of the medium was kept at 1/12.5 volume of the flask.
Optimization of the culture conditions
The effect of culture temperature and initial culture pH on isobutanol production was examined at 28–37 °C and pH 6.0–8.0, respectively. The effect of aeration on isobutanol production was examined by changing the flask rotation speed between 100 and 300 rpm (Bioshoker BR-43FL; TAITEC, Saitama, Japan).
Preparation of hydrolysate from Japanese cedar
After wood chips from Japanese cedar were milled using a cutter mill (MKCM-3; Masuko Sangyo, Saitama, Japan), the resulting particles were used as the initial raw material. Using the method of Lee et al. (2010b), hydrothermal and mechanochemical pretreatment was carried out. The resulting powders were hydrolyzed with 5 FPU/g of Acremonium cellulose (Meiji Seika Pharma, Nagoya, Japan) and 10 μL/g of Optimash BG (Genencor International, Rochester, NY, USA) in 50 mM citrate buffer (pH 5.0) at 50 °C and 120 rpm. After incubation for 24 h, the reaction mixtures were harvested by centrifugation. The supernatant was purified using Dowex Marathon WBA (Sigma-Aldrich, St. Louis, MO, USA) and was then filtered through a 0.2-μm filter (Merck Millipore, Billerica, MA, USA). Finally, the resulting solutions were used as hydrolysates. Further details of the procedure are provided elsewhere (Goshima et al. 2013).
Quantification of isobutanol and sugars
After clarifying the culture by centrifugation and filtration, the supernatant was subjected to high-performance liquid chromatography (HPLC). Isobutanol quantification was performed using an Aminex HPX-87H cationic exchange column connected with an Aminex 85H Micro-Guard Column (Bio-Rad Labs, Richmond, CA, USA). The chromatographic conditions were as follows: mobile phase, 4 mM H2SO4; flow rate, 1.0 mL min−1; and the column oven temperature, 50 °C. Isobutanol was detected using a refractive index indicator. Sugar quantification was carried out as previously described with some modification (Nakashima and Tamura 2012).
Results
Development of an isobutanol-producing strain
We previously developed E. coli strain mlc-XT7-LAFC (Nakashima et al. 2014), which is a derivative of the strain mlc-XT7 and carries an mlc* mutation (Nakashima and Tamura 2012, 2013); the PxylF-T7RNAP gene cassette in the lacZ locus; and deletions at the ldhA, adhE, pflB, and ackA-pta genes (Table 2). These deletions were introduced so that pyruvate was not processed into the fermentative pathways. Thereafter, five heterologous genes (adhA, alsS, kivd, and ilvCD) that had been placed under the control of Pt7 were knocked into the ybeM, yghX, lacY, and attB loci of the mlcXT7-LAFC strain (Table 2). These genes were used because they had been previously utilized to successfully produce isobutanol in E. coli (Atsumi et al. 2008; Baez et al. 2011; Huo et al. 2011). Note that genome editing at these knock-in loci was expected to have no serious effect on cellular function (Nakashima et al. 2014). Knocking-in adhA, alsS, and kivd was accomplished using the pSC101ts-sacB homologous recombination method with some modifications (Blomfield et al. 1991; Emmerson et al. 2006; Nakashima and Miyazaki 2014). However, ilvCD knock-in into a heterologous gene locus via homologous recombination was difficult, possibly because recombination occurred primarily at the ilvCD locus, not the heterologous locus. We therefore knocked these genes into the attB locus via site-specific recombination, which is known to be highly efficient (Diederich et al. 1992). As a result, we obtained three strains, mlcXT7-LAFC-AA (knocked-in Adh and AlsS), mlcXT7-LAFC-AAK (knocked-in Adh, AlsS, and Kivd), and mlcXT7-LAFC-AAKCD (knocked-in Adh, AlsS, Kivd, and IlvCD).
Isobutanol production using BICES
The aforementioned three strains were then cultured in M9Y medium in an effort to produce isobutanol with BICES. Screw-capped conical flasks were used as culture vessels to grow the cells under microaerobic conditions (Atsumi et al. 2008). When 40 g L−1 glucose and 8 g L−1 xylose were provided as the carbon source under the standard culture conditions (see “Materials and methods” section), strain mlcXT7-LAFC-AAKCD produced 6.8 g L−1 isobutanol in 48 h (Fig. 2). The concentration ratio between glucose and xylose had been decided based on the sugar concentrations in the lignocellulosic hydrolysate used our previous study (Goshima et al. 2013). During production, 25.2 g L−1 glucose and 5.1 g L−1 xylose were consumed. The other two strains produced little or no isobutanol because they lacked the necessary heterologous genes.

Comparison of isobutanol production among E. coli strains. These strains were cultured in M9Y medium containing 40 g L−1 glucose and 8 g L−1 xylose under standard culture conditions. Error bars indicate SE (n = 3)
Optimization of the culture conditions for isobutanol production
To optimize the culture conditions for isobutanol production by strain mlcXT7-LAFC-AAKCD, we evaluated the effects of temperature, initial culture pH, and aeration. When the effect of temperature on isobutanol production was assessed, it was found that the final product concentration increased with decreasing temperature until 32 °C (Fig. 3a). When the effect of initial culture pH was investigated, the highest product concentration was obtained at pH 6.5 (Fig. 3b). Finally, examination of the aeration conditions showed that the highest final product concentration was obtained by rotating the flask at 150 rpm (Fig. 3c). Under those optimized conditions, 8.4 g L−1 isobutanol was produced after 48 h with consumption of 25.6 g L−1 glucose and 5.2 g L−1 xylose (Fig. 3d). The productivity and theoretical yield were 1.2-fold higher than under the standard condition (Table 1), which suggests that isobutanol production could be improved through optimization.

Effects of culture conditions on isobutanol production. E. coli strain mlcXT7-LAFC-AAKCD was cultured in M9Y medium containing 40 g L−1 glucose and 8 g L−1 xylose. a Effect of the temperature on isobutanol production. b Effect of initial culture pH on isobutanol production. c Effect of aeration on isobutanol production. d Isobutanol production under optimized culture conditions. Error bars indicate SE (n = 3)
Production of isobutanol from cedar hydrolysate
In the experiments summarized above, reagent-grade glucose, xylose, and yeast extract were used. However, it is preferable to avoid using these reagents for practical production because they are always expensive (Rodrigues et al. 2006; Saha 2006). To produce isobutanol cost effectively on an industrial scale, more economical carbon, nitrogen, and mineral sources should replace the glucose, xylose, and yeast extract. We therefore assessed isobutanol production from biomass hydrolysate. In M9 medium (without yeast extract) prepared with cedar hydrolysate, the glucose and xylose concentrations were 86.4 and 15.5 g L−1, respectively. When strain mlcXT7-LAFC-AAKCD was cultured in this medium under the optimized conditions, isobutanol (3.7 g L−1) was produced after 96 h with consumption of 55.0 g L−1 glucose and 7.6 g L−1 xylose (Fig. 4). The productivity from cedar hydrolysate was therefore 0.0385 g L−1 h−1, which is 4.5 times lower than under the optimized culture conditions with glucose, xylose, and yeast extract (Table 1).

Production of isobutanol from cedar hydrolysate. The strain was cultured in M9 medium containing cedar hydrolysate without Bacto yeast extract. Error bars indicate SE (n = 3)
Discussion
In the process of optimizing the culture conditions for isobutanol production by E. coli strain mlcXT7-LAFC-AAKCD, we learned that the most important factor for isobutanol production is the culture temperature. The final product concentration increased with decreasing culture temperature until 32 °C, and the final product concentration at 37 °C was 12 % lower than at 32 °C (Fig. 3a). Several biofuels have been produced in genetically engineered E. coli at a temperature below 37 °C (Thapa et al. 2013; Lee et al. 2010a). It is known that low culture temperature can enhance the expression of a recombinant enzyme in part because it reduces formation of inclusion bodies (de Groot and Ventura 2006; Hunke and Betton 2003). In addition, low culture temperature reportedly fosters a suitable balance between protein expression and product formation in E. coli (de Groot and Ventura 2006). When the initial culture pH for the isobutanol production was examined at 37 °C, maximum product concentration was observed at pH 6.5 (Fig. 3b), perhaps at least in part because Kivd shows maximum enzymatic activity at that pH (de la Plaza et al. 2004). The effect of aeration was investigated at different flask rotation speeds, and the optimal rotation speed for isobutanol production was determined to be 150 rpm (Fig. 3c). It may be that rotating flasks at 150 rpm generate a suitable dissolved oxygen level to balance cell growth and carbon flux into the isobutanol pathway. Under aerobic conditions, cells grow rapidly and carbon preferentially flows into the tricarboxylic acid cycle, while under anaerobic conditions, cells grow poorly and carbon normally flows into the fermentative pathways. However, native fermentative genes, adhA, ldhA, and pflB, were deleted from strain mlcXT7-LAFC-AAKCD; consequently, the strain produces large amounts of isobutanol. Note that strains lacking both ldhA and pflB cannot grow under strict anaerobic conditions (Peng and Shimizu 2003). In fact, similar phenomena have often been observed during production of biofuels in genetically engineered E. coli (Abanoz et al. 2012; Cao et al. 2014).
In an earlier study, we produced isobutanol using other CCR-negative E. coli strains with expression plasmids and isopropyl β-d-1-thiogalactopyranoside (Nakashima and Tamura 2012). The final product concentration was lower in the present study, though initial glucose concentrations were different (Table 1). However, the productivity was 1.3-fold higher in the present study than in the earlier one (Table 1). This may be attributable to a reduction in stress on the E. coli, as strain mlcXT7-LAFC-AAKCD does not require the use of antibiotics to maintain plasmids and artificial inducers for gene expression; BICES may thus lift unnecessary metabolic burdens from E. coli cells.
When cedar hydrolysate was used as the carbon source, comparatively little isobutanol was produced after 96 h (Fig. 4), and the productivity was considerably lower than that achieved with reagent-grade glucose and xylose (Table 1). The E. coli cells grown in batch cultures eventually began to lose viability due to the transition from stationary phase into death phase. The loss of viability may also have been due to enzyme inhibition caused by the substances present in the hydrolysate (Mills et al. 2009). Several methods for isobutanol production from cellulose have been reported to date. For example, Clostridium cellulolyticum, a mesophilic gram-positive bacterium capable of degrading cellulose via cellulosome, was given a capacity for isobutanol production through knock-in of heterologous genes into the genome (Higashide et al. 2011; Li et al. 2014). These strains were potentially useful because isobutanol could be produced without pretreatment of the biomass, and they did not necessarily require expression plasmids and inducers. However, their productivities were low (Table 1). With our method, by contrast, the productivity from cedar hydrolysate was 4.4-fold higher than that in previously reported studies (Higashide et al. 2011; Li et al. 2014).
We are now endeavoring to improve isobutanol productivity by reinforcing the isobutanol tolerance of strain mlc-XT7-LAFC-AAKCD. The weak isobutanol tolerance of E. coli is one of the few drawbacks to isobutanol production with strain mlc-XT7-LAFC-AAKCD. Generically, E. coli cells lack tolerance to alcohols, and cell growth is inhibited by 15 g L−1 isobutanol (Atsumi et al. 2008). Recently, improved isobutanol tolerance was reported for E. coli cells expressing a heterologous cfa gene (encoding cyclopropane fatty acid synthase) (Kanno et al. 2013). In that study, Kanno and coworkers isolated a few bacteria showing a higher tolerance for butanol and isobutanol and found that this tolerance was associated with enhanced production of long-chain fatty acids and a thicker extracellular layer. This result indicates that cyclopropane fatty acid may primarily contribute to the tolerance, enabling that cfa-overexpressing E. coli could grow in the presence of 3.5 % (v/v) isobutanol.
For 1 mol of isobutanol production, 2 mol of NADPH is required (Fig. 1). Thus, another key to high isobutanol production is enhanced NADPH generation. Three potential approaches to enhancing NADPH generation are as follows. The first is to enhance carbon flux to the pentose phosphate pathway, where 2 mol of NADPH is produced through oxidation of 1 mol of glucose into 0.5 mol of glyceraldehyde 3-phosphate (Berg et al. 2002). Alternatively, native NAD-dependent glyceraldehyde 3-phosphate dehydrogenase (GAPDH; encoded by gapA) could be replaced with heterologous NADP-dependent GAPDH from Clostridium acetobuylicum (Martínez et al. 2008). And finally, a foreign NADH kinase that catalyzes the phosphorylation of NADH to NADPH could be overexpressed (Lee et al. 2013). Increasing the final product concentration and productivity through such improvements is expected to reduce production costs.
References
Abanoz K, Stark BC, Akbas MY (2012) Enhancement of ethanol production from potato-processing wastewater by engineering Escherichia coli using Vitreoscilla haemoglobin. Lett Appl Microbiol 55:436–443
Atsumi S, Hanai T, Liao JC (2008) Non-fermentative pathways for synthesis of branched-chain higher alcohols as biofuels. Nature 451:86–89
Atsumi S, Higashide W, Liao JC (2009) Direct photosynthetic recycling of carbon dioxide to isobutyraldehyde. Nat Biotechnol 27:1177–1180
Baez A, Cho KM, Liao JC (2011) High-flux isobutanol production using engineered Escherichia coli: a bioreactor study with in situ product removal. Appl Microbiol Biotechnol 90:1681–1690
Berg JM, Tymoczko JL, Stryer L (2002) The metabolism of glucose 6-phosphate by the pentose phosphate pathway is coordinated with glycolysis. In: Biochemistry, 5th edn. W.H. Freeman & Co Ltd, New York
Blombach B, Riester T, Wieschalka S, Ziert C, Youn JW, Wendisch VF, Eikmanns BJ (2011) Corynebacterium glutamicum tailored for efficient isobutanol production. Appl Environ Microbiol 77:3300–3310
Blomfield IC, Vaughn V, Rest RF, Eisenstein BI (1991) Allelic exchange in Escherichia coli using the Bacillus subtilis sacB gene and a temperature-sensitive pSC101 replicon. Mol Microbiol 5:1447–1457
Cao Y, Liu W, Xu X, Zhang H, Wang J, Xian M (2014) Production of free monounsaturated fatty acids by metabolically engineered Escherichia coli. Biotechnol Biofuels 7:59
de Groot NS, Ventura S (2006) Effect of temperature on protein quality in bacterial inclusion bodies. FEBS Lett 580:6471–6476
de la Plaza M, Fernández de Palencia P, Peláez C, Requena T (2004) Biochemical and molecular characterization of alpha-ketoisovalerate decarboxylase, an enzyme involved in the formation of aldehydes from amino acids by Lactococcus lactis. FEMS Microbiol Lett 238:367–374
Deuschle U, Kammerer W, Gentz R, Bujard H (1986) Promoters of Escherichia coli: a hierarchy of in vivo strength indicates alternate structures. EMBO J 5:2987–2994
Di Donato A, de Nigris M, Russo N, Di Biase S, D’Alessio G (1993) A method for synthesizing genes and cDNAs by the polymerase chain reaction. Anal Biochem 212:291–293
Diederich L, Rasmussen LJ, Messer W (1992) New cloning vectors for integration in the lambda attachment site attB of the Escherichia coli chromosome. Plasmid 28:14–24
Emmerson JR, Gally DL, Roe AJ (2006) Generation of gene deletions and gene replacements in Escherichia coli O157:H7 using a temperature sensitive allelic exchange system. Biol Proced Online 8:153–162
Goshima T, Tsuji M, Inoue H, Yano S, Hoshino T, Matsushika A (2013) Bioethanol production from lignocellulosic biomass by a novel Kluyveromyces marxianus strain. Biosci Biotechnol Biochem 77:1505–1510
Hahn HD, Dämbkes G, Rupprich N Bahl H, Frey GD (2013) Butanols. In: Ullmann’s Encyclopedia of Industrial Chemistry Wiley, New York, 1–13
Higashide W, Li Y, Yang Y, Liao JC (2011) Metabolic engineering of Clostridium cellulolyticum for production of isobutanol from cellulose. Appl Environ Microbiol 77:2727–2733
Hunke S, Betton JM (2003) Temperature effect on inclusion body formation and stress response in the periplasm of Escherichia coli. Mol Microbiol 50:1579–1589
Huo YX, Cho KM, Rivera JG, Monte E, Shen CR, Yan Y, Liao JC (2011) Conversion of proteins into biofuels by engineering nitrogen flux. Nat Biotechnol 29:346–351
Jia X, Li S, Xie S, Wen J (2012) Engineering a metabolic pathway for isobutanol biosynthesis in Bacillus subtilis. Appl Biochem Biotechnol 168:1–9
Kanno M, Katayama T, Tamaki H, Mitani Y, Meng XY, Hori T, Narihiro T, Morita N, Hoshino T, Yumoto I, Kimura N, Hanada S, Kamagata Y (2013) Isolation of butanol- and isobutanol-tolerant bacteria and physiological characterization of their butanol tolerance. Appl Environ Microbiol 79:6998–7005
Karabektas M, Hosoz M (2009) Performance and emission characteristics of a diesel engine using isobutanol—diesel fuel blends. Renew Energy 34:1554–1559
Kondo T, Tezuka H, Ishii J, Matsuda F, Ogino C, Kondo A (2012) Genetic engineering to enhance the Ehrlich pathway and alter carbon flux for increased isobutanol production from glucose by Saccharomyces cerevisiae. J Biotechnol 159:32–37
Lang K, Zierow J, Buehler K, Schmid A (2014) Metabolic engineering of Pseudomonas sp. strain VLB120 as platform biocatalyst for the production of isobutyric acid and other secondary metabolites. Microb Cell Factories 13:2
Lee SJ, Oh EK, Oh YH, Won JI, Han SK, Lee JW (2010a) Increased ethanol resistance in Ethanolic Escherichia coli by Insertion of heat-shock genes BEM1 and SOD2 from Saccharomyces cerevisiae. Bioprocess Biosyst Eng 15:770–776
Lee SH, Chang F, Inoue S, Endo T (2010b) Increase in enzyme accessibility by generation of nanospace in cell wall supramolecular structure. Bioresour Technol 101:7218–7223
Lee WH, Kim MD, Jin YS, Seo JH (2013) Engineering of NADPH regenerators in Escherichia coli for enhanced biotransformation. Appl Microbiol Biotechnol 97:2761–2772
Li S, Wen J, Jia X (2011) Engineering Bacillus subtilis for isobutanol production by heterologous Ehrlich pathway construction and the biosynthetic 2-ketoisovalerate precursor pathway overexpression. Appl Microbiol Biotechnol 91:577–589
Li H, Opgenorth PH, Wernick DG, Rogers S, Wu TY, Higashide W, Malati P, Huo YX, Cho KM, Liao JC (2012) Integrated electromicrobial conversion of CO2 to higher alcohols. Science 335:1596
Li Y, Xu T, Tschaplinski TJ, Engle NL, Yang Y, Graham DE, He Z, Zhou J (2014) Improvement of cellulose catabolism in Clostridium cellulolyticum by sporulation abolishment and carbon alleviation. Biotechnol Biofuels 7:25
Martínez I, Zhu J, Lin H, Bennett GN, San KY (2008) Replacing Escherichia coli NAD-dependent glyceraldehyde 3-phosphate dehydrogenase (GAPDH) with a NADP-dependent enzyme from Clostridium acetobutylicum facilitates NADPH dependent pathways. Metab Eng 10:352–359
Matsuda F, Kondo T, Ida K, Tezuka H, Ishii J, Kondo A (2012) Construction of an artificial pathway for isobutanol biosynthesis in the cytosol of Saccharomyces cerevisiae. Biosci Biotechnol Biochem 76:2139–2141
Mills TY, Sandoval NR, Gill RT (2009) Cellulosic hydrolysate toxicity and tolerance mechanisms in Escherichia coli. Biotechnol Biofuels 2:26
Nakashima N, Miyazaki K (2014) Bacterial cellular engineering by genome editing and gene silencing. Int J Mol Sci 15:2773–2793
Nakashima N, Tamura T (2012) A new carbon catabolite repression mutation of Escherichia coli, mlc∗, and its use for producing isobutanol. J Biosci Bioeng 114:38–44
Nakashima N, Tamura T (2013) Gene silencing in Escherichia coli using antisense RNAs expressed from doxycycline-inducible vectors. Lett Appl Microbiol 56:436–442
Nakashima N, Akita H, Hoshino T (2014) Establishment of a novel gene expression method, BICES (biomass-inducible chromosome-based expression system), and its application for production of 2,3-butanediol and acetoin. Metab Eng 25:204–214
Peng L, Shimizu K (2003) Global metabolic regulation analysis for Escherichia coli K12 based on protein expression by 2-dimensional electrophoresis and enzyme activity measurement. Appl Microbiol Biotechnol 61:163–178
Rodrigues LR, Teixeira JA, Oliveira R (2006) Low-cost fermentative medium for biosurfactant production by probiotic bacteria. Biochem Eng J 32:135–142
Saha BC (2006) A low-cost medium for mannitol production by Lactobacillus intermedius NRRL B-3693. Appl Microbiol Biotechnol 72:676–680
Thapa LP, Lee SJ, Yoo HY, Choi HS, Park C, Kim SW (2013) Development of glycerol-utilizing Escherichia coli strain for the production of bioethanol. Enzym Microb Technol 53:206–215
Wang BW, Shi AQ, Tu R, Zhang XL, Wang QH, Bai FW (2012) Branched-chain higher alcohols. Adv Biochem Eng Biotechnol 128:101–118
Zinoviev S, Müller-Langer F, Das P, Bertero N, Fornasiero P, Kaltschmitt M, Centi G, Miertus S (2010) Next-generation biofuels: survey of emerging technologies and sustainability issues. Chem Sus Chem 3:1106–1133
Acknowledgments
We are grateful to all members of the Bio-conversion Research Team at our institute [Biomass Refinery Research Center, National Institute of Advanced Industrial Sciences and Technology (AIST)] for their technical assistance and valuable discussion. This work was supported in part by KAKENHI (23780096) and a grant from the Japan-US cooperation on Clean Energy Technologies, METI, Japan, to N. Nakashima.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 67 kb)
Rights and permissions
About this article

Cite this article
Akita, H., Nakashima, N. & Hoshino, T. Bacterial production of isobutanol without expensive reagents. Appl Microbiol Biotechnol 99, 991–999 (2015). https://doi.org/10.1007/s00253-014-6173-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-014-6173-x