
Abstract
Despite an expanding array of molecular approaches for detecting microorganisms in a given sample, rapid and robust means of assessing the differential viability of the microbial cells, as a function of phylogenetic lineage, remain elusive. A propidium monoazide (PMA) treatment coupled with downstream quantitative polymerase chain reaction (qPCR) and pyrosequencing analyses was carried out to better understand the frequency, diversity, and distribution of viable microorganisms associated with debris collected from the crew quarters of the International Space Station (ISS). The cultured bacterial counts were more in the ISS samples than cultured fungal population. The rapid molecular analyses targeted to estimate viable population exhibited 5-fold increase in bacterial (qPCR-PMA assay) and 25-fold increase in microbial (adenosine triphosphate assay) burden than the cultured bacterial population. The ribosomal nucleic acid-based identification of cultivated strains revealed the presence of only four to eight bacterial species in the ISS samples, however, the viable bacterial diversity detected by the PMA-pyrosequencing method was far more diverse (12 to 23 bacterial taxa) with the majority consisting of members of actinobacterial genera (Propionibacterium, Corynebacterium) and Staphylococcus. Sample fractions not treated with PMA (inclusive of both live and dead cells) yielded a great abundance of highly diverse bacterial (94 to 118 taxa) and fungal lineages (41 taxa). Even though deep sequencing capability of the molecular analysis widened the understanding about the microbial diversity, the cultivation assay also proved to be essential since some of the spore-forming microorganisms were detected only by the culture-based method. Presented here are the findings of the first comprehensive effort to assess the viability of microbial cells associated with ISS surfaces, and correlate differential viability with phylogenetic affiliation.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The microbial burden of closed systems such as the International Space Station (ISS) (Castro et al. 2004), submarines (Boyden 1962; Morris 1972; Thomas et al. 2000; Upsher et al. 1994), environmental chambers (Levine and Cobet 1970; Pierson et al. 2002), and office buildings (Burge et al. 2000; Ferguson et al. 1975; Stenberg et al. 1993) has been previously evaluated using traditional culture-dependent methods. Data obtained from the Apollo (Ferguson et al. 1975), Skylab (Taylor et al. 1977), Space Shuttle (Koenig and Pierson 1997; Pierson 2001), and Russian space station Mir (Kawamura et al. 2001) demonstrated the capability to provide and maintain space environments compatible with human occupation. Lessons learned from these culture-based studies (ISS, Space Shuttle, Skylab, Mir, etc.) were implemented into the ISS to ensure an environment capable of supporting human habitation for many years (Pierson 2001). For example, high-efficiency particulate air filters were incorporated into the air handling system of the ISS to reduce levels of airborne bacteria, fungi, and particulates (Pierson 2001), and such measures yielded levels of airborne bacteria consistently below the 1 × 104 colony-forming unit (CFU)/m3 acceptability limit (Pierson 2001). However, use of state-of-the-art molecular microbiological techniques will reveal presence of viable but yet-to-be cultivated microbes that will enable the development of appropriate countermeasures.
Reports pertaining to the molecular microbial diversity of ISS drinking water (La Duc et al. 2003), integrated thermal coolant system (Benardini et al. 2005), and ISS mock-up habitat (Moissl et al. 2007) revealed the presence and predominance of certain microbes (such as Ralstonia, Afipia, Propionibacterium) that are problematic to both crew and habitat. Traditional culture-based analysis only would not have revealed the presence of these opportunistic pathogens, nor a presence of metal-loving microbes that could deteriorate habitat subsystems. Although some studies have been carried out on the molecular microbial communities associated with indoor office environments (Fierer et al. 2010), hospital surgical rooms (Edmiston et al. 2005), National Aeronautics and Space Administration (NASA) clean rooms (Newcombe et al. 2008), and ultraclean spacecraft surfaces (Venkateswaran et al. 2012), molecular methods were not previously used to examine the microbial community profile of ISS environmental surfaces.
The results of the molecular microbial diversity study of ISS’s closed systems and atmosphere can be used to establish the criteria necessary for providing an environment promoting the health, safety, and productivity of crewmembers. The National Research Council (NRC) Committee for the Decadal Survey on Biological and Physical Sciences in Space recommended that NASA “capitalize on the technological maturity, low cost, and speed of genomic analyses and the rapid generation time of microbes to monitor the evolution of microbial genomic changes in response to the selective pressures present in the spaceflight environment” (NRC 2011). Furthermore, NRC reported, “microbial species that are uncommon, or that have significantly increased or decreased in number, can be studied [in a microbial observatory] on the ISS.” In response to the NRC Committee’s recommendation to study microbial observatory (ISS environmental microbiome), microbial characterization of accumulated dust on the crew cabin utilizing traditional and state-of-the art molecular methodologies was carried out.
The objectives of this study, a part of ISS environmental microbiome, were to microbiologically characterize samples collected from vacuum cleaner bags by employing traditional culture-based techniques and state-of-the art molecular analyses. This is the first report about the utilization of next-generation sequencing technologies to measure molecular microbial diversity of the dust collected from the ISS. In addition to the total microbial burden and microbial diversity, viable microbial populations were estimated using an ATP assay (Venkateswaran et al. 2003). In addition, samples were treated with propidium monoazide (PMA), which blocks DNA (thus only viable cells), was coupled with downstream quantitative polymerase chain reaction (qPCR) and pyrosequencing analyses to better understand the abundance, frequency, diversity, and distribution of viable bacteria associated with dust particles returned from ISS (Vaishampayan et al. 2013).
Materials and methods
History of the investigation
During a period spanning portions of ISS Expeditions 30 and 31 (December 2011-July 2012), crewmember reports cited differences in the cabin environment compared to earlier experience as well as allergic responses to the cabin environment. One of the noted observations was a high level of visible dust in the Node 3 cabin of the ISS, to the extent it was sticking to the walls. Flight surgeons indicated that this had been reported not just in Node 3, but also throughout the U.S. On-Orbit Segment, and expressed a concern for crew health. Dust on ISS is expected, with humans being major contributors (via skin shedding, eating, exercising, etc.) and other sources include on-orbit maintenance activities that can release dust from sources such as payloads and systems, clothing, and visiting vehicles. As a precautionary measure, in the middle of 2012, an investigation was launched to define and mitigate dust sources, and to determine if exposure to dust might elicit an adverse effect on crew health. As a result of these crewmember reports, particulate and fiber debris samples were collected during ISS Expedition 31 in a vacuum cleaner bag and returned to Earth aboard Soyuz flight 29S in early July 2012 for further analyses. A detailed NASA in-house report (not publicly available) was generated based on culture-based microbiological analyses and concluded that there were no obvious microbiological agents that could be directly correlated with allergic reactions to humans. However, there was no molecular study conducted to comprehensively elucidate the presence of viable but yet to be cultured microorganisms in these samples that might pose problems to crew health.
Sample collection
Following ISS Expedition 31, several dust samples were delivered to the Johnson Space Center (JSC) Microbiology Laboratory for sample allocation. “Prime” vacuum cleaner bag content (ISS-debris) and visible accumulations of fibers and other materials associated with the vacuum bag (ISS-lint) returned on Soyuz flight 29S (see Fig. S1) were subjected to a variety of microbiological and next-generation molecular techniques to elucidate composition of cultured, viable, and total microorganisms. Details of the novel “Prime” vacuum cleaner developed for NASA habitats were reported elsewhere (Katherine et al. 2010). Portions of the samples were aseptically collected for microbiological testing at JSC, with the remaining bag and its contents repacked in a biological hood, sealed, and shipped to the Marshall Space Flight Center (MSFC) for particle size testing, as well as to the Jet Propulsion Laboratory (JPL) to determine their molecular microbiological composition. The vacuum cleaner bag was shipped via FedEx at ambient temperature with instructions that the samples not be irradiated in transport. For comparison, particles collected from two cleanroom (Class 100 K) floors using a Nilfisk GM80CR vacuum cleaner (disposable bag part # is 81620000; Morgantown, PA) were analyzed. These samples were from (a) the JPL spacecraft assembly facility (SAF) cleanroom floor where various Mars mission spacecraft were built and (b) JPL building #103 where non-mission critical activities were conducted. The SAF and 103 samples were characterized for the molecular microbial burden estimation at JPL and were not subjected to molecular microbial diversity analyses.
Particle sizing and characterization
The vacuum cleaner bag debris was evaluated for size classification using the following standard test sieves (ASTM-E11 1995): No. 14 (1,400 μm), No. 18 (1,000 μm), No. 35 (500 μm), No. 60 (250 μm), No. 100 (150 μm), No. 140 (106 μm), No. 200 (75 μm), No. 270 (53 μm), and No. 500 (25 μm). The mass fraction determination of the particles in each size class range was determined using a laboratory digital scale and analytical balance and physical properties were assessed using physical separation, optical microscopy, and photography methods (Perry and Coston 2014).
Traditional culture-based microbial examination
Bacterial and fungal cultures were processed, as detailed below. Approximately 1 g of each sample (debris or lint) was weighed and placed into a sterile tube containing 25 ml of sterile phosphate buffered saline (PBS) and mixed using vortex for 1 min. For the bacterial cultures, 100 μl was spread onto four plates of R2A media (Difco) and four plates of Tryptic Soy Agar (TSA, Difco). Plates were incubated at 35 °C for 48 h. For the fungal cultures, 100 μl was spread onto two plates of Sabouraud dextrose agar, one plate of Sabouraud dextrose agar with chloramphenicol, and one plate of potato dextrose agar, with incubation at 30 °C, for 5 days. Bacterial and fungal counts were calculated, as defined in the work instructions, and reported as CFU/g of material. Bacterial identifications were performed using standard biochemical analysis (VITEK2 Compact, bioMérieux, Inc.) (Castro et al. 2004) and 16S ribosomal genetic sequencing (La Duc et al. 2007). Fungal colonies were identified using classical morphological and microscopic examination (Castro et al. 2004).
Sample processing for molecular analysis
Independent to the samples taken to cultivate bacteria and fungi for analysis, subsamples from the same “Prime” vacuum cleaner bag were taken and DNA extracted. For debris, 1 g dry weight of sample was aseptically weighed and for lint samples, the vacuum bag that contains dust particles was cut into pieces (25 cm2) before DNA extraction. Briefly, each sample was placed in 15 ml of sterile PBS and sample volumes extracted. The biological materials were further concentrated using Amicon Ultra-50 Ultracel centrifugal filter tubes (Millipore, Billerica, MA, USA). Each filter unit has a molecular mass cutoff of 50 kDa, which facilitates the concentration of microbial cells, spores, and exogenous nucleic acid fragments greater than 100 bp in a final volume of 2.5 ml. All filtered samples were then divided into three separate aliquots: one of the aliquot (1,000 μl) was subjected to PMA pretreatment (viability assessment), the second (1,000 μl) was an untreated environmental sample (viable + nonviable; total DNA), and the third (500 μl) was used for ATP analysis (see below).
One 1,000 μl aliquot of filter-concentrated sample suspension was treated with 12.5 μl of PMA (2 mM; Biotium, Inc., Hayward, CA, USA) to a final concentration of 25 μM (Nocker et al. 2010; Rawsthorne et al. 2009), followed by thorough mixing and incubation in the dark for 5 min at room temperature. The sample was exposed to PhAST blue-Photo activation system for tubes (GenIUL, S.L., Terrassa, Spain) for 15 min (in parallel with the non-PMA treated sample). Samples were then split in half and one half was subjected to bead beating with the Fastprep-24 bead-beating instrument (MP Biomedicals, Santa Ana, CA, USA) with parameters set at 5 m/s for 60 s. The second half of the unprocessed sample was then combined with the mechanically aberrated counterpart before DNA was extracted via the Maxwell 16 automated system (Promega, Madison, WI, USA), in accordance with manufacture instructions, and resulting DNA suspensions (100 μl each) were stored at −20 °C.
Quantitation of total and viable microorganisms using molecular methods
ATP assay
A bioluminescence assay was performed to determine the total ATP and intracellular ATP from all samples using the CheckLite HS kit (Kikkoman), as described previously (Venkateswaran et al. 2003). Briefly, to determine total ATP (dead and viable microbes), 100-μl sample aliquots (four replicates) were each combined with 100 μl of a cell lysing detergent (benzalkonium chloride), then incubated at room temperature for 1 min prior to the addition of 100 μl of a luciferin–luciferase reagent. The sample was mixed, and the resulting bioluminescence was measured with a luminometer (Kikkoman). To determine intracellular ATP (viable microbes), 50 μl of an ATP-eliminating reagent (apyrase, adenosine deaminase) was added to a 500-μl portion of the sample and allowed to incubate for 30 min to remove any extracellular ATP, after which the assay for ATP was carried out, as described above, in four replicates including sterile PBS as a negative controls. As previously established, 1 Relative Light Unit (RLU), the unit of measurement of ATP, was assumed to be approximately equal to 1 CFU (La Duc et al. 2007).
qPCR assay
Real-time qPCR assay, targeting the 16S rRNA gene, was performed in triplicate with a CFX-96 thermal cycling Instrument (Bio-Rad, California, USA) to measure bacterial burden. Universal bacterial primers targeting the 16S rRNA gene, 1369 F (5′-CGG TGA ATACGT TCY CGG-3′) and modified 1492R (5′-GGW TAC CTTGTT ACG ACT T-3′), were used for this analysis (Suzuki et al. 2000). Each 25-μl reaction consisted of 12.5 μl of 2× iQ SYBR Green Supermix (Bio-Rad, Hercules, CA, USA), 1 μl each of forward and reverse oligonucleotide primers (10 μM each), and 1 μl of template DNA. Purified DNA from B. pumilus cells as positive controls and DNase/RNase-free molecular-grade distilled water (UltraPure, Gibco) as negative controls were included in all qPCR runs. Reaction conditions were as follows: a 3-min 95°C denaturation, followed by 35 cycles of denaturation at 95°C for 15 s, and a combined annealing and extension at 55°C for 35 s.
Molecular microbial diversity analysis using next-generation sequencing methods
Pyrosequencing conditions
Bacterial primers 28 F (5′-GAG TTT GAT CNT GGC TCA G-3′) and 519R (5′-GTN TTA CNG CGG CKG CTG-3′) were used to amplify ~500-bp fragments spanning the V1 – V3 hypervariable regions of the bacterial 16S rRNA gene. Archaeal primers 341 F (5′-GYG CAS CAG KCG MGA AW-3′) and 958R (5′-GGA CTA CVS GGG TAT CTA AT-3′) were used to amplify ~600-bp fragments spanning the V3–V5 hypervariable regions of the archaeal 16S rRNA gene. A fungal primer set ITS1F (5′-CTT GGT CAT TTA GAG GAA GTA A-3′) and ITS4R (5′-TCC TCC GCT TAT TGA TAT GC-3′) was employed to amplify ~600 bp fragments of the fungal ITS region. These primer pairs were tailored for pyrosequencing by adding a fusion linker and a proprietary 8-nt barcode sequence at the 5′ end of the forward primer, and a biotin and fusion linker sequence at the 5′ end of the reverse primer (Dowd et al. 2008). A HotStarTaq Plus master mix kit (Qiagen, Valencia, CA, USA) was used to catalyze the PCR under the following thermal cycling conditions: initial denaturing at 95 °C for 5 min, followed by 35 cycles of denaturing at 95 °C for 30 s, annealing at 54 °C for 40 s, and extension at 72 °C for 1 min, finalized by a 10-min elongation at 72 °C. Resulting PCR products were purified via Diffinity Rapid Tip (Diffinity Genomics, Inc., West Henrietta, NY, USA) chemistry, and were then pooled accordingly. Small fragments were removed with Agencourt Ampure Beads (Beckman Coulter, Brea, CA).
In preparation for FLX-Titanium sequencing (Roche, Nutley, NJ, USA), resulting PCR amplicon fragment size and concentration were accurately measured with DNA 1000 chips using a Bioanalyzer 2100 automated electrophoresis station (Agilent, Santa Clara, CA, USA) and a TBS-380 Fluorometer (Turner Biosystems, Sunnyvale, CA, USA). The total volume of initial PCR product used for subsequent emulsion PCR was 2 μl for strong positives (>10 ng/μl), 5 μl for weak positives (5–10 ng/μl), and 20 μl for samples that failed to yield PCR products (<5 ng/μl). This normalization step helped to ensure minimal bias favoring downstream amplification from initially strong PCR products. Approximately 9.6 × 106 molecules of ~600-bp double-stranded DNA were combined with 9.6 × 106 DNA capture beads, and then subjected to emulsion PCR conditions. Following recovery and enrichment, bead-attached DNA molecules were denatured with NaOH and sequencing primers were annealed. A four-region 454 pyrosequencing run was performed on a GS PicoTiterPlate (PTP) using the Genome Sequencer FLX System, in accordance with manufacturer instructions (Roche, Nutley, NJ). Twenty-four to 30 tagged samples were applied to each quarter region of the PTP. All pyrosequencing procedures were performed at the Research and Testing Laboratory (Lubbock, TX, USA) in accordance with well-established protocols (Dowd et al. 2008).
Bioinformatic analyses of bacterial pyrosequences
High-throughput 16S rRNA sequencing data was processed and analyzed using the QIIME (Quantitative Insights Into Microbial Ecology) pipeline (Caporaso et al. 2010). Sequences were quality filtered by removing sequences that (a) did not contain the primer sequence, (b) contained an uncorrectable barcode, (c) were <250 nt in length, (d) had homopolymers longer than 8 nt, or (e) had a quality score of <25, and then demultiplexed using the respective sample nucleotide barcodes. These sequences were searched against the Greengenes reference database (DeSantis et al. 2006; McDonald et al. 2012), and clustered into OTU based on their sequence similarity (97 %) with uclust (Edgar 2010). A representative sequence was picked from each OTU and taxonomic classification was assigned using MOTHUR's Bayesian classifier (Schloss et al. 2011) and Greengenes training sequences and taxonomy (DeSantis et al. 2006; McDonald et al. 2012). An OTU table was constructed using the taxonomic assignments and the OTU abundance in each sample. A heat map was constructed using OTU with more than ten sequences in at least one sample, where the counts are colored based on the abundance of each OTU.
Bioinformatic analyses of fungal pyrosequences
The ITS1 sequences were quality filtered and then demultiplexed using the respective sample nucleotide barcodes. These sequences were searched (BLAST) against a custom ITS1 database generated from the NCBI, and clustered into OTU based on their sequence similarity (97 %). An entry was deemed similar to a corresponding sequence (or identified to a OTU) if it produced a ≥97 % match over the full length of the sequence to a fully identified reference sequence, whose name was not contradicted by other, equally good matches. The corresponding values for tentative genus and order phylogenetic affiliation were 85 % and 70 %, respectively; they were deliberately set to be high to avoid false-positive inclusions. In the case of competing names for which neither synonymy nor anamorph–telemorph relationships could be established through MycoBank (Crous et al. 2004), the least inclusive parent nomenclatural level was used (e.g., Penicillium sp.). A subset of the sequence database was subjected to BLAST searches using the NCBI nucleotide database. For some of the sequences, the highest e-value and percent identity were most consistent with uncultured fungi (usually from metagenomic studies). These were recorded as such, but taxonomic information from the top ten closest sequences was used to approximate the identity of the sequence found in the ISS samples. Both bacterial and fungal pyrosequences were deposited in NCBI Sequence Read Archive (SRA) and the accession number is SRP035601.
Results
Particle characterization
The items returned aboard Soyuz flight 29S for ground-based analysis included a vacuum cleaner bag containing mixed debris and the vacuum cleaner HEPA filter element with Kapton® tape over the inlet face as shown by Fig. S2A–B. Blue-grey lint was the predominant material in the vacuum bag. The bulk material is best described as blue-grey fibrous debris matrix with human hair, food, paper, plastic, and miscellaneous granular debris mixed within it. The vacuum bag containing the debris weighed 169.2 g as received. The empty vacuum cleaner bag weighed 93.2 g. Based on the bag’s weight difference when full and empty, the total debris weight was approximately 76 g. After establishing the total debris weight, standard test sieves and forceps were used to separate the debris into different size and type fractions and each fraction was weighed. Table S1a summarizes the observed weights for each size and type fraction to <500 μm. Figure S2C–F shows material retained on the No. 35 test sieve.
The material that passed through the No. 35 test sieve consisted of a uniform tan-colored, powdery mixture of granular material and short human hair fragments (Fig. S2G). The granular material was similar in color to the dried skin fragments and nail clipping found in the larger sized granular material fractions. This material was subjected to further size classification using a test sieve series with mechanical agitation initially for 5 min with successive 1-min agitation periods for a total of 10 min. The sieve series consisted of a No. 60 test sieve (250 μm aperture), No. 100 test sieve, 150 μm aperture, No. 140 test sieve, 106 μm aperture, No. 200 test sieve, 75 μm aperture, No. 270 test sieve, 53 μm aperture, and No. 500 test sieve, 25 μm aperture. Table S2 summaries the size classification results for the debris <500 μm in size which passed through the No. 35 test sieve. The total material weight after removing it from a storage bag and completing the sieving operation was 3.984 g compared to the earlier 4.0 g that was retained on the No. 35 test sieve. The mass accountability was 99.6 % for this sieving operation (Table S1b).
The particles associated with debris were more of solid materials when compared to the fine particles (lint) scraped from the HEPA filter of the vacuum bag. From the visual examination, it was presumed that the materials associated with ISS-debris contained more of solid matter (such as peanuts, paper) compared to the fibrous materials associated with ISS-lint (Fig. S1). Hence, when weighed for microbiological assays, bulk of the weight of debris might be of non-biological origin. The density of the particles collected in the vacuum cleaner bag would be directly related to the efficiency, age, overall usage of the instrument employed, as well as cleanliness and maintenance of the closed environmental surfaces. The “Prime” vacuum cleaner system was used only once in the ISS, whereas the SAF and 103 vacuum cleaner instruments were used for 2 and 6 months, respectively. Even though the vacuum cleaner system used in ISS was different from SAF and 103 clean rooms, all three vacuum cleaners were fitted with HEPA filter bags.
Microbial burden
Total and viable microbiological burden of ISS and other Earth control vacuum cleaner bags, as estimated by conventional and rapid molecular methods, are given in Table 1. The duration in collecting particles (single time to 6 months) varied among all three vacuum cleaners and hence no definitive trend in microbial population was noticed (Table 1). Since all three assays performed during this study independently assess different kinds of populations, cross comparison of various methods and associated microbial population was not attempted.
The bacteria capable of showing growth at 35 °C in nutrient rich media were in the range of 106 CFU/g in ISS-debris sample where as the SAF and 103 debris samples contain at least 2 logs less than the ISS-debris sample. The cultured bacteria were in the range of 105 CFU/g for all lint samples. When compared to the ISS-debris sample, the cultured bacterial population was ~50 % less in ISS-lint samples. In contrast, the SAF and 103 samples exhibited 1 log more cultured bacteria in lint than their debris samples. The cultured fungal population was 9.3 × 104 CFU/g in ISS-debris and 7.3 × 103 CFU/g in ISS-lint samples, which shows that the readily cultured fungal population was only ~7 % in desiccated ISS-lint samples when compared to the ISS-debris. The fungal burden in ISS and SAF samples were similar, but despite collecting for 6 months, the 103 debris samples exhibited 1-log less fungal burden compared to others.
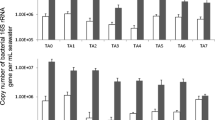
The qPCR-based estimate for total bacterial population (dead and alive; PMA-untreated) in the ISS-debris sample (9.5 × 106 16S rRNA copies/g) was 2 logs less than measured in the ISS-lint sample (8.9 × 108 16S rRNA copies/g). The 16S rRNA copy number was 1-log decreased for the viable bacterial population (PMA-treated) in these samples. As depicted in Table 1, ~12 % of the total bacteria were viable in ISS-debris, whereas only 2.6 % of bacterial population was viable in the ISS-lint sample. The percentage of viable population was higher in SAF (42 % for SAF-debris and 75 % for SAF-lint) compared to the ISS counterparts.
When the samples were subjected to ATP assay, the total microbial population (dead and alive) was 5-fold more in the ISS-debris sample (5.8 × 107 RLU/g) than measured in the ISS-lint sample (1.2 × 107 RLU/g). Unlike qPCR measurements, the ATP content was less in the ISS-lint sample when compared to the ISS-debris sample, which was also observed in the cultured assay. Approximately 85 % of the total microorganisms as measured by ATP were viable in the ISS-debris, whereas 34 % of microorganisms were viable in the ISS-lint sample. The SAF-lint and 103-lint samples possessed 1-log more total microbial load than their debris samples where as the viable microbial burden was almost similar in both SAF and 103 samples.
Culture-based microbial diversity
Bacterial strains subjected to 16S rRNA-based identification revealed the presence of either spore-forming bacteria or human commensals (Table 2). Bacillus and Staphylococcus species were common in both ISS-debris and ISS-lint samples, whereas the ISS-lint sample contained additional spore-forming bacterial species, Paenibacillus and Brevibacillus. Among the fungal strains tested via morphological and microscopic techniques, members belonging to the fungal phyla, ascomycota (Aspergillus, Penicillium) and basidiomycota (Rhodotorula), were isolated.
Pyrosequencing-derived bacterial diversity
In total, the next-generation sequencing procedures carried out in this study yielded 65,826 bacterial 16S rRNA gene sequences >350-bp in length from the four sample sets examined. When these sequences were processed with the bioinformatics software MOTHUR, ~21 % of the sequences were omitted from consideration due to the aforementioned quality control criteria. The remaining 79 % of sequences (51,570; Table 3) whose minimum length was 250 bp were aligned and subjected to cluster analyses to reveal their phylogenetic affiliations. A breakdown of the number of pyrosequences and OTU observed in the various samples examined over the course of this study is provided in Table 3. Even though equivalent materials (weight/volume) were analyzed from the ISS-debris and ISS-lint samples, the ISS-debris contained many more pyrosequence reads than the ISS-lint sample. The ISS-debris sample gave rise to 86 % of total pyrosequence reads (44,479 sequences), whereas the ISS-lint sample had only 7,271 sequences. The PMA-untreated ISS-debris (27,072 sequences) possessed 5.9-fold more bacterial pyrosequences than the ISS-lint samples (4,595 sequences), whereas the OTU numbers were roughly equivalent in both samples (118 in ISS-debris vs. 94 in ISS-lint). Both the samples examined harbored OTU affiliated with physiologically resistant bacteria (Actinobacteria and Firmicutes) and, to some extent, proteobacterial OTU were also present. A closer examination of the pyrosequence reads resulting from the ISS-debris and ISS-lint samples indicated a predominance of members of 36 genera (Table 4). It was particularly apparent that members of a few bacterial genera (Corynebacterium, Propionibacterium, Staphylococcus, Streptococcus) were present in great abundance in these samples. Collectively, sequences arising from the members of these abundant genera constituted >90 % of the total in samples that were not treated with PMA.
Regardless of sample type, PMA-treated sample (viable) fractions consistently yielded considerably fewer pyrosequences than their untreated counterparts. Overall, the effect of PMA treatment was significantly higher in both ISS-debris (61 % sequence reduction) and ISS-lint (63 % sequence reduction) samples, which indicated the presence of a large number of dead cells or extraneous DNA. Similar to the sequence abundance, the reduction in bacterial OTU numbers was even more in PMA-treated samples for ISS-debris (reduced from 118 to 23 OTU) and ISS-lint samples (reduced from 94 to 12 OTU). Many of the sequences of physiologically recalcitrant bacteria observed in the PMA-untreated samples were absent or scarce in the PMA-treated fractions of the same samples. Interestingly, the pyrosequences of the viable members of the three abundant genera were >97 % in both of the PMA-treated samples. The viable bacterial species of Corynebacterium (seven species) and Propionibacterium (three species) required special growth conditions; hence the conventional culture approach employed during this study did not reveal the presence of these bacterial species. However, the members of the staphylococcal species isolated on the TSA growth medium (Table 5) were also observed in the next-generation sequencing approach (Staphylococcus hominis and S. epidermidis).
The presence of viable Propionibacterium acnes pyrosequences was greater in PMA-treated samples, when compared to untreated matrices of both ISS-debris (7-fold increase) and ISS-lint (1.4-fold increase) samples. This might be due to the availability of more beads when the competing DNA template was lower in pyrosequencing. Similarly, more pyrosequences of viable S. epidermidis (1.9-fold), S. pettenkoferi (3.7-fold), and an unidentified Staphylococcus species (2.5-fold) were observed in the ISS-debris sample; these sequences were not noticed in high numbers in the ISS-lint sample (Table S2). Some bacterial species (Clostridium, Sphingomonas, Delftia, and Pseudomonas) were observed only in PMA-treated samples and none of these sequences, or very few sequences (<1 %), were retrieved from the PMA-untreated samples (Table S2).
Incidence of viable bacterial species elucidated by traditional culture and next-generation molecular methods is depicted in Table 5. In total, sequences of 36 viable bacterial OTU were recovered, of which 23 and 12 OTU were observed in ISS-debris and ISS-lint samples, respectively. Among these 36 OTU, C. kroppenstedtii, Corynebacterium sp., P. acnes, Rothia mucilaginosa, S. pettenkoferi, and Staphylococcus sp. sequences were retrieved from both ISS-debris and ISS-lint samples. Even though high numbers of Streptococcus sequences (9 OTU, Table S2) were noticed in these samples, which were reported to be common human commensals, none of them were viable. Unlike members of the phylum actinobacteria, viable members of the other phyla encountered during this study did not overlap between ISS-debris and ISS-lint samples. Presence of anaerobic members (Clostridium species) in the ISS-debris sample might be due to the microaerophilic conditions maintained in the clumps of the ISS-debris, when compared to the ISS-lint sample. Pyrosequences belonging to spore-forming aerobic bacteria were absent; however, these Bacillus and related spore-forming bacterial species were successfully isolated by the traditional culture method. Even though spore-forming bacteria were readily isolated, the molecular method did not retrieve pyrosequences from these samples and warrants further study. When the incidence of cultured members of the bacterial species was compared, bacterial species other than S. hominis and S. epidermidis were not common. The only bacterial species that was either cultured or had its sequence retrieved by pyrosequencing method was S. epidermidis.
When the samples were subjected to the archaeal characterization, both qPCR and next-generation sequencing methods did not generate amplification of archaeal products (data not shown). It is presumed that the presence of archaea in these samples was either at a very low concentration (<100 gene copies per PCR reaction) where the employed method was not able to detect (La Duc et al. 2012), or materials associated with dust samples collected might have inhibited the archaeal DNA amplification. The latter was confirmed as not true since spiking purified archaeal DNA in these samples before archaeal PCR amplification did exhibit appropriate band (data not shown).
Pyrosequencing-derived fungal diversity
Resulting fungal pyrosequence abundance and OTU designations associated with the various samples examined are given in Table 6. In total, 18,635 high-quality fungal pyrosequences comprising 30 distinct fungal OTU were generated in this investigation. Unlike bacterial characterization, where more than 100 OTU pyrosequences were retrieved in both samples, the fungal OTU were less predominant. Among the samples tested, fungal sequences (~8.5-fold increase) and OTU were abundant in ISS-lint (28 OTU), whereas only 1,092 fungal pyrosequences and eight OTU were present in ISS-debris samples. This was surprising since bacterial incidence was higher in ISS-debris samples than in ISS-lint samples (Table 4).
In addition to the 30 taxa listed in Table 6, an additional 11 OTU were present that were closest to sequences from uncultured fungi (most were from other metagenomic studies). Four of them had no matches above the 97 % identity cut-off value. The others were closest to sequences from species: Aureobasidium, Cladosporium, Cryptococcus, Malassezia, Phaeococcomyces, Pichia, and Rhodotorula. Of the total sequences in the data set, approximately 9 % were closest to sequences from plant pathogens, almost 15 % were closest to sequences from saprobes, 17 % were closest to those from fungi that produce allergens, and more than 32 % were closest to sequences from fungi that are either human pathogens or are opportunistic human pathogens.
Discussion
Exposures to bioaerosols in the occupational environment are associated with a wide range of health issues, including infectious diseases, acute toxic effects, allergies, and cancer (Douwes et al. 2003). Hence NASA is undertaking several approaches to utilize state-of-the art molecular techniques as per the NRC recommendations. In addition, characterizing microbiological profiles of ISS surfaces would benefit NASA to develop appropriate countermeasures and enable to better maintain closed habitats for long duration missions (NRC 2011).
Results of the culture-based analysis of the vacuum cleaner bag samples indicated that there is low probability that the health of a robust crewmember would be affected due to the bacteria (Bacillus and spore-formers) cultured, whereas the presence of Aspergillus and Penicillium might be a concern. It is interesting to note that traditional cultural conditions promoted the growth of Bacillus and related species, whereas sequences of these spore-forming species were not retrieved from the very same samples when molecular methods were employed. The spore-forming species in these samples were presumed to be present as spores whose nucleic acids might not have been released by the DNA extraction method employed. However, the previous model microbial community analysis employed showed that the automated DNA extraction method used in this study was suitable and proven to lyse open variety of microbial species including spores, actinobacteria, archaea, and fungal organisms (Kwan et al. 2011). A possible reason for the absence of spore-forming bacteria in pyrosequencing method might be that the spores of wild-type Bacillus species might be hardier than laboratory-purified bacterial spores; hence, DNA extraction might have been difficult. However, when spores sensed nutrients present in TSA and R2A, spores might have germinated and formed colonies on the plates.
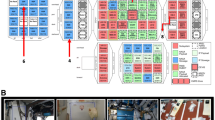
The viable microbial diversity estimated via molecular methods was far more diverse than that observed via conventional plate count assays. The differences in the abundance of OTU present in ISS-debris and lint samples were plotted as a heat map, in combination with their phylogenetic relationship and correlation with the viability (PMA treatment) (Fig. 1). As expected, the majority of the bacterial OTU were abundant in ISS-debris sample, and most of these were not viable in the samples tested. The heat map analysis further showed that OTU belonging to potential human associated microbes (such as Actinomyces, Propionibacterium, Cornybacterium, Staphylococcus, and Clostridium) were found to be abundant and viable. This might be attributed the fact that, during deep sequencing when there was no competition, the low abundant microorganisms with ease would have amplified by the pyrosequencing method. One possible explanation might be that the saturation of beads in the pyrosequencing methods employed. For example, the DNA from dead microorganisms (PMA untreated), along with viable ones (PMA-treated), would have saturated the availability of the beads (~25,000 beads), whereas the DNA fragments from viable microorganisms might have no competition and have had enough beads during emulsion PCR before the attached fragments sequenced in pyrosequencing. The results of this study also showed that pyrosequences of PMA-untreated samples were >25,000, whereas PMA-treated samples were <20,000 in ISS-debris samples (Table 3).

Heat map of OTU across ISS debris and lint sample treated with and without PMA. OTU with more than ten sequences in at least one sample were considered to construct this heat map. Blue represents low OTU abundance while red represents high abundance
Even though >90 % of the pyrosequences retrieved were associated with viable members of the actinobacteria during this study, the virulence, pathogenicity, and antibacterial resistance of these strains should be carried out after their isolation before concluding the significance of their high level of incidence in ISS-debris samples. It should be noted that the culture-based study did not reveal the presence of Actinomyces, Corynebacterium, or Propionibacterium, since cultivation of these bacteria requires different protocols than the procedures adapted during this study, whereas the molecular methods employed showed presence of these viable bacteria. The members of the above-mentioned actinobacterial genera have been reported to cause occupational asthma and pneumonitis (Hagemeyer et al. 2013; Tsapko et al. 2011).
Actinobacteria constitute a large group of Gram-positive bacteria commonly found in the soil and indoor environments (Rintala 2011). Exposure to actinobacteria in indoor environments might be continuous, since they were both common environmental bacteria (La Duc et al. 2012) and associated with humans (Ventura et al. 2007). However, the occurrence of some species of spore-forming filamentous Actinomyces has been associated with moisture damage in buildings (Huttunen et al. 2008) and health-hazardous situations such as allergic respiratory diseases (Hagemeyer et al. 2013). Hypersensitivity pneumonitis and occupational asthma were diagnosed in workers exposed in confined places while working in composting sites and packaging recycling plants (Hagemeyer et al. 2013). Fungal and actinomycetes were identified as allergens and specific antibodies to Aspergillus and Actinomyces were detected in these confined spaces (Hagemeyer et al. 2013). In vitro and in vivo studies have shown that actinobacteria are very immunoactive and, hence, potential causative agents for respiratory and other disorders (Brüggemann et al. 2004; Rintala 2011). P. acnes, an actinobacteria, was reported to be relatively slow-growing and typically aerotolerant, but this anaerobic bacterium associated with the skin condition acne causes chronic blepharitis and endophthalmitis (Dali et al. 2001). This bacterium is usually undetectable, largely commensal, and normally found in the skin and gastrointestinal tract of humans (Perry and Lambert 2011). In addition to the actinobacteria, the prevalence of S. epidermidis among the ISS samples, in both cultured and molecular methods, was not surprising since Staphylococcus species were associated with skin in healthy adults (Human Microbiome Project 2012).
Integrating research findings on fungal characteristics of indoor environments since the 1940s, a holistic interpretation emerged for sick building syndrome and suggested that in particular instances some fungi could be used as bioindicators of indoor air quality (Cabral 2010). Houses and buildings, with low indoor humidity, displayed lower degrees of indoor fungal growth and outdoor environments were dominated by Cladosporium species. On the contrary, in sick houses and buildings, high indoor humidity allowed higher degrees of fungal growth (mainly of Penicillium and Aspergillus), which was associated with the release of conidia and hyphal fragments into the atmosphere. Isolation of Penicillium and Aspergillus via both culture based analysis as well as modern molecular methods in this study suggested that appropriate precautions should be taken to maintain ISS. Retrieval of pyrosequences of Trichosporon species from the lint sample might be a concern, since invasive infections caused by Trichosporon have emerged in immuno-compromised patients with hematological malignancies, a type of cancer that affects blood, bone marrow, and lymph nodes (Hashino et al. 2013). Monitoring these bioindicator fungal species, and subsequent countermeasures, would promote improved air quality and the health and well-being of astronauts, and increase our understanding of the biology of ISS environmental microbiome.
A total of 32 % of the total number of sequences determined in this study were closest to sequences from opportunistic pathogenic fungi, such as species of Aspergillus, Candida, Cryptococcus, and Trichosporon. Additionally, several of the species indicated in this study have been associated with serious diseases, including lung inflammation caused by Penicillium spinulosum, sepsis and wound infection by Candida parapsilosis, and renal disease caused by Penicillium aurantiogriseum (Jussila et al. 2002; Mantle et al. 1991; Trofa et al. 2008). Some of these are often associated with humans, but in those with weakened immune systems, serious infections can occur. An additional 17 % of the sequences were most similar to those from fungi that are known allergens, including species of Penicillium and Aspergillus (Chaudhary and Marr 2011; Ward et al. 2010). On extended flights, the concentrations of these potential pathogens should be kept at a minimum. In addition to the human pathogenic fungi, in this study, approximately 9 % of the pyrosequences were similar to those from known plant pathogens, such as Fusarium equiseti, Penicillium digitatum and some Dothidiomycetes spp. Since researchers are at present conducting plant growth experiments in ISS (NASA 2010), there is a possibility that these plant pathogenic fungi might have originated from such plants that are coexisting in ISS with crewmembers. While it is possible to eliminate the surface contaminants from the plant seeds, and possibly from the plants, it is not practical to remove every fungal or bacterial spore from the astronauts and from the space vehicle. Therefore, there is a distinct likelihood that some of the spores would be plant pathogens that would be of major concern to those attempting to grow plants.
The Expedition 31 crewmembers were exposed constantly to the dust in a closed system, and also reported allergies. Subsequent microbiological analyses using traditional methods noticed no anomaly and also not able to predict the causative microbial agents. The next-generation sequencing study provided NASA with the ability to assess the spectrum of microorganisms associated with the ISS surfaces and would help to predict possible microbial agents that might affect crew health. The results also exhibited presence of representative of human associated (skin, gastrointestinal track, wounds, etc.) bacterial commensals and opportunistic fungal pathogens. Furthermore, based on the phylogenetic affiliation it is possible to determine whether these viable microorganisms are hardy and robust that have been previously reported to survive in oligotrophic environments (cleanrooms, hospital, commercial airliner, etc.). The study has improved our understanding of background microbial contamination, thus facilitating the development of biosensors to monitor closed habitats like ISS and future manned missions.
References
ASTM-E11 (1995) Standard Specification for Woven Wire Test Sieve Cloth and Test Sieves. In: ASTM (ed). vol 33 CFR 159.125. American Society for Testing and Materials, Washington DC
Benardini J, Ballinger J, Crawford R, Roman M, Sumner R, Venkateswaran K In: 34th International Conference on Environmental Systems, International Space Station Internal Thermal Coolant System: an initial assessment of the microbial communities within fluids from ground support and flight hardware, July 2005, Rome, Italy, 2005. SAE Technical paper series
Boyden DG (1962) The bacterial flora in fleet ballistic missile submarines during prolonged submergence. In: U.S. Naval Medical Research Laboratory Report No. 386. In: Bureau of Medicine and Surgery ND (ed). vol 21 (17)
Brüggemann H, Henne A, Hoster F, Liesegang H, Wiezer A, Strittmatter A, Hujer S, Dürre P, Gottschalk G (2004) The complete genome sequence of Propionibacterium acnes, a commensal of human skin. Science 305(5684):671–673. doi:10.1126/science.1100330
Burge HA, Pierson DL, Groves TO, Strawn KF, Mishra SK (2000) Dynamics of airborne fungal populations in a large office building. Curr Microbiol 40(1):10–16
Cabral JPS (2010) Can we use indoor fungi as bioindicators of indoor air quality? Historical perspectives and open questions. Sci Total Environ 408(20):4285–4295. doi:10.1016/j.scitotenv.2010.07.005
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Pena AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R (2010) QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7(5):335–336. doi:10.1038/nmeth.f.303
Castro VA, Thrasher AN, Healy M, Ott CM, Pierson DL (2004) Microbial characterization during the early habitation of the International Space Station. Microb Ecol 47(2):119–126
Chaudhary N, Marr KA (2011) Impact of Aspergillus fumigatus in allergic airway diseases. Clin Transl Allergy 1(1):4. doi:10.1186/2045-7022-1-4
Crous PW, Gams W, Stalpers JA, Robert V, Stegehuis G (2004) MycoBank: an online initiative to launch mycology into the 21st century. Stud Mycol 50:19–22
Dali P, Giugliano ER, Vellozzi EM, Smith MA (2001) Susceptibilities of Propionibacterium acnes ophthalmic isolates to moxifloxacin. Antimicrob Agents Chemother 45(10):2969–2970. doi:10.1128/aac.45.10.2969-2970.2001
DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, Huber T, Dalevi D, Hu P, Andersen GL (2006) Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol 72(7):5069–5072
Douwes J, Thorne P, Pearce N, Heederik D (2003) Bioaerosol health effects and exposure assessment: progress and prospects. Ann Occup Hyg 47(3):187–200. doi:10.1093/annhyg/meg032
Dowd SE, Sun Y, Secor PR, Rhoads DD, Wolcott BM, James GA, Wolcott RD (2008) Survey of bacterial diversity in chronic wounds using pyrosequencing, DGGE, and full ribosome shotgun sequencing. BMC Microbiol 8:43. doi:10.1186/1471-2180-8-43
Edgar RC (2010) Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26(19):2460–2461. doi:10.1093/bioinformatics/btq461
Edmiston CE Jr, Seabrook GR, Cambria RA, Brown KR, Lewis BD, Sommers JR, Krepel CJ, Wilson PJ, Sinski S, Towne JB (2005) Molecular epidemiology of microbial contamination in the operating room environment: is there a risk for infection? Surgery 138(4):579–582
Ferguson JK, Taylor GR, Mieszkuc BJ (1975) Microbiological investigations, pp 83–103. Scientific and Technical Information Office. National Aeronautics and Space Administration, Washington, DC
Fierer N, Lauber CL, Zhou N, McDonald D, Costello EK, Knight R (2010) Forensic identification using skin bacterial communities. PNAS 107(14):6477–6481. doi:10.1073/pnas.1000162107
Hagemeyer O, Bünger J, Kampen V, Raulf-Heimsoth M, Drath C, Merget R, Brüning T, Broding HC (2013) Occupational allergic respiratory diseases in garbage workers: relevance of molds and Actinomycetes. In: Pokorski M (ed) Neurobiology of eespiration. Adv Exp Med Biol 788:313–320, Springer Netherlands
Hashino S, Takahashi S, Morita R, Kanamori H, Onozawa M, Kawamura T, Kahata K, Kondo T, Tokimatsu I, Sugita T, Akizawa K, Asaka M (2013) Fungemia due to Trichosporon dermatis in a patient with refractory Burkitt's leukemia. Blood Res 48(2):154–156
Human Microbiome Project C (2012) Structure, function and diversity of the healthy human microbiome. Nature 486(7402):207–214. doi:10.1038/nature11234
Huttunen K, Rintala H, Hirvonen M-R, Vepsäläinen A, Hyvärinen A, Meklin T, Toivola M, Nevalainen A (2008) Indoor air particles and bioaerosols before and after renovation of moisture-damaged buildings: the effect on biological activity and microbial flora. Environ Res 107(3):291–298. doi:10.1016/j.envres.2008.02.008
Jussila J, Komulainen H, Kosma VM, Pelkonen J, Hirvonen MR (2002) Inflammatory potential of the spores of Penicillium spinulosum isolated from indoor air of a moisture-damaged building in mouse lungs. Environ Toxicol Pharmacol 12(3):137–145
Katherine T, Steve L, Rachel E (2010) Development of a modified vacuum cleaner for lunar surface systems. 40th International Conference on Environmental Systems. International Conference on Environmental Systems (ICES). American Institute of Aeronautics and Astronautics, Washington, DC
Kawamura Y, Li Y, Liu H, Huang X, Li Z, Ezaki T (2001) Bacterial population in Russian space station "Mir". Microbiol Immunol 45(12):819–828
Koenig DW, Pierson DL (1997) Microbiology of the Space Shuttle water system. Water Sci Technol 35(11–12):59–64
Kwan K, Cooper M, La Duc MT, Vaishampayan P, Stam C, Benardini JN, Scalzi G, Moissl-Eichinger C, Venkateswaran K (2011) Evaluation of procedures for the collection, processing, and analysis of biomolecules from low-biomass surfaces. Appl Environ Microbiol 77(9):2943–2953. doi:10.1128/aem.02978-10
La Duc MT, Nicholson W, Kern R, Venkateswaran K (2003) Microbial characterization of the Mars Odyssey spacecraft and its encapsulation facility. Environ Microbiol 5(10):977–985
La Duc MT, Dekas A, Osman S, Moissl C, Newcombe D, Venkateswaran K (2007) Isolation and characterization of bacteria capable of tolerating the extreme conditions of clean room environments. Appl Environ Microbiol 73(8):2600–2611. doi:10.1128/AEM.03007-06
La Duc MT, Vaishampayan P, Nilsson HR, Torok T, Venkateswaran K (2012) Pyrosequencing-derived bacterial, archaeal, and fungal diversity of spacecraft hardware destined for Mars. Appl Environ Microbiol 78(16):5912–5922. doi:10.1128/AEM.01435-12
Levine HB, Cobet AB (1970) The tektite-I dive. Mycological aspects. Arch Environ Health 20(4):500–505
Mantle PG, McHugh KM, Adatia R, Heaton JM, Gray T, Turner DR (1991) Penicillium aurantiogriseum-induced, persistent renal histopathological changes in rats; an experimental model for Balkan endemic nephropathy competitive with ochratoxin A. IARC Sci Publ 115:119–127
McDonald D, Price MN, Goodrich J, Nawrocki EP, Desantis TZ, Probst A, Andersen GL, Knight R, Hugenholtz P (2012) An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J 6(3):610–618. doi:10.1038/ismej.2011.139
Moissl C, Hosoya N, Bruckner J, Stuecker T, Roman M, Venkateswaran K (2007) Molecular microbial community structure of the Regenerative Enclosed Life Support Module Simulator air system. Int J Astrobiol 6(2):131–145
Morris JE (1972) Microbiology of the submarine environment. Proc R Soc Med 65(9):799–800
NASA (2010) Reference guide to the Inernational Space Station. In: NASA HQ (ed). vol NP-2010-09-682-HQ. National Aeronautics and Space Administration, Washington, DC
Newcombe DA, LaDuc MT, Vaishampayan P, Venkateswaran K (2008) Impact of assembly, testing, and launch operations on the airborne bacterial diversity within a spacecraft assembly facility clean-room. Int J Astrobiol 7(3–4):223–236
Nocker A, Richter-Heitmann T, Montijn R, Schuren F, Kort R (2010) Discrimination between live and dead cellsin bacterial communities from environmental water samples analyzed by 454 pyrosequencing. Int Microbiol 13(2):59–65
NRC (2011) Committee for the decadal survey on biological physical sciences in space: recapturing a future for space exploration: life and physical sciences research for a new era. The National Academies Press
Perry JL, Coston JE (2014) Analysis of particulate and fiber debris samples returned from the International Space Station. In: 44th International Conference on Environmental Systems, Tucson, AZ, July 13–17, 2014. Curran Associates, Inc
Perry A, Lambert P (2011) Propionibacterium acnes: infection beyond the skin. Expert Rev Anti-Infect Ther 9(12):1149–1156. doi:10.1586/eri.11.137
Pierson DL (2001) Microbial contamination of spacecraft. Gravit Space Biol Bull 14(2):1–6
Pierson DL, Ott CM, Groves TO (2002) Characterization of microbial activity in the chamber systems and environment. Univelt, San Diego, pp 229–259
Rawsthorne H, Dock CN, Jaykus LA (2009) PCR-based method using propidium monoazide to distinguish viable from nonviable Bacillus subtilis spores. Appl Environ Microbiol 75(9):2936–2939. doi:10.1128/AEM.02524-08
Rintala H (2011) Actinobacteria in indoor environments: exposures and respiratory health effects. Front Biosci (Sch Ed) 3:1273–1284
Schloss PD, Gevers D, Westcott SL (2011) Reducing the effects of PCR amplification and sequencing artifacts on 16S rRNA-based studies. PLoS One 6(12):e27310. doi:10.1371/journal.pone.0027310
Stenberg B, Eriksson N, Hansson M. K., Ho¨o¨g J, Sandstro¨m M, Sundell J, Wall S (1993) The Office Illness Project in northern Sweden. An interdisciplinary study of the “sick building-syndrome” (SBS). In: Indoor Air'93, Helsinki, Finland
Suzuki MT, Taylor LT, DeLong EF (2000) Quantitative analysis of small-subunit rRNA genes in mixed microbial populations via 5′-nuclease assays. Appl Environ Microbiol 66(11):4605–4614
Taylor GR, Graves RC, Brockett RM, Ferguson JK, Mieszkuc BJ (1977) Skylab environmental and crew microbiological studies. In: Johnston R, Dietlein LF (eds) Biomedical results from Skylab. Scientific and Technical Information Office, National Aeronautics and Space Administration, Washington, DC, pp 53–63
Thomas TL, Hooper TI, Camarca M, Murray J, Sack D, Mole D, Spiro RT, Horn WG, Garland FC (2000) A method for monitoring the health of US Navy submarine crewmembers during periods of isolation. Aviat Space Environ Med 71(7):699–705
Trofa D, Gacser A, Nosanchuk JD (2008) Candida parapsilosis, an emerging fungal pathogen. Clin Microbiol Rev 21(4):606–625. doi:10.1128/cmr.00013-08
Tsapko VG, Chudnovets AJ, Sterenbogen MJ, Papach VV, Dutkiewicz J, Skórska C, Krysińska-Traczyk E, Golec M (2011) Exposure to bioaerosols in the selected agricultural facilities of the Ukraine and Poland — a review. Ann Agric Environ Med AAEM 18(1):19–27
Upsher JF, Fletcher LE, Upsher CM (1994) Microbiological conditions on Oberon submarines. Department of Defence, Defence Science and Technology Organisation, Melbourne
Vaishampayan P, Probst AJ, La Duc MT, Bargoma E, Benardini JN, Andersen GL, Venkateswaran K (2013) New perspectives on viable microbial communities in low-biomass cleanroom environments. ISME J 7(2):312–324 doi:http://www.nature.com/ismej/journal/v7/n2/suppinfo/ismej2012114s1.html
Venkateswaran K, Hattori N, La Duc MT, Kern R (2003) ATP as a biomarker of viable microorganisms in clean-room facilities. J Microbiol Methods 52(3):367–377
Venkateswaran K, La Duc MT, Vaishampayan P (2012) Genetic inventory task: final report, JPL Publication 12–12, vol 1. Jet Propulsion Laboratory, California Institute of Technology, Pasadena, pp 1–117
Ventura M, Canchaya C, Tauch A, Chandra G, Fitzgerald GF, Chater KF, van Sinderen D (2007) Genomics of Actinobacteria: tracing the evolutionary history of an ancient phylum. Microbiol Mol Biol Rev 71(3):495–548. doi:10.1128/mmbr.00005-07
Ward MD, Chung YJ, Copeland LB, Doerfler DL (2010) A comparison of the allergic responses induced by Penicillium chrysogenum and house dust mite extracts in a mouse model. Indoor Air 20(5):380–391. doi:10.1111/j.1600-0668.2010.00660.x
Acknowledgments
Part of the research described in this publication was carried out at the Jet Propulsion Laboratory, California Institute of Technology, under a contract with NASA. This research was funded by a 2012 Space Biology NNH12ZTT001N grant # 19-12829-26 under Task Order NNN13D111T award to K. Venkateswaran. J. Cisneros, a Louis Stokes Alliance for Minority Participation – Bridge to the Doctorate (LSAMP-BD) fellow, was supported by the LSAMP-BD (Cohort X) program (National Science Foundation grant # HRD-1246662). The authors gratefully acknowledge ISS Expedition 31 crew for sample collection and D. Eisenman, JPL for effective coordination of sample procurement. We would also like to thank JSC Microbiology Laboratory members in performing culture-based analysis and Y. Sun (Research and Testing Laboratory) for trouble shooting discussion on the pyrosequencing method. The authors are indebted to M. Jones and C. Guethe from JPL for critical review of this manuscript. © 2014 California Institute of Technology. Government sponsorship acknowledged.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 1761 kb)
Rights and permissions
About this article
Cite this article
Venkateswaran, K., Vaishampayan, P., Cisneros, J. et al. International Space Station environmental microbiome — microbial inventories of ISS filter debris. Appl Microbiol Biotechnol 98, 6453–6466 (2014). https://doi.org/10.1007/s00253-014-5650-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-014-5650-6