
Abstract
Shikimate and 3-dehydroshikimate are useful chemical intermediates for the synthesis of various compounds, including the antiviral drug oseltamivir. Here, we show an almost stoichiometric biotransformation of quinate to 3-dehydroshikimate by an engineered Gluconobacter oxydans strain. Even under pH control, 3-dehydroshikimate was barely detected during the growth of the wild-type G. oxydans strain NBRC3244 on the medium containing quinate, suggesting that the activity of 3-dehydroquinate dehydratase (DHQase) is the rate-limiting step. To identify the gene encoding G. oxydans DHQase, we overexpressed the gox0437 gene from the G. oxydans strain ATCC621H, which is homologous to the aroQ gene for type II DHQase, in Escherichia coli and detected high DHQase activity in cell-free extracts. We identified the aroQ gene in a draft genome sequence of G. oxydans NBRC3244 and constructed G. oxydans NBRC3244 strains harboring plasmids containing aroQ and different types of promoters. All recombinant G. oxydans strains produced a significant amount of 3-dehydroshikimate from quinate, and differences between promoters affected 3-dehydroshikimate production levels with little statistical significance. By using the recombinant NBRC3244 strain harboring aroQ driven by the lac promoter, a sequential pH adjustment for each step of the biotransformation was determined to be crucial because 3-dehydroshikimate production was enhanced. Under optimal conditions with a shift in pH, the strain could efficiently produce a nearly equimolar amount of 3-dehydroshikimate from quinate. In the present study, one of the important steps to convert quinate to shikimate by fermenting G. oxydans cells was investigated.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Shikimate and 3-dehydroshikimate (3-DHS) are common synthetic intermediates of aromatic amino acids in the shikimic acid pathway in plants, fungi, and bacteria. The interconversion of these intermediates is catalyzed by shikimate dehydrogenase. Both chemicals are used as raw materials for the synthesis of compounds, such as the antiviral drug oseltamivir, because each molecule has a six-member carbon ring with two asymmetric carbons (three asymmetric carbons for shikimate), which are difficult to chemically synthesize. In addition, microbial fermentation to produce 3-DHS is useful because 3-DHS is an antioxidant (Richman et al. 1996).
Naturally, shikimate can be obtained by extracting the fruits of the Chinese plant star anise (Illicium vernum; "tou-shikimi" in Japanse) or produced from d-glucose using a native shikimate-producing bacterial isolate, Citrobacter freundii (Tripathi et al. 2013). The high demand for drug development has driven the development of a more efficient fermentation process using a recombinant Escherichia coli strain that overexpresses the feedback resistant 3-deoxy-d-arabino-heptulosonic acid 7-phosphate synthase (AroFFBR), shikimate dehydrogenase, transketolase, and phosphoenolpyruvate synthase (Chandran et al. 2003).
Alternatively, the Gluconobacter oxydans strain NBRC3244 (formerly G. oxydans IFO3244) has the potential to transform quinate to shikimate using the following three enzymes that are essential for this biotransfomation: membrane-bound quinate dehydrogenase (QDH) (Adachi et al. 2003b), 3-dehydroquinate dehydratase (DHQase), and NADP+-dependent shikimate dehydrogenase (Fig. 1). NAD+- and NADP+-independent oxidations of quinate by Gluconobacter spp. were reported for the first time in 1967 (Whiting and Coggins 1967). A similar enzyme, QDH, has been identified as a pyrroloquinoline quinone (PQQ)-dependent enzyme in Acinetobacter sp. (van Kleef and Duine 1988). We purified the PQQ-dependent QDH from G. oxydans NBRC3244 and cloned the gene for QDH of NBRC3244 to heterologously express it in Pseudomonas sp. (Vangnai et al. 2004, 2010). The biotransformation of quinate to 3-dehydroquinate (3-DHQ) by QDH has been reported for growing G. oxydans NBRC3244, G. oxydans NBRC3292, G. oxydans (formerly Gluconobacter melanogenus) NBRC3294, and Gluconacetobacter liquefaciens (formerly G. liquefaciens) NBRC12388 cells (Adachi et al. 2003a). Furthermore, the biotransformation of 3-DHQ to 3-DHS is catalyzed by DHQase. To make this biotransformation step occur easily, several processes were developed using either Ca2+-alginate-immobilized Gluconobacter cells, dried Gluconobacter membranes, or Ca2+-alginate-immobilized Gluconobacter membranes (Adachi et al. 2003a, 2006a, b, 2010). Even from quinate, 3-DHS is produced by dried Gluconobacter cells (Adachi et al. 2003a). Finally, 3-DHS is quantitatively reduced to shikimate in vitro using one of the following methods: purified shikimate dehydrogenase and NADPH, coupled with an NADPH-regeneration system, consisting of an NADP+-dependent glucose dehydrogenase of G. oxydans and d-glucose (Adachi et al. 2006b, c); or immobilized enzymes (Adachi et al. 2010). Because quinate is readily available as a natural plant extract, it can be recovered from chlorogenate in coffee pulp waste via a reaction with of fungus-derived chlorogenate hydrolase (Adachi et al. 2008a). Thus, an efficient and sustainable shikimate bioproduction process can be developed using a combination of biotransformation by Gluconobacter spp. and an industrial waste utilization strategy (Fig. 1).

Biotransformation of chlorogenate to shikimate by Aspergillus chlorogenate hydrolase and several Gluconobacter enzymes. Of all the whole cell reactions used to produce shikimate, the part described in this study is highlighted in gray. QDH quinate dehydrogenase, 3-DHQ 3-dehydroquinate, DHQase 3-dehydroquinate dehydratase, 3-DHS 3-dehydroshikimate, DHSase 3-dehydroshikimate dehydratase, SKDH shikimate dehydrogenase
Even though the formation of 3-DHS has been shown using the enzymatic reactions of Gluconobacter spp., 3-DHS has not been clearly detected in growing Gluconobacter cells on the medium containing quinate, presumably due to poor catalysis of the wild type cells under the conditions used. Here, we engineered a Gluconobacter strain to overexpress DHQase. By characterizing the recombinant strain and optimizing fermentation conditions, quantitative 3-DHS production from quinate was achieved under conditions with strict pH control. This study represents the first attempt to develop a shikimate fermentation process from industrial waste.
Materials and methods
Materials
3-DHQ and 3-DHS were prepared according to the method described previously (Adachi et al. 2006a). Restriction endonucleases and modification enzymes for genetic engineering were either gifts from Toyobo (Osaka, Japan) or purchased from Agilent Technologies (Santa Clara, CA, USA). Yeast extract was also a generous gift from Oriental Yeast (Osaka, Japan). All other materials were commercially available and of a guaranteed grade.
Bacterial strains, plasmids, and growth conditions
The bacterial strains and plasmids used in this study are listed in Table 1. G. oxydans strains NBRC3244, NBRC3292, NBRC3293, NBRC3294, and ATCC621H were used in this study. A broad host range vector pBBR1MCS-4 was used for the overexpression of the aroQ gene in G. oxydans. Gluconobacter spp. were grown on ∆P (5 g glucose, 20 g glycerol, 10 g polypeptone, and 10 g yeast extract per liter), QA-I (1 g glycerol, 1 g polypeptone, 3 g yeast extract per liter, and 50 mM sodium quinate), and QA-II (2 g glycerol, 1 g polypeptone, 3 g yeast extract, 2.5 g MgSO4 · 7H2O per liter, 50 mM potassium phosphate [pH 7.0], and 26 mM sodium quinate) media at 30 °C with vigorous shaking. When necessary, the pH of the medium was adjusted with either an NaOH or HCl solution. A stock solution of sodium quinate at 0.5 M was prepared by dissolving quinic acid, adjusting the pH to 6–7 with NaOH, and filter sterilizing (0.22 μm) the resulting solution. The resulting solution was stored at −20 °C until further use. Ampicillin was used at a final concentration of 100 μg ml−1.
E. coli DH5α was used for plasmid construction (Bethesda Research Laboratories 1986). E. coli HB101 harboring pRK2013 was used as a helper strain for conjugative plasmid transfer using a triparental mating method (Figurski and Helinski 1979). An E. coli BL21 (DE3) strain was used to harbor the pET vector (Studier and Moffatt 1986). E. coli strains were grown on modified Luria–Bertani medium, consisting of 10 g polypeptone, 5 g yeast extract, and 5 g NaCl per liter, adjusted pH to 7 with NaOH (Sambrook and Russel 2001). Kanamycin and ampicillin were used at final concentrations of 50 and 50 μg ml−1, respectively.
Construction of plasmids
The synthetic oligonucleotides used in this study are listed in Table S1. The gox0437 gene of G. oxydans 621H was amplified using a pair of primers dqdpET_F and dqdpET_R (Table S1). The 0.5-kb PCR product was ligated to pGEM-T Easy Vector (Promega, Madison, WI, USA). The 0.5-kb DNA fragment containing gox0437 was excised with NdeI and XhoI restriction enzymes and inserted into their corresponding sites in pET-21a (Novagen, Merck Millipore, Darmstadt, Germany) to construct pET-GOX0437.
The aroQ gene of G. oxydans NBRC3244 (NBRC3244_0516; MM, SN, TA, and KM, unpublished) was amplified using the pair of primers dqdpMCS-F3-Eco1 and dqdpMCS-R2-Pst1 (Table S1). The 0.6-kb PCR product was ligated into pBBR1MCS-4 (Kovach et al. 1995) behind the lac promoter and pSHO8 (Kawai et al. 2013) treated with EcoRI and PstI restriction enzymes to construct pIS01 and pIS03, respectively. The DNA region containing the aroQ (AB852565) gene and its putative promoter region of G. oxydans NBRC3244 were amplified using the pair of primers dqdpMCS-F4-Eco1 (Table S1) and dqdpMCS-R2-Pst1. The 0.9-kb PCR product was ligated into pBBR1MCS-4 (Kovach et al. 1995) treated with EcoRI and PstI restriction enzymes to construct pIS02. All nucleotide sequences used in cloning were confirmed using cycle sequencing techniques according to the manufacturer's instruction (Applied Biosystems, California, USA).
Construction of an aroQ-overexpressing G. oxydans
The G. oxydans NBRC3244 strain was transformed with plasmids, such as pBBR1MCS-4 derivatives, via a triparental mating method using the E. coli HB101 strain harboring pRK2013 (Figurski and Helinski. 1979). Acetic acid was added to the media for selection at a final concentration of 0.1 % (wt. vol−1) to eliminate E. coli growth. Acetic acid- and ampicillin-resistant conjugant colonies were screened twice on ∆P agar medium containing 0.1 % (wt. vol−1) acetic acid and 100 μg ml−1 ampicillin. Finally, the transconjugants were screened in liquid ∆P medium containing 100 μg ml−1 ampicillin.
Expression of the genes and preparation of cell-free extracts
Modified LB medium containing 50 μg ml−1 ampicillin was inoculated with E. coli BL21 (DE3) transformants and incubated overnight using a shaking incubator at 37 °C; Seed cultures were transferred to fresh LB medium in Erlenmeyer flask at a ratio of 1 % and incubated at the specified temperature with shaking. When the turbidity of the culture at 600 nm reached 0.2 OD units, isopropyl-β-d-thiogalactopyranoside was added to a final concentration of 1 mM to induce gox0437 gene expression. After 5 h, cells were collected by centrifugation at 9,000 rpm for 10 min at 4 °C.
The collected cells were resuspended in 20 mM Tris–HCl (pH 8.0) and disrupted by French pressure cell press at 14,000 lb in−2. Cell debris was removed by centrifugation at 9,000 rpm for 10 min and the resulting supernatant was used as crude enzyme preparation for determining DHQase activity.
Gluconobacter cells were cultivated in 100 ml of QA-I medium, containing 100 μg ml−1 ampicillin, to late exponential growth phase. Cells were collected by centrifugation at 8,000 × g for 10 min, washed twice with 5 mM potassium phosphate (pH 7.2) containing 5 mM 2-mercaptoethanol, and resuspended with the same solution. The cell suspension was passed through a French pressure cell press at 16,000 lb in−2 twice. Cell-free extracts were prepared by removing cell debris.
Detection of 3-DHQ and 3-DHS
3-DHQ and 3-DHS were detected by paper chromatography as previously described (Adachi et al. 2003a). Otherwise, these compounds were analyzed by HPLC using a chromatography column for organic acids (Shodex RSpak KC-811, 8 × 300 mm; Showa Denko, Tokyo, Japan). Chromatography was performed at 60 °C using 0.03 % (vol vol−1) perchloric acid solution as a mobile phase at a flow rate of 0.7 ml min−1. Culture supernatants were diluted (10- to 100-fold) with distilled and deionized water and then filtered by a hydrophilic polytetrafluoroethylene filter (Millipore Millex-LH; 0.45 μm). The compounds were detected by diode array detector (SPD-M20A, Shimazu, Kyoto, Japan) at 210, 190, 234, 213, and 275 nm, which are the absorption peaks for quinate, 3-DHQ, 3-DHS, shikimate, and protocatechuate, respectively.
Enzyme assays
DHQase activity was determined by monitoring the formation of 3-DHS at 234 nm at 25 °C as previously described (Adachi et al. 2008b). One unit of enzyme was defined as the amount of enzyme that produces 1 μmol of 3-DHS per min, which was calculated using the molecular absorption coefficient of 3-DHS at 234 nm (ε = 1.2 × 104 M−1 cm−1; Chaudhuri et al. 1986). Protein content was determined using the modified Lowry method, using bovine serum albumin as the standard protein (Dulley and Grieve 1975).
3-DHS production by growing Gluconobacter cells
Wild-type and recombinant Gluconobacter strains were cultivated on ∆P medium containing 100 μg ml−1 ampicillin when necessary. These seed cultures were transferred at a 1:100 ratio to 2 ml QA-II medium in 15-mm (i.d.) test tubes, 50 ml of QA-II medium in 500-ml Erlenmeyer flask, or 500 ml of QA-II medium in a 3-l baffled flask, and incubated for 23 h or 40 h at 30 °C. For some experiments, the pH of cultures was adjusted to 7.3 using an NaOH solution and the culture was incubated for an additional 20 or 30 h.
Amino acid sequence alignments
Multiple alignment analysis of amino acid sequences was performed using the software Clustal X version 2.0.12 (Larkin et al. 2007).
Nucleotide sequence accession number
The nucleotide sequence and its predicted amino acid sequence of the aroQ gene of G. oxydans NBRC3244 have been deposited at DDBJ/EMBL/GenBank under the accession number AB852565.
Results
Growing wild-type G. oxydans NBRC3244 cells poorly produce 3-DHS
First, we examined the biotransformation of quinate to 3-DHS by the growing cells of wild-type G. oxydans strains NBRC3244, NBRC3292, NBRC3293, and NBRC3294 in QA-I medium. The pH of the medium decreased to approximately 5 during the stationary phase of growth for all bacterial strains. Under these conditions, 3-DHS was not detected, although most quinate was observed to be completely converted to 3-DHQ (data not shown). The decrease in the pH of the medium during cell growth can be a major factor that inhibits the transformation of 3-DHQ to 3-DHS because DHQase has no activity when the pH is less than 5.0 (Adachi et al. 2008b). Therefore, we examined whether controlling the pH could improve the productivity of 3-DHS in the growing cells of the G. oxydans strains using an initial concentration of 50 mM quinate (Fig. S1). When the initial pH of the medium was adjusted to 7.0 with 50 mM phosphate buffer as described in the “Materials and methods” section, the final pH of the medium for all bacterial strains tested ranged from a pH of 6.5–6.7. Further investigation with medium with an initial pH at 7.5 showed that the growth of G. oxydans NBRC3244 was slightly slower than that at pH 7.0 (data not shown). However, the cells could still transform quinate to 3-DHQ with a higher efficiency compared to the other bacterial strains, suggesting that NBRC3244 can be used for the biotransformation of quinate over a wider pH range than other strains. Increasing the initial pH of the medium (e.g., pH 8.0) inhibited the cell growth of all G. oxydans strains (data not shown). Even though, the pH of the medium remained neutral when the initial pH was 7.5, 3-DHS formation could be detected at only trace concentrations (0.5–0.6 mM) in NBRC3244 cultures after a 50-h period. We hypothesized that the DHQase level in G. oxydans cells is a rate-limiting step for the production of 3-DHS. Therefore, before trying to construct a DHQase-overexpression derivative of NBRC3244, we examined a candidate gene for DHQase in G. oxydans strains.
Expression of G. oxydans DHQase in E. coli
Even though the N-terminal amino acid sequence of membrane-bound DHQase purified from NBRC3244 corresponds with that of the GOX0437 protein from G. oxydans 621H strain (Adachi et al. 2008b), the in vitro activity of this protein candidate needed to be confirmed. Thus, we constructed a pET-21a plasmid harboring the gox0437 gene and transformed E. coli BL21 (DE3) with this plasmid. When the E. coli BL21 (DE3)/pET-GOX0437 cells were grown at 37 °C, a large part of the 20-kDa protein corresponding to DHQase was detected in the cell debris fraction when analyzed by SDS-PAGE, suggesting that most of the expressed DHQase formed inclusion bodies (Fig. S2). Because high growth temperatures promote the formation of inclusion bodies of the recombinant protein (Strandberg and Enfors 1991), transformant cells were cultivated at 30 °C to mitigate the problem. SDS-PAGE analysis revealed that the 20-kDa protein was more soluble at this temperature and the amount of protein in the debris fraction was reduced (Fig. S2). The DHQase activity of cell-free extracts of transformants grown at 30 °C was 11 units (mg protein)−1, which was 4-fold higher than that at 37 °C (Table 2). After confirming that the product of the gox0437 gene had DHQase activity, we aimed to construct a G. oxydans NBRC3244 strain overexpressing the GOX0437 protein. As described below, we tried homologous overexpression of the gene for DHQase in NBRC3244.
Construction of aroQ-overexpression strains of G. oxydans NBRC3244
Recently, a draft genome sequence of G. oxydans NBRC3244 was successfully annotated (MM, SN, TA, and KM, unpublished). We successfully found a gox0437 ortholog in NBRC3244 by mining the genome. Based on its sequence similarity and classification as a DHQase, the gox0437 ortholog likely belongs to a type II DHQase known as aroQ in most cases (Garbe et al. 1991). Hereafter, we will refer to the gene for G. oxydans DHQase as G. oxydans aroQ.
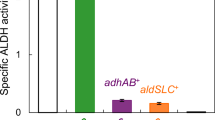
In order to optimize the gene expression level, we constructed three aroQ-expression plasmids containing the following promoters: E. coli lac promoter (P lac ), putative promoter region of G. oxydans PQQ-dependent alcohol dehydrogenase subunits I and II (P adhAB ), and putative aroQ-own promoter region (P aroQ ) as described in the “Materials and methods” section. The biotransformation of quinate by each G. oxydans NBRC3244 transformant was monitored in 2 ml QA-II medium containing 26 mM quinate (Fig. 2). When the wild-type and all recombinant NBRC3244 strains were grown for 48 h, quinate was not detected but appeared to be oxidized to 3-DHQ, which was detected in the culture medium. On the contrary, a significant amount of 3-DHS accumulated only in the culture medium of transformants harboring the aroQ gene with any kind of promoter. Although differences in the promoter among the three aroQ-expression plasmids had a less significant impact on 3-DHS productions (0.03 < p < 0.1, Student t test), the transformant harboring P lac -aroQ with a relatively higher 3-DHS yield was selected for further investigation. DHQase activity in cell-free extracts of NBRC3244/pIS01 (P lac -aroQ) was 0.62 ± 0.07 units mg−1, which is much higher than that of the control parental strain (<0.002 units mg−1; less than detection limit). Thus, it is reasonable to assume that the overexpression of aroQ elevated the level of DHQase activity, resulting in high 3-DHS production in NBRC3244/pIS01.

The production of 3-DHS by growing G. oxydans cells. G. oxydans NBRC3244 and its transformants were grown in 2 ml QA-II medium in tubes for 48 h. The amounts of 3-DHQ (gray bars) and 3-DHS (black bars) in the culture supernatant were determined by HPLC. With the exception of wild type, the data shown represent mean values with standard deviations (error bars) from triplicate experiments. WT wild-type NBRC3244, vector NBRC3244/pBBR1MCS-4, P lac -aroQ NBRC3244/pIS01, P aroQ -aroQ NBRC3244/pIS02, P adhAB -aroQ NBRC3244/pIS03
Quantitative production of 3-DHS by maintaining neutral pH
Although 3-DHS production increased by overexpressing the aroQ gene, the transformation efficiency of approximately 23 % mol yield was still unsatisfactory. We hypothesized that a gap between the optimum pH for DHQase activity (Adachi et al. 2008b) and the pH of the culture medium may limit 3-DHS production. To examine this hypothesis and enhance 3-DHS production, we adjusted the pH of the culture medium during fermentation as described under the “Materials and Methods” section. NBRC3244 and NBRC3244/pIS01 (P lac -aroQ) were grown in QA-II medium (50 ml) in a 500-ml Erlenmeyer flask for 23 h, after which the biotransformation of quinate to 3-DHQ was expected to proceed by monitoring the pH of the medium. Then, the pH of the culture medium shifted from 6.6 to 7.3, which is closer to the optimum pH for DHQase, and the culture was grown for an additional 20 h to produce more 3-DHS (Fig. 3). The pH adjustment resulted in almost a 3-fold enhancement of 3-DHS production in NBRC3244/pIS01 compared to cultures with uncontrolled pH.

The production of 3-DHS by growing G. oxydans cells in midi-scale culture experiments. Wild-type G. oxydans NBRC3244 and NBRC3244 harboring pIS01 were grown in 50 ml of QA-II medium in a 500-ml Erlenmeyer flask. After 23 h of cultivation, the pH of the medium was adjusted from 6.6 to 7.3 with an NaOH solution and then grown for an additional 20 h (indicated by " + "). Control experiments without pH adjustment were also conducted (indicated by " − "). The amounts of quinate, 3-DHQ (gray bars), and 3-DHS (black bars) in the culture supernatant were determined by HPLC. Quinate was not detected in all four samples. WT wild-type NBRC3244, aroQ ++ NBRC3244/pIS01; black diamonds, final pH of the culture media
Because controlling the pH increased 3-DHS production in NBRC3244/pIS01, we validated the efficiency of the pH adjustment by monitoring the amounts of compounds over longer periods than the preceding experiment using 500 ml of QA-II medium in a 3-l baffled flask. In this experiment, the pH of the culture was adjusted from 6.5 to 7.3 during an incubation time of 40 h, and the culture continued to grow for 30 h (Fig. 4). Both the oxidation of quinate and the subsequent dehydration of 3-DHQ proceeded quantitatively, and 3-DHS was stoichiometrically produced from quinate over an incubation period of 70 h.

Time course of the amounts of quinate, 3-DHQ, and 3-DHS accumulating in the culture supernatant of recombinant G. oxydans. G. oxydans NBRC3244 harboring pIS01 was grown in 500 ml of QA-II medium in a 3-L baffled flask. After 40 h of cultivation, the pH of the medium was adjusted from 6.5 to 7.3 using an NaOH solution, and the culture was grown for an additional 30 h. a Cell growth (OD660, open circles) and the pH of the medium (closed circles) were plotted as a function of cultivation periods. b The amounts of quinate (open circles), 3-DHQ (open triangles), and 3-DHS (closed circles) in the culture supernatant were determined by HPLC
Discussion
We previously reported that 3-DHQ could be produced from quinate by growing G. oxydans cells (Adachi et al. 2003a). 3-DHQ can be further transformed to 3-DHS by Ca2+-alginate-immobilized or dried Gluconobacter cells as well as immobilized or dried Gluconobacter membranes (Adachi et al. 2003a, 2006a, b, 2010). Here, we report using the growing cells of Gluconobacter strains to produce 3-DHS from quinate. We demonstrated that under growing cell conditions, the recombinant Gluconobacter strain overexpressing aroQ under control of the lac promoter could produce 3-DHS quantitatively from quinate by adjusting the pH of the culture medium. This is the first step towards developing a method to ferment shikimate from quinate. In our biotransformation experiments, more 3-DHS accumulated in large baffled flasks (Fig. 4) than in small Erlenmeyer flasks (Fig. 3). Although it is difficult to compare them directly with each other, we anticipate that longer incubation periods and/or possibly increasing the fitness of cells with higher aeration may account for higher 3-DHS production. The latter is presumably implemented by changing the expression profile in response to a changing oxygen supply. Hanke et al. (2012) recently reported that oxygen limitations affect the global gene expression of G. oxydans 621H. However, this oxygen limitation is likely to be more drastic compared to the conditions used in the present study.
We previously purified two kinds of DHQases, soluble and membrane-bound, and suggested that the latter is responsible for the extracellular formation of 3-DHS (Adachi et al. 2008b). Therefore, we constructed an expression plasmid for the GOX0437 protein of G. oxydans 621H, in which the N-terminal amino acid sequence is the same as that of membrane-bound DHQase of NBRC3244. We then overexpressed the protein in E. coli BL21 (DE3). A large fraction of the GOX0437 protein formed inclusion bodies in cells grown at 37 °C (Fig. S2). Elevated growth temperature is one parameter that promotes inclusion body formation due to hydrophobic interactions taking place between polypeptide chains (Schein and Noteborn 1988). A higher temperature increases hydrophobic interactions by exposing hydrophobic stretches of amino acids that are not normally exposed (Strandberg and Enfors 1991). Based on our multiple sequence alignment of bacterial and fungal DHQases (Roszak et al. 2002), GOX0437 has an extended N terminus, which presumably functions as a signal peptide for transit to the periplasmic space (Adachi et al. 2008b; Fig. S3). A block in the protein translocation channel could be another explanation for the large fraction of insoluble DHQase observed.
Because the growing cells of wild-type Gluconobacter strains produced a low level of 3-DHS from quinate (Fig. S1), we tried to develop a strain that efficiently transformed quinate to 3-DHS by overexpressing DHQase of NBRC3244 for use in a single-pot fermentation. By using a broad-host range plasmid containing the lac promoter, we successfully constructed the NBRC3244 derivative that expresses more than 100-fold higher DHQase than the parental strain. The engineered Gluconobacter strain produced a significant amount of 3-DHS without adjusting the pH and an almost equimolar amount of 3-DHS from quinate by adjusting the pH of the culture medium to 7.3.
We have developed an in vitro system to produce shikimate consisting of Gluconobacter membranes and enzymes (Adachi et al. 2006b, 2010). Gluconobacter cells are known to produce shikimate from quinate, and we have already reported that trace amounts of shikimate are produced from 3-DHQ by wild-type Gluconobacter cells (Adachi et al. 2003a). Therefore, our next target was to develop of a single-pot fermentation to produce quantitative yields of shikimate from quinate. This fermentation system required both oxidation and reduction steps, which are essential for this biotransformation. Nevertheless, the feasibility of such process was proven by Suzuki et al. (2002), in which oxidation and reduction reactions using growing Gluconobacter cells resulted in a nearly stoichiometric epimerization of d-arabitol to xylitol. Therefore, it is possible to develop a fermentation system containing both oxidation and reduction steps, though the cofactor preference of shikimate dehydrogenase on NADPH is another issue to be considered. G. oxydans likely produces enough NADPH, because, based on its genome, it lacks a gene for phosphofructokinase but has genes involved in the pentose phosphate pathway (Prust et al. 2005). Still, there are several issues to be solved toward shikimate production. Because QDH also oxidizes shikimate with a low affinity (Vangnai et al. 2004), it has to be considered modification of the substrate specificity of QDH. Not only enhancement of shikimate dehydrogenase activity but also uncharacterized properties of the Gluconobacter cell could be key factors to overcome the next bottleneck in the conversion of quinate to shikimate, e.g., import of 3-DHS into the cell, export of shikimate, and branching metabolic pathways from shikimate.
References
Adachi O, Tanasupawat S, Yoshihara N, Toyama H, Matsushita K (2003a) 3-dehydroquinate production by oxidative fermentation and further conversion of 3-dehydroquinate to the intermediates in the shikimate pathway. Biosci Biotechnol Biochem 67:2124–2131
Adachi O, Yoshihara N, Tanasupawat S, Toyama H, Matsushita K (2003b) Purification and characterization of membrane-bound quinoprotein quinate dehydrogenase. Biosci Biotechnol Biochem 67:2115–2123
Adachi O, Ano Y, Toyama H, Matsushita K (2006a) Enzymatic preparation of metabolic intermediates, 3-dehydroquinate and 3-dehydroshikimate, in the shikimate pathway. Biosci Biotechnol Biochem 70:3081–3083
Adachi O, Ano Y, Toyama H, Matsushita K (2006b) High shikimate production from quinate with two enzymatic systems of acetic acid bacteria. Biosci Biotechnol Biochem 70:2579–2582
Adachi O, Ano Y, Toyama H, Matsushita K (2006c) Purification and properties of NADP-dependent shikimate dehydrogenase from Gluconobacter oxydans IFO 3244 and its application to enzymatic shikimate production. Biosci Biotechnol Biochem 70:2786–2789
Adachi O, Ano Y, Akakabe Y, Shinagawa E, Matsushita K (2008a) Coffee pulp koji of Aspergillus sojae as stable immobilized catalyst of chlorogenate hydrolase. Appl Microbiol Biotechnol 81:143–151
Adachi O, Ano Y, Toyama H, Matsushita K (2008b) A novel 3-dehydroquinate dehydratase catalyzing extracellular formation of 3-dehydroshikimate by oxidative fermentation of Gluconobacter oxydans IFO 3244. Biosci Biotechnol Biochem 72:1475–1482
Adachi O, Ano Y, Shinagawa E, Yakushi T, Matsushita K (2010) Conversion of quinate to 3-dehydroshikimate by Ca-alginate-immobilized membrane of Gluconobacter oxydans IFO 3244 and subsequent asymmetric reduction of 3-dehydroshikimate to shikimate by immobilized cytoplasmic NADP-shikimate dehydrogenase. Biosci Biotechnol Biochem 74:2438–2444
Bethesda Research Laboratories (1986) BRL pUC host: E. coli DH5a competent cells. Bethesda Res Lab Focus 8:9–12
Boyer HW, Roulland-Dussoix D (1969) A complementation analysis of the restriction and modification of DNA in Escherichia coli. J Mol Biol 41:459–472
Chandran SS, Yi J, Draths KM, von Daeniken R, Weber W, Frost JW (2003) Phosphoenolpyruvate availability and the biosynthesis of shikimic acid. Biotechnol Prog 19:808–814
Chaudhuri S, Lambert JM, McColl LA, Coggins JR (1986) Purification and characterization of 3-dehydroquinase from Escherichia coli. Biochem J 239:699–704
Dulley JR, Grieve PA (1975) A simple technique for eliminating interference by detergents in the Lowry method of protein determination. Anal Biochem 64:136–141
Figurski DH, Helinski DR (1979) Replication of an origin-containing derivative of plasmid RK2 dependent on a plasmid function provided in trans. Proc Natl Acad Sci U S A 76:1648–1652
Garbe T, Servos S, Hawkins A, Dimitriadis G, Young D, Dougan G, Charles I (1991) The Mycobacterium tuberculosis shikimate pathway genes: evolutionary relationship between biosynthetic and catabolic 3-dehydroquinases. Mol Gen Genet 228:385–392
Hanke T, Richhardt J, Polen T, Sahm H, Bringer S, Bott M (2012) Influence of oxygen limitation, absence of the cytochrome bc 1 complex and low pH on global gene expression in Gluconobacter oxydans 621H using DNA microarray technology. J Biotechnol 157:359–37
Kawai S, Goda-Tsutsumi M, Yakushi T, Kano K, Matsushita K (2013) Heterologous overexpression and characterization of a flavoprotein–cytochrome c complex fructose dehydrogenase of Gluconobacter japonicus NBRC3260. Appl Environ Microbiol 79:1654–1660
Kovach ME, Elzer PH, Hill DS, Robertson GT, Farris MA, Roop RM 2nd, Peterson KM (1995) Four new derivatives of the broad-host-range cloning vector pBBR1MCS, carrying different antibiotic-resistance cassettes. Gene 166:175–176
Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23:2947–2948
Prust C, Hoffmeister M, Liesegang H, Wiezer A, Fricke WF, Ehrenreich A, Gottschalk G, Deppenmeier U (2005) Complete genome sequence of the acetic acid bacterium Gluconobacter oxydans. Nat Biotechnol 23:195–200
Richman JE, Chang YC, Kambourakis S, Draths KM, Almy E, Snell KD, Strasburg GM, Frost JW (1996) Reaction of 3-dehydroshikimic acid with molecular oxygen and hydrogen peroxide: products, mechanism, and associated antioxidant activity. J Am Chem Soc 118:11587–11591
Roszak AW, Robinson DA, Krell T, Hunter IS, Fredrickson M, Abell C, Coggins JR, Lapthorn AJ (2002) The structure and mechanism of the type II dehydroquinase from Streptomyces coelicolor. Structure 10:493–503
Sambrook J, Russel DW (2001) Molecular cloning: a laboratory manual, 3rd edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor
Schein CH, Noteborn MHM (1988) Formation of soluble recombinant proteins in Escherichia coli is favored by lower growth temperature. Nat Biotechnol 6:291–294
Strandberg L, Enfors SO (1991) Factors influencing inclusion body formation in the production of a fused protein in Escherichia coli. Appl Environ Microbiol 57:1669–1674
Studier FW, Moffatt BA (1986) Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. J Mol Biol 189:113–130
Suzuki S, Sugiyama M, Mihara Y, Hashiguchi K, Yokozeki K (2002) Novel enzymatic method for the production of xylitol from d-arabitol by Gluconobacter oxydans. Biosci Biotechnol Biochem 66:2614–2620
Tripathi P, Rawat G, Yadav S, Saxena RK (2013) Fermentative production of shikimic acid: a paradigm shift of production concept from plant route to microbial route. Bioprocess Biosyst Eng 36:1665–1673
van Kleef MA, Duine JA (1988) Bacterial NAD(P)-independent quinate dehydrogenase is a quinoprotein. Arch Microbiol 150:32–36
Vangnai AS, Toyama H, De-Eknamkul W, Yoshihara N, Adachi O, Matsushita K (2004) Quinate oxidation in Gluconobacter oxydans IFO3244: purification and characterization of quinoprotein quinate dehydrogenase. FEMS Microbiol Lett 241:157–162
Vangnai AS, Promden W, De-Eknamkul W, Matsushita K, Toyama H (2010) Molecular characterization and heterologous expression of quinate dehydrogenase gene from Gluconobacter oxydans IFO3244. Biochemistry (Mosc) 75:452–459
Whiting GC, Coggins RA (1967) The oxidation of d-quinate and related acids by Acetomonas oxydans. Biochem J 102:283–293
Acknowledgments
This work was supported in part by Grant-in-Aid from the Japan Society for the Promotion of Science.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 1180 kb)
Rights and permissions
About this article
Cite this article
Nishikura-Imamura, S., Matsutani, M., Insomphun, C. et al. Overexpression of a type II 3-dehydroquinate dehydratase enhances the biotransformation of quinate to 3-dehydroshikimate in Gluconobacter oxydans . Appl Microbiol Biotechnol 98, 2955–2963 (2014). https://doi.org/10.1007/s00253-013-5439-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-013-5439-z