
Abstract
Milbemycins A3/A4 are important 16-membered macrolides which have been commercialized and widely used as pesticide and veterinary medicine. However, similar to other milbemycin producers, the production of milbemycins A3/A4 in Streptomyces bingchenggensis is usually accompanied with undesired by-products such as C5-O-methylmilbemycins B2/B3 (α-class) and β1/β2 (β-class) together with nanchangmycin. In order to obtain high yield milbemycins A3/A4-producing strains that produce milbemycins A3/A4 as main components, milD, a putative C5-O-methyltransferase gene of S. bingchenggensis, was biofunctionally investigated by heterologous expression in Escherichia coli. Enzymatic analysis indicated that MilD can catalyze both α-class (A3/A4) and β-class milbemycins (β11) into C5-O-methylmilbemycins B2/B3 and β1, respectively, suggesting little effect of furan ring formed between C6 and C8a on the C5-O-methylation catalyzed by MilD. Deletion of milD gene resulted in the elimination of C5-O-methylmilbemycins B2/B3 and β1/β2 together with an increased yield of milbemycins A3/A4 in disruption strain BCJ13. Further disruption of the gene nanLD encoding loading module of polyketide synthase responsible for the biosynthesis of nanchangmycin led to strain BCJ36 that abolished the production of nanchangmycin. Importantly, mutant strain BCJ36 (∆milD∆nanLD) produced milbemycins A3/A4 as main secondary metabolites with a yield of 2312 ± 47 μg/ml, which was approximately 74 % higher than that of the initial strain S. bingchenggensis BC-109-6 (1326 ± 37 μg/ml).
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
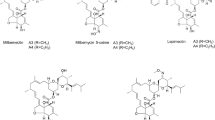
Milbemycins are a group of macrolides chemically related to the avermectins and were firstly isolated from Streptomyces hygroscopicus subsp. aureolacrimosus (Takiguchi et al. 1980). They have attracted considerable attention and developed as acaricides, insecticides, and anthelmintics due to the extremely high activity against various insects and parasites, a special action mode, low toxicity to mammals, and environmentally benign characteristics. The commercial product Milbemectin (a mixture of milbemycins A3 and A4) has been developed as an acaricide for the control of mites in 1990 (Pluschkell et al. 1999). Since then, other commercial products related to milbemycins A3 and A4, such as lepimectin, latidectin, and milbemycin oxime, were marketed and used in the fields of animal health and agriculture (Jung et al. 2002; McCall 2005; Yadav and Singh 2011).
Apart from S. hygroscopicus subsp. aureolacrimosus and Streptomyces griseochromogenes (Takiguchi et al. 1980; Nonaka et al. 2000), another newly milbemycin-producing strain Streptomyces bingchenggensis was isolated by our research team and currently used as an industrial producer of milbemycins (Wang et al. 2009b, 2010). S. bingchenggensis produces milbemycins A3, A4, and four α-class and β-class C5-O-methylmilbemycins (B2, B3, β1, and β2) as its major components together with the polyether ionophore nanchangmycin and a trace of new milbemycin analogs (Xiang et al. 2007, 2008, 2009a, 2009b; Wang et al. 2009a). Due to the outstanding activity of milbemycins A3 and A4, great efforts have been made to improve the production of these two compounds, yielding a mutant strain of S. bingchenggensis with enhanced production of milbemycins A3 and A4 by rational screening (Wang et al. 2009b). Compared to the classical strain improvement technology, the rational screening strategy has a large improvement in the efficiency of the selection process; however, it is still laborious and time-consuming. On the other hand, the development of molecular microbiology and recombinant DNA technology has led to a number of strategies for rational strain improvement known collectively as metabolic engineering, which largely depends on the knowledge and understanding of the biosynthesis of the secondary metabolite and its related metabolic fluxes (Chen et al. 2010; Baltz 2011; Mo et al. 2013). Recently, the biosynthetic gene cluster of milbemycin has been identified by analyzing the genome of S. bingchenggensis (Wang et al. 2010). The biosynthetic gene cluster of milbemycin (SBI00709-SBI00790) in S. bingchenggensis contains four large ORFs (milA1-milA4), some regulatory genes and genes encoding tailoring enzymes, which are homologous to those of avermectin. However, the milbemycin biosynthetic gene cluster is divided into two separate parts (milA1 and milA2-milA4) by a 60-kb region that is not required for milbemycins production. This gene organization is distinct from the biosynthetic gene cluster of avermectin but similar to that of meilingmycin (Ikeda et al. 1999; He et al. 2010). The investigation of the biosynthesis of avermectin and meilingmycin facilitates the understanding of the biosynthetic pathway of milbemycins, which is believed to contribute to the titer improvement of milbemycins via metabolic engineering. In the milbemycin biosynthetic gene cluster, a putative C5-O-methyltransferase gene, milD, shows high homology to aveD and meiD that locates in the biosynthetic gene cluster of avermectin and meilingmycin, respectively, suggesting that MilD is possibly responsible for the methylation of the hydroxyl group on C-5 of milbemycins A3/A4 and β11/β4 to afford B2/B3 and β1/β2, respectively (Fig. 1). As another major secondary metabolite, nanchangmycin is biosynthesized by polyketide synthase (PKS) PKSNan using malonyl-CoA and methylmalonyl-CoA as common precursors, implying a biosynthetic competition between milbemycins and nanchangmycin. Therefore, milD and pksNan are suitable targets for gene disruption to improve the production of milbemycins A3 and A4.

Proposed pathway of milbemycins (Nonaka et al. 2000)
Here, the function of MilD was investigated by heterologous expression in E. coli, demonstrating C5-O-methyltransferase activity toward α-class and β-class milbemycins. The disruption of milD and nanLD encoding the loading module of PKSNan abolished the production of C5-O-methylmilbemycins B2/B3 and β1/β2 together with nanchangmycin, leading to an increase in the yield of milbemycins A3/A4.
Materials and methods
Strains, vectors, reagents, and cultivation
All bacterial strains and plasmids used in this study are listed in Table 1. The wild-type strain S. bingchenggensis has been deposited at the China General Microbiology Culture Collection Center (Accession no. CGMCC1734), Institute of Microbiology, Chinese Academy of Science. The 16S rDNA sequence of S. bingchenggensis was deposited in GenBank (Accession no. DQ449953). Primers (Table 2) were synthesized in Sangon Biotech (Shanghai, China). Restriction endonucleases, DNA ligase, and DNA polymerases were purchased from TaKaRa Biotechnology (Dalian, China). DNA sequencing was performed by GenScript (Nanjing, China). Mannitol soya flour (MS) medium (Kieser et al. 2000) was used for S. bingchenggensis BC-109-6 sporulation and conjugation between Escherichia coli and Streptomyces. Yeast extract-malt extract (YEME) medium (Kieser et al. 2000) containing 25 % sucrose was used to grow mycelia for the isolation of total DNA. Luria–Bertani (LB) medium was used for E. coli propagation (Sambrook and Russell 2001). All E. coli procedures were performed according to standard protocols (Sambrook and Russell 2001). Isolation of genomic DNA from S. bingchenggensis and agarose gel electrophoresis were performed according to Kieser et al. (2000). DNA fragments were purified from agarose gels with the Geneclean kit II (BIO101, Beijing, China).
Cloning, expression, purification, and in vitro assay of MilD
The gene milD was amplified from genomic DNA of S. bingchenggensis BC-109-6 using a pair of primers milD-1 and milD-2. Amplification reaction was conducted by standard procedure with an annealing temperature of 57.5 °C. The PCR products were digested with BamHI and HindIII and inserted into pET-30a(+) to give the expression plasmid pET-30a-milD. After sequence confirmation, the constructed plasmid was transformed into E. coli BL21 (DE3). After the cells harboring pET-30a-milD were grown in LB medium at 37 °C to an absorbance at OD600 of about 0.6 and induced by the addition of 0.5 mM isopropyl-β-d-thiogalactoside (IPTG), the cells were incubated at 30 °C for an additional 4 h. The cells were harvested by centrifugation at 6,000 × g for 10 min at 4 °C, washed with 25 ml of 50 mM Tris–HCl buffer (pH 7.4), and then resuspended in 1 ml of the same buffer. The obtained cell suspension was sonicated and the inclusion body was collected by centrifugation (12,000 × g, 15 min) at 4 °C. The pellet was then resuspended in 20 mM Tris–HCl buffer (pH 8.0) containing 2 M urea, 2 mM EDTA, and 1 % Triton X-100 and centrifuged again as described above. This step was repeated twice. The MilD inclusion bodies were washed twice with buffer without urea and stored at −20 °C.
The inclusion bodies were solubilized by incubation at room temperature for at least 1 h in solubilization buffer (100 mM NaH2PO4, 10 mM Tris–HCl, 6 M urea, pH 8.0) and clarified by centrifugation at 12,000 × g for 20 min at 4 °C, and the supernatant was retained for analysis. His-tagged MilD fusion proteins were purified by Ni2+ affinity chromatography (Invitrogen) according to the instructions of the manufacturer. The proteins were eluted by the use of a series of elution buffers of imidazole (10, 50, 100, and 150 mM) in the above Tris–HCl buffer. The pure fractions were dialyzed with a storage buffer (50 mM Tris–HCl buffer, pH 7.4) at 4 °C. Refolding of MilD was performed as described by Clark et al. (1999). The purity of the protein was ascertained by sodium monododecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE).
Methylation of milbemycins by MilD was carried out in a reaction mixture containing 100 mM Tris–HCl (pH 7.4), 1 mM dithiothreitol (DTT), 1 mM MgCl2, 100 μM of each substrate (milbemycin A3, milbemycin A4, or milbemycin β11), 0.5 mM S-adenosylmethionine (SAM) (Sigma), and 100–200 μg MilD to a total volume of 0.5 ml. The reaction was incubated at 30 °C for 15 min. The reaction was terminated using an equal volume of ethyl acetate and the reactants were vigorously shaken and centrifuged at 12,000 × g for 5 min. The ethyl acetate phase was carefully transferred into a new tube, freeze-dried and redissolved in 20 μl of methanol for liquid chromatography-mass spectrometry (LC-MS) experiment. The electrospray ionization ESI-MS analysis was carried out by using Agilent HPLC 1200 system equipped with a mass spectrometer (Q-TOF Micro LC-MS-MS spectrometer, Waters, Milford, MA, USA). The HPLC conditions were as follows: column, Eclipse × DB-C18 column (4.6 × 150 mm, 5 μm); detection at 242 nm; flow rate, 1 ml/min; eluent, CH3OH/H2O (90:10, v/v). Mass spectra were acquired in positive ion mode.
Construction of ∆milD mutant strain
In order to construct ∆milD mutant, the in-frame deletion strategy was employed. Using genomic DNAs of S. bingchenggensis as template, a 985-bp fragment upstream of the milD was amplified with primers milD-L1 and milD-L2. The amplified fragment was digested with HindIII/XbaI and ligated into corresponding sites of pUC19 to generate pBC1577. Then, a 1,117-bp fragment downstream of the milD was amplified with milD-R1 and milD-R2. The amplified fragment was excised by XbaI/EcoRI and then ligated into corresponding sites of pBC1577 to generate pBCJ1397. The 2.1-kb fragment containing the upstream and downstream of milD was obtained after the digestion of pBCJ1397 with HindIII/EcoRI and subsequently inserted into the same sites of pKC1139 to generate pBC3784. After the verification by PCR amplification and restriction digestion analysis, pBC3784 was transformed into the non-methylating E. coli ET12567/pUZ8002. The conjugations were performed using the spores of S. bingchenggensis BC-109-6 according to the literature (Kieser et al. 2000) and exconjugants were selected using MS agar containing apramycin. These cultures were grown for 2 days at 28 °C, then for 7–10 days at 39 °C. The colonies that were apramycin-resistant at 39 °C were identified as the integrating mutants, in which a single-crossover homologous recombination event took place. Insertion mutants were confirmed by PCR analysis and inoculated on nonselective MS plates at 28 °C for a second round of recombination. Double-crossover mutants were screened by replica from the colonies grown on the MS medium. Mutants that lost resistance to apramycin were selected for further screening and genotypic confirmation by PCR using milD-V1 and milD-V2 as primers. The obtained double-crossover mutants were designated as S. bingchenggensis BCJ13 with the disruption of milD.
Construction of ∆milD∆nanLD mutant strain
To make the ∆milD∆nanLD mutant strain, an 844-bp upstream fragment amplified with primers nan-L1 and nan-L2 was cloned into the XbaI/HindIII site of pUC19 to give pBCN-1. Then, a 926-bp downstream fragment amplified with nan-R1 and nan-R2 was cloned into the EcoRI/XbaI sites of pBCN-1 to give pBCN-2. A thiostrepton resistance gene (1.29-kb) cassette was amplified from pHZ1358 using tsr1 and tsr2 as primers. The resultant PCR products were digested with XbaI and then ligated to the corresponding sites of pBCN-2 to yield pBCN-3. The 3.06-kb insert was recovered from pBCN-3 by digesting with HindIII/EcoRI and inserted into the same sites of pKC1139 to generate pBC8559. Following the procedure described above, pBC8559 was introduced into strain S. bingchenggensis BCJ13 for double-crossover recombination. The resulting strain BCJ36, which is apramycin-sensitive but thiostrepton-resistant, was then subjected to PCR amplification to validate the genotype using nan-V1 and nan-V2 as primers.
Fermentation and HPLC analysis of antibiotic production
S. bingchenggensis BC-109-6 and all mutant strains were fermented in the same culture condition. The strains were firstly cultured in seed medium (sucrose 1 %, yeast extract 0.5 %, peptone 0.35 %, skimmed milk powder 0.1 %, and K2HPO4 0.05 %, pH 7.0) at 28 °C for 42 h on a rotary shaker at 250 rpm. Then 2.0 ml of the culture was transferred into 250-ml Erlenmeyer flasks containing 25 ml of the fermentation medium consisting of sucrose 80.0 g/l, soybean powder 20.0 g/l, skimmed milk powder 1.0 g/l, CaCO3 3.0 g/l, K2HPO4 1.0 g/l, and FeSO4 · 7H2O 0.1 g/l, pH 7.2. Fermentation was carried out at 28 °C for 8 days on a rotary shaker at 250 rpm. After finishing the fermentation, the broths were mixed with an equal volume of methanol. The resultant mixture was then centrifuged at 12,000 × g for 20 min, and the supernatant was filtered through a 0.22-μm membrane filter and analyzed by HPLC. HPLC was performed with a Shimadzu LC-2010CHT system (Shimadzu, Koyoto, Japan) by using a NOVA-PAKR C18 column (3.9 × 150 mm, 5 μm, Waters) at a flow rate of 1.0 ml/min with a linear gradient from 0 to 100 % of solvent B in 15 min (solvent A: MeCN-H2O-MeOH (350:50:100, v/v/v); solvent B, MeOH) and detected at 242 nm.
Results
Cloning, expression and functional analysis of MilD
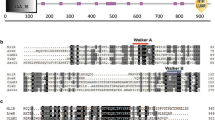
Analysis of milbemycin biosynthetic gene cluster in S. bingchenggensis led to the identification of gene milD (GenBank accession no. FJ531497.2), the product of which exhibits 92.66 %, 87.90 % and 56.34 % homology with that of meiD (Streptomyces nanchangensis NS3226, accession no. ADC45587.1), milD (S. griseochromogenes, accession no. AAR15334.1) and aveD (Streptomyces avermitilis MA-4680, accession no. NP_822112.1), respectively. The amino acid sequence alignment of MilD with its homologous proteins is shown in Fig. 2 and a conserved LDVGxGxG motif was observed, suggesting that MilD is a S-adenosylmethionine-dependent methyltransferase (Ibrahim et al. 1998). Therefore, MilD was considered to be involved in the C5-O-methylation during the biosynthesis of milbemycins. To confirm this speculation, recombinant MilD was prepared and used for in vitro assay.

Sequence alignment of MilD from Streptomyces bingchenggensis with its most similar homologs. MilD1 from S. bingchenggensis, MeiD from Streptomyces nanchangensis NS3226, MilD2 from Streptomyces griseochromogenes and AveD from Streptomyces avermitilis MA-4680
The milD gene was cloned into the pET-30a(+) vector for protein expression in E. coli BL21 (DE3). His-tagged MilD recombinant enzyme was expressed in a form of inclusion body (Fig. 3). After the purification under denaturing conditions and the following renaturation, recombinant soluble MilD (molecular mass of 30.3 kDa) was obtained with a concentration of 0.07 mg/ml. To characterize the substrate specificity, the recombinant MilD was incubated with individual substrate (α-class milbemycin A3, milbemycin A4 and β-class milbemycin β11) and the reaction mixture was analyzed by LC-MS. When MilD was incubated with milbemycin A3, a new peak with a retention time of 8.75 min and a molecular ion at m/z = 542.3 [M + H]+ was detected (Fig. 4a–c), implying the bioconversion of milbemycin A3 to B2. When milbemycin A4 or β11 was used as substrate, the formation of B3 or β1 was, respectively, detected and validated using the same method mentioned above (Fig. 4d–i). To investigate substrate specificity, the relative activity of MilD toward different substrates was calculated. As listed in Table 3, no obvious difference in the relative activity of MilD toward α-class (A3/A4) and β-class milbemycins (β11) was observed, which suggests that the formation of furan ring between C6 and C8a has no significant influence on the C5-O-methylation catalyzed by MilD.

SDS-PAGE analysis of MilD expressing in Escherichia coli BL21 (DE3) before (a) and after (b) purification. a Lane 1: protein molecular weight marker; Lane 2: the supernatant of cell lysate (pET-30a) after sonication; Lane 3: the supernatant of cell lysate (pET-30a-milD) after sonication; Lane 4: the pellet of cell lysate (pET-30a) after sonication; Lane 5: the pellet of cell lysate (pET-30a-milD) after sonication. b Lane 1: protein molecular weight marker; Lane 2: solubilized protein after 6 M urea treatment of inclusion body; Lane 3: the purified recombinant protein MilD after affinity chromatography

LC-MS analysis of the reaction mixtures of selected substrates with MilD after incubation for 15 min. a Reaction of MilD with milbemycin A3. b Reaction of MilD with milbemycin A3 and SAM. c Mass spectrum of the new peak corresponding to milbemycins B2. d Reaction of MilD with milbemycin A4. e Reaction of MilD with milbemycin A4 and SAM. f Mass spectrum of the new peak corresponding to milbemycins B3. g Reaction of MilD with milbemycin β11. h Reaction of MilD with milbemycin β11 and SAM. i Mass spectrum of the new peak corresponding to milbemycin β1
Enhancement of milbemycins A3/A4 production by deletion of milD
As described above, MilD can catalyze the methylation of milbemycins A3/A4 and β11 to B2/B3 and β1, respectively. Therefore, we hypothesized that deletion of milD may contribute to increase the yield of milbemycins A3/A4. Thus, a high milbemycins-producing strain BC-109-6 was used as the initial strain to construct milD disruption mutant. As shown in Fig. 5, a 2.38-kb product and a 3.14-kb product was obtained from the genome of BCJ13 and BC-109-6, respectively, using milD-V1 and milD-V2 as primers. The result suggested that a 771 bp internal fragment in milD was successfully deleted in the disruption mutant strain BCJ13. Compared to the BC-109-6, BCJ13 did not produce methylated milbemycins B2, B3, β1, and β2, while milbemycins A3 and A4 were still produced (Fig. 6a, b). Thus, MilD was believed to be responsible for the methylation of the hydroxyl group on C-5 of milbemycins β1, β2, B2, and B3 in vivo, which was consistent with the results of in vitro bioassay described above and the proposed biosynthetic pathway of milbemycins described in the previous literature (Fig. 1; Nonaka et al. 2000). Moreover, the yield of milbemycins A3/A4 in BCJ13 (2237 ± 54 μg/ml) was higher than that in the BC-109-6 (1326 ± 37 μg/ml), implying a positive effect of the deletion of milD on the production of milbemycins A3/A4.

Gene disruption in S. bingchenggensis BC-109-6. a Schematic description of milD gene deletion. b PCR analysis with genomic DNA from S. bingchenggensis BC-109-6 and mutant strain BCJ13, using primers milD-V1 and milD-V2. Lane 1: DNA ladder; Lane 2: BC-109-6; Lane 3: BCJ13. c Schematic description of nanLD gene deletion. d PCR analysis with genomic DNA from mutant strain BCJ13 and BCJ36, using primers nan-V1 and nan-V2. Lane 1: DNA ladder; Lane 2: BCJ36; Lane 3: BCJ13

The HPLC profiles of metabolites produced by the S. bingchenggensis BC-109-6 and its deletion mutants. a BC-109-6. b BCJ13. c BCJ36
Inactivation of nanLD gene to eliminate the biosynthesis of nanchangmycin
The biosynthesis of milbemycins and that of nanchangmycin share the same precursors such as malonyl-CoA and methylmalonyl-CoA as building-block units for their respective polyketide backbones. Therefore, we attempted to abolish the biosynthesis of nanchangmycin to further enhance the production of milbemycins A3 and A4 by constructing a mutant strain BCJ36, in which the gene nanLD encoding the loading module of PKSNan responsible for the biosynthesis of nanchangmycin was replaced by a thiostrepton resistance gene cassette. Due to the increased yield of milbemycins A3/A4 and failure to produce milbemycins β1/β2 and B2/B3, strain BCJ13 was used as the parent strain to construct a double mutant strain BCJ36, which was then verified by PCR amplification using nan-V1 and nan-V2 as primers. As shown in Fig. 5, an expected 1.95-kb product was amplified from the genomic DNA of BCJ36 and an expected 3.39-kb product was amplified from the genomic DNA of parent strain BCJ13, suggesting that nanLD was successfully replaced by the thiostrepton resistance gene cassette. Comparison of the metabolic HPLC spectrum of BCJ36 with that of BCJ13 clearly showed that BCJ36 lost the ability to produce nanchangmycin (Fig. 6b, c). The yield of milbemycins A3/A4 produced by BCJ36 and BCJ13 in shaking-flask fermentation was 2312 ± 47 and 2237 ± 54 μg/ml, respectively, implying little influence of the disruption of nanchangmycin biosynthetic gene cluster on the production of milbemycins.
Stability of high-yield mutant BCJ36
The genetic stability of mutant BCJ36 was evaluated by five successive subcultivation tests. The shaking flask experiments and HPLC analysis showed that the mutants still did not produce C5-O-methyl derivatives, β1, β2, B2, B3 and nanchangmycin, and the yield of milbemycins A3/A4 among five generations ranged from 2193 ± 26 to 2321 ± 34 μg/ml. These results suggested that the mutant strain BCJ36 was genetically stable and could be useful for industrial production of milbemycins A3/A4.
Discussion
Although avermectin and meilingmycin possess a similar structure, their biosynthetic gene clusters show a distinct difference in gene organization (Ikeda et al. 1999; He et al. 2010). Especially, a 55-kb large region that not required for meilingmycin biosynthesis is located in meilingmycin biosynthetic gene cluster and meiD encoding C5-O-methyltranferase separates from gene meilF encoding C5-ketoreductase (He et al. 2010). Recently, the biosynthetic gene cluster of milbemycin (SBI00709-SBI00790) was identified from S. bingchenggensis by genome analysis (Wang et al. 2010), demonstrating a high homology and similar gene organization with the gene cluster of meilingmycin in S. nanchangensis. However, the post-PKS modification in the biosynthesis of milbemycin and meilingmycin may be different. For example, the C5-O-methylation usually occurs in β-class meilingmycins without furan ring formed between C6 and C8a, such as meilingmycins D and E, implying that the formation of furan ring may have influence on the C5-O-methylation (He et al. 2010). Whereas this phenomenon of selective catalysis seems to not occur during the biosynthesis of milbemycins in S. bingchenggensis, because α-class and β-class C5-O-methylated milbemycins (B2, B3, β1, and β2) are co-produced as major secondary metabolites by S. bingchenggensis. Interestingly, the C5-O-methylation of avermectins was considered to take place after the formation of furan ring (Ikeda and Ōmura 1997), making whether formation of furan ring affects the C5-O-methylation more complicated. To address this issue, MilD was overexpressed in E. coli BL21 (DE3) as N-terminal His6-tagged fusion protein and its biochemical characteristic was investigated. In the in vitro assay, C5-O-methylated derivatives β1, B2, and B3 were detected after the incubation of recombinant MilD with C5-hydroxylated milbemycins β11, A3, and A4, respectively (Fig. 4). According to the proposed biosynthetic pathway of milbemycins as shown in Fig. 1, there are two routes to yield milbemycins A3/A4: route 1 is milbemycins β7/β6 → β12/β5 → β11/β4 → A3/A4, and route 2 is β7/β6 → J/K → A3/A4 (Nonaka et al. 1999). In route 1, β11/β4 can be catalyzed to yield A3/A4 by a cytochrome P450 hydroxylase or β1/β2 by a C5-O-methyltransferase. In fact, removing genes coding enzymes that transform the metabolite into a different one has been proved to be an efficient approach to improve the yield of desired compounds such as daunorubicin, doxorubicin, clavulanic acid, kanamycin B, platensimycin, and platencin (Scotti and Hutchinson 1996; Lomovskaya et al. 1998, 1999; Mosher et al. 1999; Paradkar et al. 2001; Olano et al. 2008; Smanski et al. 2011; Ni et al. 2011). Recently, it has been reported that the disruption of meiD led to the elimination of C5-O-methylated nanchangmycins D and E (He et al. 2010) and the deletion of aveD in S. avermitilis resulted in a mutant strain that only produces C5-hydroxylated avermectins (Hong et al. 2001). Therefore, milD encoding C5-O-methyltransferase is a suitable target for gene disruption to construct mutant strain that abolishes the production of C5-O-methylated milbemycins. As expected, the disruption of milD in S. bingchenggensis results in the abolishment of β1/β2 and B2/B3, which is consistent with the results obtained from in vitro assay mentioned above. Additionally, an increased yield of A3/A4 was observed in mutant strain BCJ13. There are two possible reasons to explain this phenomenon: firstly, the deletion of C5-O-methyltransferase activity may result in accumulation of β11/β4, which in turn increases the yield of A3/A4; secondly, the disruption of milD blocks the C5-O-methylation of A3/A4 to generate B2/B3.
Nanchangmycin is another major secondary metabolite produced by S. bingchenggensis. It is biosynthesized by polyketide synthase encoding by pksNan (SBI08394-SBI08428) using malonyl-CoA and methylmalonyl-CoA as common precursors, implying a biosynthetic competition between milbemycins and nanchangmycin. In principle, actinomycetes that produce more than one secondary metabolite usually encounter competition for the same precursors, ultimately limiting the potential yield of the most desired compounds (Komatsu et al. 2010; Baltz 2011). Thus, eliminating the production of undesired secondary metabolites to change the metabolic flux is a practical approach to improve the yield of compounds of interest (Gao et al. 2010; Lee et al. 2012). For example, selective deletion of other PKS-containing clusters in S. nanchangensis can improve the yield of nanchangmycin (Sun et al. 2002). In the case of S. avermitilis, abolishment of avermectin production usually leads to the increased yield of oligomycin (Cropp et al. 2001; Wei et al. 2006; Tanaka et al. 2009; Yu et al. 2012). Furthermore, in order to diminish the precursor competition, some genome-minimized Streptomyces strains deleted for many secondary metabolite gene clusters were developed and successfully used as heterologous hosts to improve the yield of desired compounds (Komatsu et al. 2010; Gao et al. 2010; Baltz 2011; Gomez-Escrlbano and Bibb 2011; Zhou et al. 2012; Lee et al. 2012). Therefore, the nanchangmycin production in S. bingchenggensis was abolished by inactivation of the gene nanLD encoding the initial loading module of PKS responsible for the biosynthesis of nanchangmycin. Unfortunately, the yield of milbemycins A3/A4 was not further significantly increased in strain BCJ36 as expected. The similar phenomenon was also observed in the mutant strains of S. nanchangensis and S. avermitilis, in which the yield of meilingmycin and avermectins was not enhanced by blocking the biosynthesis of nanchangmycin and oligomycin, respectively (Sun et al. 2002; Zhang et al. 2004). Notably, S. bingchenggensis BC-109-6 used to construct ∆milD∆nanLD double mutant strain is a high milbemycin-producer (Wang et al. 2009b). Therefore, we hypothesize that enough amounts of malonyl-CoA and methylmalonyl-CoA can be supplied in S. bingchenggensis BC-109-6 under laboratory conditions, leading to the abolishment of precursor competition between the biosynthesis of milbemycins and nanchangmycin. Thus, the yield of milbemycins would not increase even that the biosynthesis of nanchangmycin was blocked.
In conclusion, the function of milD in S. bingchenggensis was characterized according to in vitro and in vivo assay and a mutant strain BCJ36 with enhanced yield of milbemycins A3/A4 and less by-products was constructed by deletion of milD and nanLD. The elimination of C5-O-methylmilbemycins (B2, B3, β1, and β2) and nanchangmycin not only increases the yield of milbemycins A3/A4, but avoids the negative effect of undesired by-products on the purification of milbemycins A3/A4. Furthermore, the genetic stability of the mutant strain suggests the potential use in industry to produce milbemycins A3/A4.
References
Baltz RH (2011) Strain improvement in actinomycetes in the postgenomic era. J Ind Microbiol Biotechnol 38:657–666
Chen Y, Smanski MJ, Shen B (2010) Improvement of secondary metabolite production in Streptomyces by manipulating pathway regulation. Appl Microbiol Biotechnol 86:19–25
Clark EDB, Schwarz E, Rudolph, R (1999) Inhibition of aggregation side reactions during in vitro protein folding. In: Ronald W (ed) Methods in enzymology. Academic, pp 217–236
Cropp A, Chen S, Liu H, Zhang W, Reynolds KA (2001) Genetic approaches for controlling ratios of related polyketide products in fermentation processes. J Ind Microbiol Biotechnol 27:368–377
Gao H, Zhuo Y, Ashforth E, Zhang L (2010) Engineering of a genome-reduced host: practical application of synthetic biology in the overproduction of desired secondary metabolites. Protein Cell 1:621–626
Gomez-Escrlbano JP, Bibb MJ (2011) Engineering Streptomyces coelicolor for heterologous expression of secondary metabolite gene clusters. Microb Biotechnol 4:207–215
He Y, Sun Y, Liu T, Zhou X, Bai L, Deng Z (2010) Cloning of separate meilingmycin biosynthesis gene clusters by use of acyltransferase–ketoreductase didomain PCR amplification. Appl Environ Microbiol 76:3283–3292
Hong YS, Lee JH, Kim HS, Kim KW, Lee JJ (2001) Targeted gene disruption of the avermectin B O-methyltransferase gene in Streptomyces avermitilis. Biotechnol Lett 23:1765–1770
Ibrahim RK, Bruneau A, Bantignies B (1998) Plant O-methyltransferases: molecular analysis, common signature and classification. Plant Mol Biol 36(1):1–10
Ikeda H, Ōmura S (1997) Avermectin biosynthesis. Chem Rev 97:2591–2609
Ikeda H, Nonomiya T, Usami M, Ohta T, Ōmura S (1999) Organization of the biosynthetic gene cluster for the polyketide anthelmintic macrolide avermectin in Streptomyces avermitilis. Proc Natl Acad Sci U S A 96:9509–9514
Jung M, Saito A, Buescher G, Maurer M (2002) Milbemycin oxime. In: Vercruysse J, Rew RS (eds) Macrocyclic lactones in anti-parasitic therapy. CAB International, pp 51–74
Kieser T, Bibb MJ, Buttner MJ, Chater KF, Hopwood DA (2000) Practical streptomyces genetics. John Innes Foundation Norwich, UK
Komatsu M, Uchiyama T, Omura S, Cane DE, Ikeda H (2010) Genome-minimized Streptomyces host for the heterologous expression of secondary metabolism. Proc Natl Acad U S A 107:2646–2651
Lee HN, Kim HJ, Kim P, Lee HS, Kim ES (2012) Minimal polyketide pathway expression in an actinorhodin cluster-deleted and regulation-stimulated Streptomyces coelicolor. J Ind Microbiol Biotechnol 39:805–811
Lomovskaya N, Doi-Katayama Y, Filippini S, Nastro C, Fonstein L, Gallo M, Colombo AL, Hutchinso CR (1998) The Streptomyces peucetius dpsY and dnrX genes govern early and late steps of daunorubicin and doxorubicin biosynthesis. J Bacteriol 180:2379–2386
Lomovskaya N, Otten SL, Doi-Katayama Y, Fonstein L, Liu XC, Takatsu T, Inventi-Solari A, Filippini S, Torti F, Colombo AL, Hutchinson CR (1999) Doxorubicin overproduction in Streptomyces peucetius: cloning and characterization of the dnrU ketoreductase and dnrV genes and the doxA cytochrome P-450 hydroxylase gene. J Bacteriol 181:305–318
McCall JW (2005) The safety-net story about macrocyclic lactone heartworm preventives: a review, an update, and recommendation. Vet Parasitol 133:197–206
Mo SJ, Lee SK, Jin YY, Oh CH, Suh JW (2013) Application of a combined approach involving classical random mutagensis and metabolic engineering to enhance FK506 production in Streptomyces sp. RM7011. Appl Microbiol Biotechnol 97:3053–3052
Mosher RH, Paradkar AS, Anders C, Barton B, Jensen SE (1999) Genes specific for the biosynthesis of clavam metabolites antipodal to clavulanic acid are clustered with the gene for clavaminate synthase 1 in Streptomyces clavuligerus. Antimicrob Agents Chemother 43:1215–1224
Ni X, Li D, Yang L, Huang T (2011) Construction of kanamycin B overproducing strain by genetic engineering of Streptomyces tenebrarius. Appl Microbiol Biotechnol 89:723–731
Nonaka K, Kumasaka C, Okamoto Y, Maruyama F, Yoshikawa H (1999) Bioconversion of milbemycin-related compounds: biosynthetic pathway of milbemycins. J Antibiot 52:109–116
Nonaka K, Tsukiyama T, Okamoto Y, Sato K, Kumasaka C, Yammoto T, Maruyama F, Yoshikawa H (2000) New milbemycins from Streptomyces hygroscopicus subsp. aureolacrimosus: fermentation, isolation and structure elucidation. J Antibiot 53:694–704
Olano C, Lombó F, Méndez C, Salas JA (2008) Improving production of bioactive secondary metabolites in actinomycetes by metabolic engineering. Metab Eng 10:281–292
Paradkar AS, Mosher RH, Anders C, Griffin A, Griffin J, Hughes C, Greaves P, Barton B, Jensen SE (2001) Applications of gene replacement technology to Streptomyces clavuligerus strain development for clavulanic acid production. Appl Environ Microbiol 67:2292–2297
Pluschkell U, Horowitz AR, Ishaaya I (1999) Effect of milbemectin on the sweet potato whitefly, Bemisia tabaci. Phytoparasitica 27:183–191
Sambrook J, Russell DW (2001) Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory, Cold Spring Harbor, New York
Scotti C, Hutchinson CR (1996) Enhanced antibiotic production by manipulation of the Streptomyces peucetius dnrH and dnmT genes involved in doxorubicin (adriamycin) biosynthesis. J Bacteriol 178:7316–7321
Smanski MJ, Yu Z, Casper J, Lin S, Peterson RM, Chen Y, Wendt-Pienkowski E, Rajski SR, Shen B (2011) Dedicated ent-kaurene and ent-atiserene synthases for platensimycin and platencin biosynthesis. Proc Natl Acad Sci U S A 108:13498–13503
Sun Y, Zhou X, Liu J, Bao K, Zhang G, Tu G, Kieser T, Deng Z (2002) ‘Streptomyces nanchangensis’, a producer of the insecticidal polyether antibiotic nanchangmycin and the antiparasitic macrolide meilingmycin, contains multiple polyketide gene clusters. Microbiology 148:361–371
Takiguchi Y, Mishima H, Okuda M, Terao M, Aoki A, Fukuda R (1980) Milbemycins, a new family of macrolide antibiotics: fermentation, isolation and physico-chemical properties. J Antibiot 33:1120–1127
Tanaka Y, Komatsu M, Okamoto S, Tokuyama S, Kaji A, Ikeda H, Ochi K (2009) Antibiotic overproduction by rpsL and rsmG mutants of various actinomycetes. Appl Environ Microbiol 75:4919–4922
Wang XJ, Wang JD, Xiang WS, Zhang J (2009a) Three new milbemycin derivatives from Streptomyces bingchenggensis. J Asian Nat Prod Res 11:597–603
Wang XJ, Wang XC, Xiang WS (2009b) Improvement of milbemycin-producing Streptomyces bingchenggensis by rational screening of ultraviolet-and chemically induced mutants. World J Microbiol Biotechnol 25:1051–1056
Wang XJ, Yan YJ, Zhang B, An J, Wang JJ, Tian J, Jiang L, Chen YH, Huang SX, Yin M, Zhang J, Al G, Liu CX, Zhu ZX, Xiang WS (2010) Genome sequence of the milbemycin-producing bacterium Streptomyces bingchenggensis. J Bacteriol 192:4526–4527
Wei X, Yunxiang L, Yinghua Z (2006) Enhancement and selective production of oligomycin through inactivation of avermectin’s starter unit in Streptomyces avermitilis. Biotechnol Lett 28:911–916
Xiang WS, Wang JD, Wang XJ, Zhang J, Wang Z (2007) Further new milbemycin antibiotics from Streptomyces bingchenggensis. J Antibiot 60:608–613
Xiang WS, Wang JD, Fan HM, Wang XJ, Zhang J (2008) New seco-milbemycins from Streptomyces bingchenggensis: fermentation, isolation and structure elucidation. J Antibiot 61:27–32
Xiang WS, Wang JD, Wang XJ, Zhang J (2009a) Bingchamides A and B, two novel cyclic pentapeptides from the Streptomyces bingchenggensis: fermentation, isolation, structure elucidation and biological properties. J Antibiot 62:501–505
Xiang WS, Wang JD, Wang XJ, Zhang J (2009b) A novel macrolide compound from Streptomyces bingchenggensis: fermentation, isolation, structure elucidation and biological properties. J Antibiot 62:229–231
Yadav P, Singh R (2011) A review on anthelmintic drugs and their future scope. Int J Pharm Pharmaceut Sci 3:17–21
Yu Q, Bai LQ, Zhou XF, Deng ZX (2012) Inactivation of the positive LuxR-type oligomycin biosynthesis regulators OlmRI and OlmRII increases avermectin production in Streptomyces avermitilis. Chinese Sci Bull 57:869–876
Zhang X, Chen Z, Zhao J, Song Y, Wen Y, Li J (2004) Deletion analysis of oligo mycin PKS genes (olmA) in Streptomyces avermitilis. Chinese Sci Bull 49:350–354
Zhou M, Jing X, Xie P, Chen W, Wang T, Xia H, Qin Z (2012) Sequential deletion of all the polyketide synthase and nonribosomal peptide synthetase biosynthetic gene clusters and a 900-kb subtelomeric sequence of the linear chromosome of Streptomyces coelicolor. FEMS Microbiol Lett 33:169–179
Acknowledgments
This research was supported by the National Key Technology R&D Program (2010CB126102), the Outstanding Youth Foundation of China (31225024), the National Natural Science Foundation of China (30971937), the Program for New Century Excellent Talents in University (NCET-08-0668, 1154-NCET-002), the Outstanding Youth Foundation of Heilongjiang Province (JC200706), and the Program for New Teachers in University (20092325120007).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Zhang and An contributed equally to this work.
Rights and permissions
About this article
Cite this article
Zhang, J., An, J., Wang, JJ. et al. Genetic engineering of Streptomyces bingchenggensis to produce milbemycins A3/A4 as main components and eliminate the biosynthesis of nanchangmycin. Appl Microbiol Biotechnol 97, 10091–10101 (2013). https://doi.org/10.1007/s00253-013-5255-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-013-5255-5