
Abstract
Efficient and highly enantioselective hydrolysis of 2-carboxyethyl-3-cyano-5-methylhexanoic acid ethyl ester (CNDE) is the most crucial step in chemoenzymatic synthesis of Pregabalin. By using site-saturation mutagenesis and high-throughput screening techniques, lipase Lip from Thermomyces lanuginosus DSM 10635 was engineered to improve its activity towards CNDE. The triple mutant, S88T/A99N/V116D exhibited a 60-fold improvement in specific activity for CNDE (2.35 U/mg) over the wild-type Lip (0.039 U/mg). Modeling and docking studies demonstrated that the mutant could more effectively stabilize oxygen anions in transition states and the lid of Lip in the open conformation. Additionally, the kinetic resolution of CNDE catalyzed by Escherichia coli cell overexpressing S88T/A99N/V116D mutant afforded (3S)-2-carboxyethyl-3-cyano-5-methylhexanoic acid in 42.4 % conversion and 98 % ee within 20 h with a substrate loading of 1 M (255 g/l). These results demonstrated that a novel and promising biocatalyst was created for efficient chemoenzymatic manufacturing of Pregabalin.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
(S)-3-(Aminomethyl)-5-methylhexanoic acid (Pregabalin) is a lipophilic 4-aminobutyric acid (GABA) analogue and has been developed to a new blockbuster drug for the treatment of several central nervous system disorders including neuropathic pain, epilepsy, anxiety and social phobia (Silverman 2008). Compared to chemical catalysis, biocatalysis provides tremendous advantages such as high efficiency, high selectivity and “green” reaction conditions (Pollard and Woodley 2007; Wenda et al. 2011; Woodley 2008). In recent years, biocatalysts have been increasingly applied in practical synthesis of chiral compounds. Consequently, several biocatalysts such as nitrilase (Xie et al. 2006), lipase and esterase (Felluga et al. 2008; Martinez et al. 2008; Lambertus 2009; Hu et al. 2011; Zheng et al. 2012, 2013) were employed for the preparation of chiral intermediates of Pregabalin. The most successful and virtually the only effective scalable chemoenzymatic strategy for Pregabalin involved lipase-catalyzed resolution of 2-carboxyethyl-3-cyano-5-methylhexanoic acid ethyl ester (CNDE) to produce the desired (S)-mono acid enantiomer in high yields and enantioselectivity (Hu et al. 2011; Martinez et al. 2008). Only the biocatalyst Lipolase®, a commercially available lipase supplied by Novozymes, was reported for efficient kinetic resolution of CNDE up to date. Accordingly, it will be of great significance to develop a novel biocatalyst for the chemoenzymatic route to Pregabalin.
TLL (lipase from Thermomyces lanuginosus) was the enzyme responsible for the lipolytic activity of Lipolase® and Lipozyme TL IM®, the commercial soluble and immobilized form of lipase supplied by Novozymes. Although TLL initially oriented toward the food industry, it has been applied in many different industrial areas from biodiesel production to fine chemicals (mainly in enantio- and regioselective or specific processes) (Fernandez-Lafuente 2010). Recently, another thermophilic lipase LN with 78.4 % amino acid sequence identity to TLL has been cloned from T. lanuginosus HSAUP03 80006 (Zheng et al. 2011). Therefore, there is substantial interest in development of lipase LN or its homologous proteins for use in chemical industries.
Enzymes were usually required to catalyze non-natural substrates under conditions that favor an efficient and economically viable process in industrial application (Dalby 2011). However, in most cases, wild enzymes may not be suitable for practical applications due to the differences between the cellular environment and the industrial setting (Wang et al. 2012b). Therefore, it is crucial to search for a relatively simple and cost-effective method to solve this problem. The widely used strategy is to engineer existing biocatalysts to be compatible with the target industrial process via directed evolution (Dalby 2011; Reetz 2012; Turner 2009; Wang et al. 2012b). Directed evolution has been a powerful tool used to engineer proteins with diverse desired properties, such as activity (Gerstenbruch et al. 2012; Martinez et al. 2013; Wang et al. 2012a), enantioselectivity (Cambon et al. 2010; De Groeve et al. 2010; Reetz et al. 2010), stability (Hoelsch et al. 2013; Mordukhova et al. 2008), and tolerance against organic solvents (Li et al. 2012). The most frequently used strategies and techniques for directed evolution libraries are error-prone polymerase chain reaction (epPCR), DNA shuffling and site-saturation mutagenesis (Reetz et al. 2010). Nevertheless, site-saturation mutagenesis is an efficient and fast method in the directed evolution of enzymes, because in all cases small enzyme libraries means the screening effort can be reduced significantly (Reetz et al. 2001; Sanchis et al. 2008).
In this report, the lipase Lip, which has highly homologous to lipase LN, was cloned from T. lanuginosus DSM 10635 and overexpressed in Escherichia coli. The site-saturation mutagenesis at oxyanion hole and the lid hinge region for enhancement of the specific activity was described. Furthermore, the mutant S88T/A99N/V116D with the highest hydrolytic activity towards CNDE was investigated by kinetic analysis, structural modeling and docking studies. In addition, the kinetic resolution of CNDE by using the whole cell of recombinant E. coli as biocatalyst was described.
Materials and methods
Chemicals
Restriction endonucleases and T4 DNA ligase were purchased from Fermentas (Shenzhen, China). Pfu DNA polymerase, Taq DNA polymerase and PrimeSTAR HS DNA polymerase were purchased from Takala (Dalian, China). The DNA gel extraction, plasmid extraction and PCR product purification kits were purchased from Axygen (Hangzhou, China). Kanamycin and isopropyl-β-d-thiogalacto- pyranoside (IPTG) was purchased from Sigma. CNDE was kindly provided by Zhejiang Apeloa Medical Technology Co. Ltd. (Jinhua, China). Lipase from T. lanuginosus (TLL; commercial name: Lipolase®) was obtained from Novozymes (Denmark) and purified as previously described (Peters et al. 1998). The other chemicals used in this work were of analytical grade from local suppliers.
Strains, culture conditions and plasmids
Strain T. lanuginosus DSM 10635 was purchased from German Collection of Microorganisms and Cell Cultures (DSMZ). T. lanuginosus DSM 10635 was cultivated in a liquid medium containing (g/l): soluble starch 20.0, yeast extract 50.0, K2HPO4 5.0, MgSO4 · 7H2O 1.0, CaCl2 0.15, and olive oil 18.0. T. lanuginosus DSM 10635 were grown in 500-ml Erlenmeyer flasks containing 50 ml of culture medium. Flasks were incubated on a rotary shaker at 180 rpm at 50 °C for 72 h. The E. coli JM109, BL21 (DE3) (Novagen, USA), plasmid pMD18-T (TaKaRa, Dalian, China) and pET-28b(+) (Novagen, USA) were used for gene cloning and recombinant proteins expression experiments, respectively. E. coli cells were grown in Luria–Bertani (LB) medium with an appropriate antibiotic if necessary as described by Sambrook and Russell (2001). All media were solidified with 1.5 % agar.
DNA manipulation and plasmid construction
The total RNA from T. lanuginosus DSM 10635 was extracted and taken as template for cDNA synthesis using PrimeScript TM 1st Strand cDNA Synthesis Kit (TaKaRa), according to the manufacturer's instructions. The mature protein coding sequence (lip) of lipase Lip was obtained by PCR. The primers Lip-F and Lip-R (Table 1) designed from the available T. lanuginosus lipase sequence (GenBank EU004196) and cDNA template were used for PCR amplification. The PCR products were analyzed by 0.9 % agarose gel electrophoresis. The approximately 0.8-kb fragment was recovered by DNA gel Extraction kit (Axygen), then cloned into T/A vector pMD18-T, and the recombinant plasmid pMD18-T-lip was transformed into E. coli JM109. The recombinant plasmid DNA was sequenced on both strands with an Applied Biosystems Model 377 automatic DNA sequencer, and a dye-labeled terminator sequencing kit. Subsequently, the sequences were analyzed using the software DNAMAN Version 5.2 (Lynnon Biosoft, Quebec, Canada). The recombinant vector pMD18-T-lip was double digested by NcoI/HindIII. The double digestion product was purified using DNA gel Extraction kit, and ligated into a previously NcoI/HindIII linearised pET-28(b) vectors. The ligation mixture containing 100 ng of vector and 200 ng of insert DNA using 1 U of T4 DNA ligase in a final volume of 10 μl, which was incubated overnight at 16 °C. The recombinant plasmid pET28-lip was transformed into E. coli BL21 (DE3) for protein overexpression. The transformed colonies were screened by colony PCR followed by double digestion for verification.
Construction of saturation mutagenesis libraries
Site-saturation mutagenesis was carried out using a protocol based on PCR (Chronopoulou and Labrou 2011). Saturation mutagenesis libraries were created at sites A (Ser88, Trp94, and Asn97), B (Ser88 and Leu152), C (Val91, Ala96, Ala99 and Ala100), and D (Val116) using corresponding forward primers and reverse primers (Table 1) according to an improved PCR-based method for creating saturation mutagenesis libraries. Thirty PCR reactions were performed with PrimeSTAR HS DNA polymerase using these primers and plasmid (pET28-lip-WT or pET28-lip-mutant isolated from E. coli BL21 [DE3]) as the template DNA. The temperature program used was 3 min at 98 °C followed by 30 cycles of 10 s at 98 °C, 10 s at 60 °C and 6 min at 72 °C and finished with 10 min at 72 °C. PCR amplification products were digested with DpnI to remove parent plasmid (2 h at 37 °C with 10 units of DpnI), then the reaction mixtures were transformed into chemical competent E. coli BL21 (DE3) and plated on LB agar plates containing 50 μg/ml Kanamycin.
Screening of variants for improved lipase activity
The constructed mutation clone libraries were screened for lipase activity using a pH-based high-throughput screening that developed by Moris-Varas et al. (1999) in a 96-well plate format with some modifications. All clones were transferred to individual wells in 96-well, 2.2-ml growblock (Axygen) that containing 1 ml LB medium. The plates were incubated at 37 °C, 200 rpm and induced with 0.1 mM isopropyl-β-d-1-thiogalactopyranoside (IPTG) until OD600 reached 0.5–0.6. After incubated at 28 °C for 10 h, cells were harvested by centrifugation (4 °C, 3,000 × g, 15 min). The cell pellets were resuspended in 200 μl resuspension buffer (10 mM phosphate buffer, pH 7.2). Aliquots (20 μl) of the supernatant were transferred to two new microtiter plates containing 180 μl of the substrate solution (10 mM phosphate buffer (pH 7.2), 0.01 % Bromothymol blue, 100 mM CNDE). The microtiter plates were incubation at 30 °C for 0.5–3 h. Several control experiments were carried out. Parent E. coli cell supernatant and 10 mM phosphate buffer (pH 7.2) were used as positive and negative control in the reaction, respectively. A change from blue to yellow was an indicator of substrate hydrolysis. The positive clones were marked and further confirmed by GC.
Expression and purification of lipase variants
Recombinant strains E. coli BL21 (DE3) harboring either pET28-lip-WT or pET28-lip-mutant were grown at 37 °C in LB medium supplemented with 50 μg/ml kanamycin. IPTG was added to a final concentration of 0.1 mM for induction when the OD600 was about 0.6 measured by spectrophotometer. After 10 h, induction at 28 °C, the cells were harvested by centrifugation at 12,000 × g for 10 min at 4 °C.
Recombinant E. coli cells were suspended in 50 mM Tris–HCl buffer (pH 8.0) and disrupted for 20 min using an ultrasonic oscillator. The cell-free extract was fractionated with solid ammonium sulfate. The precipitate obtained with 35–55 % saturation of ammonium sulfate was collected by centrifugation at 12,000 × g for 20 min and dissolved in 1.0 M ammonium sulfate in Tris–HCl (50 mM, pH 8.0). The enzyme solution was loaded onto a Phenyl Sepharose 6 Fast Flow column (1.6 × 20 cm, GE) equilibrated with 1.0 M ammonium sulfate in Tris–HCl (50 mM, pH 8.0) and followed by elution with a linear gradient of ammonium sulfate from 1.0 to 0 M, in distilled water at a flow rate of 0.5 ml min−1.The fractions containing lipase activity were dialyzed against Tris–HCl (50 mM, pH 8.0). The dialyzed component was loaded onto a DEAE-Sepharose FF column (1.6 × 20 cm, GE) previously equilibrated with Tris–HCl (50 mM, pH 8.0). The adsorbed components were eluted with a linear gradient of NaCl from 0 to 0.5 M at a flow rate of 1.0 ml min−1. The fractions containing lipase activity were pooled together, dialyzed overnight against the same buffer, lyophilized and resuspended in Tris–HCl (50 mM, pH 8.0). The concentrations of proteins were measured according to the method of Bradford (1976) using bovine serum albumin (BSA) as the standard.
Activity assay and analytical methods
Lipase activity of pure enzyme was determined by monitoring the hydrolysis of CNDE. Lipase activity was measured at 40 °C in 10 ml reaction system containing properly pure enzyme, Tris–HCl buffer (100 mM, pH 7.5), 50 mM calcium acetate and 100 mM CNDE. One unit of lipase activity (U) was defined as the amount of enzyme releasing 1 μmol 2-carboxyethyl-3-cyano-5-methylhexanoic acid/min under the assay conditions.
Enantiomeric compositions of residual ester and the corresponding acid in reaction system were determined by GC-14C gas chromatography (Shimadzu, Japan) equipped with FID detector and chiral capillary column Astec CHIRALDEX™ G-TA (30 m × 0.25 mm, 0.25 μm film thickness) using helium as carrier gas. The injector and detector temperatures were set at 220 °C. The column temperature was set at 135 °C for 42 min. The conversion (C) and enantiomeric ratio (E) were calculated based on ee S and ee P as the method developed by Rakels et al. (1993).
Determinations of kinetics parameters
Kinetic parameters of the wild-type and mutant Lip towards CNDE were determined in Tris–HCl buffer (100 mM, pH 7.5) at 40 °C by increasing substrate concentrations ranging from 1 to 100 mM with purified enzyme. The value of parameter k cat was obtained by using the equation k cat = V max/[E], where [E] is the molar concentration of the enzymes.
Molecular homology modeling and docking
The three-dimensional homology models of Lip and variants were generated using Build Homology Models (MODELER), one of the modules in Discovery Studio 2.1 (Accelrys Software, San Diego, CA, USA), using crystal structure of TLL at 2.2 Å resolution (PDB accession code 1GT6) as the template (Yapoudjian et al. 2002). The generated model was then subjected to molecular mechanics optimization using CHARMM27 force-field. Energy minimization (geometry optimization) was performed until the gradient of 0.01 kcal/(Å mol) was reached. The geometry qualities of the final models were checked using Procheck program (Laskowski et al. 1993). Finally, the best quality model was chosen for further calculations, molecular modeling, and docking studies by Autodock 4.0 (Morris et al. 2009). The visualization was performed with PyMOL program version 1.2r1 (http://www.pymol.org).
Whole-cell biocatalysis
The bioconversion of CNDE to (3S)-2-carboxyethyl-3-cyano-5-methylhexanoic acid using recombinant E. coli as a whole-cell biocatalysts was studied. E. coli cells were harvested by centrifugation at 12,000 × g for 10 min at 4 °C after expression of lipase and washed with saline twice. The reaction mixture (250 ml) contains 1 % E. coli cell (10 g dry cell weight/l), 150 mM calcium acetate and 1 M CNDE (255 g/l). In order to be compared, the experiment using the wild-type Lip whole-cell and 0.3 M CNDE was performed. The hydrolysis reaction was carried out on a stirred reactor (500 rpm) at 30 °C and neutral pH (maintaining neutral pH using 1 M NaOH). Samples were taken periodically to check the enantiomeric excess of the product and conversion of the substrate by chiral-GC analysis.
Nucleotide sequence accession number
The mature protein coding sequence (lip) of lipase Lip from T. lanuginosus DSM 10635 was deposited in the GenBank database under accession number KC479729.
Results
Heterologous expression of lip gene in E. coli
The cDNA (lip, 825 bp) obtained by RT-PCR from T. lanuginosus DSM 10635 using primers Lip-F and Lip-R encoded a 274-amino-acid polypeptide of mature lipase Lip. Sequence analysis indicated that the lip gene (GenBank accession No. KC479729) shared the highest similarity of 99.9 % with the lipase LN from T. lanuginosus HSAUP03 80006 (GenBank accession No. EU004197) (Zheng et al. 2011). There was only one base different in nucleotide sequence, but Lip and LN shared the same amino acid sequence.
The lip gene was cloned into the expression vector pET-28b, and overexpressed in E. coli BL21 (DE3) under the control of T7 promoter. Protein expression was induced with 0.1 mM IPTG and the amount of recombinant Lip expression product was more than 20 % of total bacterial proteins. The induced crude extract containing the lipase were analyzed by SDS-PAGE (Fig. 1a). Most of them existed in a form of soluble protein. After purified by ammonium sulfate precipitation, hydrophobic interaction chromatography and ion-exchange chromatography, a band (about 34 kDa) corresponding to the lipase with a yield of 25.6 % was purified from cell disruption supernatant (Fig. 1b). The kinetic resolution of CNDE by purified Lip suggested that the enzyme showed high enantioselectivity (E>200) for CNDE, but low specific activity (0.039 U/mg; Table 2).

SDS-PAGE analysis of soluble fractions obtained from expression experiments (a) and purified Lip from E. coli BL21 (DE3) transformant (b). Lane M protein markers, lane 1 recombinant E. coli cell without induced by IPTG, lane 2 recombinant E. coli cell induced by IPTG, lane 3 precipitation of cell extract, lane 4 supernatant of the cell extract, lane 5 ammonium sulfate precipitate, lane 6 Phenyl Sepharose pool, lane 7 DEAE-Sepharose pool
Building of three dimensional molecular model of Lip
The amino acid sequence of Lip, deduced from the cDNA, was 80 % and 78.4 % identical to that of TTL (Talaromyces thermophilus lipase; NCBI accession No. JF414585) (Romdhane et al. 2012) and TLL (NCBI accession No. ABV69591), respectively. The crystal structure data of TTL has not been reported, while seven crystal structure data of TLL have been reported in the PDB database by the end of March 2012. 1GT6 with open-lid conformation and highest resolution (2.20 Å) was selected as the model template. After refinement, the best quality model (Fig. 2) was chosen for further calculations, molecular modeling and docking studies.

The overall 3-D structure of Lip obtained by homology modelling. Ser151, Asp206, and His263 form the catalytic triad (represented by sticks models; red). The oxyanion hole was consisted of Ser88 and Leu152 (represented by sticks models; blue). The α-helix span between Val91 to Asn97 formed the lid (magenta) and the hinge regions at both sides of the lid (magenta)
Similar to most basic features of microbial lipase, the model structure shows a common α–β hydrolase fold and a catalytic triad composed of a Ser151, which is activated via hydrogen bonds as part of a charge relay system, along with the His263 and Asp206. The active-site Ser residue is contained in the consensus sequence G–X–S–X–G. The α-helix fragment from Val91 to Asn97, which is known as a lid, is located in the top left corner of the active center, and plus eight amino acids (87GSRS90 and 98LAAD101) as two hinges around the lid. The amino acid sequence of the lid and two hinge regions shares high identity with TTL and TLL. The oxyanion hole consisted of Ser88 and Leu152, which donate their backbone amide protons to stabilize the tetrahedral intermediate transiently formed in the hydrolysis reaction, and Ser88 was neighboured to a conserved glycine residue (Shu et al. 2009).
The flexible docking studies between Lip and ligand (S-CNDE) were performed with AutoDock software (version 4.0) using Lamarckian genetic algorithm (Morris et al. 1998). The docking centre was located at the catalytic residue Ser151 and the docking box was settled to contain all desired active-site residues. As shown in Fig. 3, Ser88, Trp94, and Asn97 were located near the S-CNDE, and there were two hydrogen bonds between Ser88 and S-CNDE. The docking result suggested that Ser88, Trp94, and Asn97 and the hydrogen bonds between Ser88 and S-CNDE are important for the hydrolysis reaction.

Three-dimensional representation of substrate S-CNDE in the Lip model active site by docking (AutoDock 4.0). The substrate and the key amino acid residues were described in stick. The catalytic triad Ser151–His263–Asp206 (red), S-CNDE (green), and three amino acid residues Ser88, Trp94, and Asn 97 were located near the S-CNDE (orange)
Construction and screening of saturation mutagenesis libraries of Lip
The saturation mutagenesis libraries were screened using the high-throughput colorimetric activity assay on a 96-well plate format. After four cycles of saturation mutagenesis at sites A and B (Ser88, Trp94, Asn97 and Leu152), 36 positive colonies were screened from 800 colonies. Further evaluation of activity and enantioselectivity by GC, eight colonies which displayed higher catalytic activity were obtained. Sequencing results showed that all eight variants were S88T, and S88T-5 was chosen for further investigation due to the highest activity (0.36 U/mg). No positive mutant with improved activity was isolated at position 94, 97 and 152. Then a combinatory saturation mutagenesis library at sites C (the amino acids in the lid and hinge regions, V91, A96, A99 and A100) was constructed by choosing the best variant S88T as a template, and a double mutant S88T/A99N with higher activity (1.03 U/mg) was screened. Finally, a triple mutant (S88T/A99N/V116D) with higher activity (2.35 U/mg) was screened in saturation mutagenesis library at sites D (V116) by using the variant S88T/A99N as a template. The variants were purified according to the same method used in the wild-type Lip purification. The specific activities of the wild-type Lip, its variants and TLL for CNDE are listed in Table 2. The most active variant (S88T/A99N/V116D) demonstrated a 60-fold improvement over the wild-type Lip in specific activity for CNDE, which almost matches TLL's performance (2.40 U/mg).
Kinetics parameters analysis
The kinetic parameters for both of the purified wild-type enzyme, the Lip variants and TLL towards CNDE were determined. The kinetic parameters K m, V max and k cat of wild-type and Lip variants are summarized in Table 3. Variant S88T was found to have a V max value of 0.431 U mg−1, about 5.8-fold higher than that of the wild-type Lip (0.074 μmol mg−1 min−1), and the K m value of variant S88T (19.3 mM) was lower than that of wild type (36.6 mM), which implied that substrate-binding affinity of the mutant has been significantly improved. The specificity (k cat/K m) of variant S88T is 0.64 min−1 mM−1, indicating that variant S88T has 10.5-fold higher specificity for CNDE. The K m value of variant S88T/A99N (18.7 mM) and S88T/A99N/V116D (18.2 mM) only slightly changed compared to the variant S88T, which suggested that the mutants of A99N and V116D had little effect on the substrate-binding affinity. Meanwhile, the V max value of variant S88T/A99N (1.423 μmol mg−1 min−1) and S88T/A99N/V116D (2.711 μmol mg−1 min−1) improved significantly compared to the variant S88T and wild-type Lip, which implied that the mutants of A99N and V116D have strong positive effect on V max. The specificity (k cat/K m) of variant S88T/A99N and variant S88T/A99N/V116D is 2.28 and 4.47 min−1 mM−1. The results suggested that the specificity of the best variant S88T/A99N/V116D almost match the TLL's specificity for substrate CNDE.
Biosynthesis of chiral intermediate of Pregabalin by the whole-cell biocatalyst
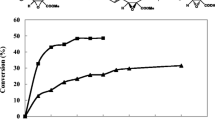
To verify the potentiality of the mutant in enzymatic hydrolysis of CNDE, the kinetic resolution was performed with recombinant E. coli as a whole-cell biocatalyst in a 250-ml reaction mixture. The reaction profile was shown in Fig. 4. (3S)-2-carboxyethyl-3-cyano-5-methylhexanoic acid was produced in 424 mM with a substrate loading of 1 M after 20 h. Moreover, the optical purity of product increased from 95 % to 98 % with proceeding of the reaction and was maintained at high level (>98 %) after 6 h. In contrast, only 120 mM product was formed with 0.3 M CNDE after 50 h bioconversion by the wild-type Lip whole-cell, and it would be inhibited at substrate loading higher than 500 mM. These results suggested that the mutant S88T/A99N/V116D whole-cell was an efficient biocatalyst for manufacturing of Pregabalin.

Progress curves of the kinetic resolution of CNDE in 250 ml reaction mixture using mutant S88T/A99N/V116D and wild-type Lip whole-cell as biocatalyst, respectively. The Filled triangle and filled square denote the ee value and concentration of (3S)-2-carboxyethyl-3-cyano-5-methylhexanoic acid using mutant S88T/A99N/V116D whole-cell as biocatalyst, respectively; empty triangle and empty square denote the ee value and concentration of (3S)-2-carboxyethyl-3-cyano-5-methylhexanoic acid using wild-type Lip whole-cell as biocatalyst, respectively
Discussion
Pregabalin (Lyrica®) is one of the fastest growing drugs in the market. The most successful scalable chemoenzymatic strategy for Pregabalin involved Lipolase®-catalyzed resolution of CNDE. Up until now, except for the commercial available lipase, no other robust biocatalyst has been reported. Therefore, development of novel biocatalyst with high process efficiency will be of great significance for Pregabalin production. The lipase Lip from T. lanuginosus DSM 10635, which was heterologous expressed in E. coli, has showed great potentiality as biocatalyst candidate for its high (S)-enantioselectivity to CNDE (E>200). However, the catalytic activity of the recombinant Lip is too low to fulfill the requirements of the industrial application. The site-saturation mutagenesis technique was used to improve the hydrolytic activity of Lip towards CNDE.
The key to the success of site-saturation mutagenesis is the selection of the mutation site. The flexible docking studies indicated Ser88, Trp94, and Asn97 lying in the proximity of the S-CNDE. Consequently, Ser88, Trp94, and Asn97 were selected as the mutation points in the mutagenesis experiment. The catalytic activity of enzymes is commonly attributed to their ability to stabilize transition states (Simon and Goodman 2010). Oxyanion holes stabilize oxygen anions in transition states of lipase and substrate. Ser88 and Leu152 constituting the oxyanion hole were also selected as the mutation points. On the other hand, it is widely believed that activation of lipase involves the movement of the lid and the unmasking or restructuring of the active site of lipases through conformational changes (from close conformation to open conformation). Therefore, Val91, Ala96, Ala99, and Ala100 in the lid and hinges were selected as the mutation points. Furthermore, Brzozowski et al. (2000) demonstrated that the lid of TLL in the maximally open position must be stabilized by hydrogen bonds between Asp62–Asn88, Asn88–Gly61, and Asp96–Asp111. The alignment of amino acid sequence between Lip and TLL suggested that Val116 in Lip corresponding to Asp111 in TLL. Consequently, Val116 was also selected as the mutation point.
The lipase from T. lanuginosus was classified into Rhizomucor mihei lipase homologous family, which was also called as Mucoral lipase family. The Lipase Engineering Database (Pleiss et al. 2000) has collected 123 lipases in homologous family abH23.01 (Rhizomucor mihei lipase like). Although the 123 fungi lipases vary in length from 130 to 809 amino acid residues, the amino acid residues forming the catalytic triad and oxyanion hole are highly conserved. Oxyanion holes of Rhizomucor mihei lipase homologous family consist of three hydrogen bond donors (NH of a Leu, NH and OH of a Ser or Thr), orienting toward a central oxygen atom in the substrate (Holzwarth et al. 1997; Norin et al. 1994; Schmid and Verger 1998). Our study found that mutation of Ser88 to any other amino acids, except for Thr, caused a loss of activity. These results suggested that the hydrogen bond between β-OH of Ser88 and S-CNDE was necessary for its function. The increased activity for S88T might be explained by the increase in steric bulk introduced. Threonine substituted Serine, which meant that a-H of β-carbon atom substituted by –CH3 with much larger steric hindrance, leading to the conformation alterative of the β-hydroxy. Therefore, modeling studies of the structure of wild-type Lip and mutant S88T complex with CNDE were carried out in an attempt to verify the hypothesis. Figure 5 shows that the hydrogen bonds between NH of Leu152 and Ser88 (wild-type Lip) or Thr88 (mutant S88T) and the carbonyl oxygen of CNDE did not changed. The side-chain OH of Ser88 or Thr88 contributed to the oxyganion hole, and the β-OH of Thr88 in the mutant S88T interacted tightly with carbonyl oxygen of substrates (at a distance of 2.2 Å; Fig. 5b), while the β-OH of Ser88 in the wild-type Lip with carbonyl oxygen of substrates was at a distance of 3.2 Å (Fig. 5a). Shortening of the distance means that oxyanion holes can more effectively stabilized oxygen anions in transition states in the hydrolytic reaction. Therefore, the mutant S88T exhibited higher hydrolytic activity towards CNDE. Analysis of kinetics parameters also implied that the binding affinity of S88T to CNDE has been significantly improved.

Models of oxyanion holes and catalytic triad with CNDE of wild-type Lip (a) and mutant S88T (b). Hydrogen bonds between donors (Ser88 and Leu152 [wild type, a] and Thr88 and Leu152 [mutant, b]) and the carbonyl oxygen of substrates are represented by red dashed lines. The side-chain OH of Ser88 or Thr88 contributes to the oxyganion hole, and the β-OH of Thr88 in the mutant S88T interacts tightly with carbonyl oxygen of substrates (at a distance of 2.2 Å, b), while the β-OH of Ser88 in the wild-type Lip with carbonyl oxygen of substrates at a distance of 3.2 Å (a)
Lipase activity is greatly increased at the oil–water interface, a phenomenon known as interfacial activation (Berg et al. 1998; Verger 1997). The activation of lipases involves the movement of the lid and the conformational changes. Carriere et al. (1997) designed a lipase mutant with a permanent open conformation by domain exchange between the classical human pancreatic lipase and the guinea pig pancreatic lipase related protein 2, which showed the importance of the hinge regions at both sides of the lid domain to the interfacial activation. In order to explain why the substitution of A99N and V116D exhibited higher hydrolytic activity towards CNDE, the model structures of wild-type Lip and mutant S88T/A99N/V116D were analyzed. Figure 6 showed that Gly66, Asp67, Ser90, Asn93, Asn99, Asp101, and Asp116 had a specific function in Lip in stabilization of the lid in the open conformation. In mutant S88T/A99N/V116D, three new hydrogen bonds were formed to stabilize the lid in the maximally open position, one hydrogen bond was between Asp101 and Asp116, and the other two was between Asn99 and Asp116 (Fig. 6b). However, the three newly formed hydrogen bonds did not exist in the wild-type Lip (Fig. 6a). Compared to wild-type lipase, the higher hydrolytic activity of A99N/V116D variant could be attributable to a higher fraction of enzyme molecules present in the open conformation. The hydrogen bonds between Asp67–Ser90 and Asn99–Asp116 might play important roles in stabilizing the lid in the open conformation. The activity was only one-tenth of the parent lipase when a site-specific mutagenesis D67A was occurred (data not shown).

Comparison of the open conformation of the wild type (a) and mutant S88T/A99N/V116D (b). Amino acid backbones are represented by ribbon cartoon model, lid and hinge regions are shown in red. Gly66, Asp67, Ser90, Asn93, Ala99, Asp101, Val116 (wild type, a) and Gly66, Asp67, Ser90, Asn93, Asn99, Asp101, Asp116 (S88T/A99N/V116D, b) are represented by sticks models. In mutant S88T/A99N/V116D, three new hydrogen bonds, one was between Asp101 and Asp116, the other two hydrogen bonds between Asn99 and Asp116, were formed to stabilize the lid in open conformation
Furthermore, we successfully applied the created E. coli whole-cell biocatalyst in the kinetic resolution of CNDE. The biocatalyst showed high activity even at high substrate loading (255 g/l CNDE). After 20 h, a conversion of 42.4 % with an ee P of 98 % was achieved. Utilizing the whole-cell instead of the purified enzymes or the immobilized enzymes as the biocatalyst for Pregabalin production is a potential way to reduce the cost of biocatalysis. There is a concern that the need of overcoming the cell wall for the substrate to reach the enzyme(s) may decrease the reaction rate when using the whole cells (de Carvalho 2011). However, such impact can be negligible in this reaction since both the substrate (CNDE) and product ((3S)-2-carboxyethyl-3-cyano-5-methylhexanoic acid) are liposoluble substances. Predictably, the mutant S88T/A99N/V116D whole-cell biocatalyst can be an attractive and competitive alternative to Lipolase® to be used in chemoenzymatic route to Pregabalin. Obviously, enzyme immobilization or cell immobilization is another potential way to reduce the cost of biocatalysis. What is more, a proper immobilization technique may not only permit users to obtain a reusable biocatalyst; it may also be a powerful tool to improve enzyme properties, including stability, activity, specificity and inhibitions (Garcia-Galan et al. 2011; Hernandez and Fernandez-Lafuente 2011).
Lipase Lip from T. lanuginosus DSM 10635 is one of the potential biocatalyst candidates for the kinetic resolution of CNDE into (3S)-2-carboxyethyl-3-cyano-5-methylhexanoic acid. However, the catalytic activity of the wild-type Lip cannot match the industrial applications. In this work, site-saturation mutagenesis has been successfully used to develop a triple-mutated variant (S88T/A99N/V116D) of lipase Lip. Compared to wild-type lipase, the developed lipase variant showed a 60-fold increase in hydrolytic activity toward CNDE. Further insight into the mechanism was gained by structural modeling and docking analysis. The results showed that the mutation S88T contributes to the stabilization of oxygen anions of CNDE in transition states, and the higher hydrolytic activity of A99N/V116D variant might be due to a higher fraction of enzyme molecules present in the open conformation. Furthermore, a highly robust whole-cell biocatalytic process was developed for the S-enantioselective kinetic resolution of CNDE.
References
Berg OG, Cajal Y, Butterfoss GL, Grey RL, Alsina MA, Yu BZ, Jain MK (1998) Interfacial activation of triglyceride lipase from Thermomyces (Humicola) lanuginosa: kinetic parameters and a basis for control of the lid. Biochemistry 37:6615–6627
Bradford M (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–252
Brzozowski AM, Savage H, Verma CS, Turkenburg JP, Lawson DM, Svendsen A, Patkar S (2000) Structural origins of the interfacial activation in Thermomyces (Humicola) lanuginosa lipase. Biochemistry 39:15071–15082
Cambon E, Piamtongkam R, Bordes F, Duquesne S, Andre I, Marty A (2010) Rationally engineered double substituted variants of Yarrowia lipolytica lipase with enhanced activity coupled with highly inverted enantioselectivity towards 2-bromo phenyl acetic acid esters. Biotechnol Bioeng 106:852–859
Carriere F, Thirstrup K, Hjorth S, Ferrato F, Nielsen PF, WithersMartinez C, Cambillau C, Boel E, Thim L, Verger R (1997) Pancreatic lipase structure–function relationships by domain exchange. Biochemistry 36:239–248
Chronopoulou EG, Labrou NE (2011) Site-saturation Mutagenesis: a powerful tool for structure-based design of combinatorial mutation libraries. Dunn B (ed) Current protocols in protein science. John Wiley, New York, 63: 26.6.1–26.6.10
Dalby PA (2011) Strategy and success for the directed evolution of enzymes. Curr Opin Struc Biol 21:473–480
de Carvalho CCCR (2011) Enzymatic and whole cell catalysis: finding new strategies for old processes. Biotechnol Adv 29:75–83
De Groeve MRM, Remmery L, Van Hoorebeke A, Stout J, Desmet T, Savvides SN, Soetaert W (2010) Construction of cellobiose phosphorylase variants with broadened acceptor specificity towards anomerically substituted glucosides. Biotechnol Bioeng 107:413–420
Felluga F, Pitacco G, Valentin E, Venneri CD (2008) A facile chemoenzymatic approach to chiral non-racemic beta-alkyl-gamma-amino acids and 2-alkylsuccinic acids. A concise synthesis of (S)-(+)-Pregabalin. Tetrahedron-Asymmetr 19:945–955
Fernandez-Lafuente R (2010) Lipase from Thermomyces lanuginosus: uses and prospects as an industrial biocatalyst. J Mol Catal B-Enzym 62:197–212
Garcia-Galan C, Berenguer-Murcia A, Fernandez-Lafuente R, Rodrigues RC (2011) Potential of different enzyme immobilization strategies to improve enzyme performance. Adv Syn Catal 353(16):2885–2904
Gerstenbruch S, Wulf H, Mussmann N, O'Connell T, Maurer KH, Bornscheuer UT (2012) Asymmetric synthesis of d-glyceric acid by an alditol oxidase and directed evolution for enhanced oxidative activity towards glycerol. Appl Microbiol Biotechnol 96:1243–1252
Hernandez K, Fernandez-Lafuente R (2011) Control of protein immobilization: coupling immobilization and site-directed mutagenesis to improve biocatalyst or biosensor performance. Enzyme Microb Technol 48(2):107–122
Hoelsch K, Suhrer I, Heusel M, Weuster-Botz D (2013) Engineering of formate dehydrogenase: synergistic effect of mutations affecting cofactor specificity and chemical stability. Appl Microbiol Biotechnol 97:2473–2481
Holzwarth HC, Pleiss J, Schmid RD (1997) Computer-aided modelling of stereoselective triglyceride hydrolysis catalyzed by Rhizopus oryzae lipase. J Mol Catal B-Enzym 3:73–82
Hu SH, Martinez CA, Tao JH, Tully WE, Patrick K, Dumond Y (2011) Preparation of Pregabalin and related compounds. US8134023B2.
Lambertus T (2009) Processes for making Pregabalin and intermediates therefor. WO2009149928
Laskowski RA, Macarthur MW, Moss DS, Thornton JM (1993) Procheck — a program to check the stereochemical quality of protein structures. J Appl Crystallogr 26:283–291
Li ZW, Roccatano D, Lorenz M, Schwaneberg U (2012) Directed evolution of subtilisin E into a highly active and guanidinium chloride- and sodium dodecylsulfate-tolerant protease. ChemBioChem 13:691–699
Martinez CA, Hu SH, Dumond Y, Tao JH, Kelleher P, Tully L (2008) Development of a chemoenzymatic manufacturing process for pregabalin. Org Process Res Dev 12:392–398
Martinez R, Jakob F, Tu R, Siegert P, Maurer KH, Schwaneberg U (2013) Increasing activity and thermal resistance of Bacillus gibsonii alkaline protease (BgAP) by directed evolution. Biotechnol Bioeng 110:711–720
Mordukhova EA, Lee HS, Pan JG (2008) Improved thermostability and acetic acid tolerance of Escherichia coli via directed evolution of homoserine o-succinyltransferase. Appl Environ Microbiol 74:7660–7668
Moris-Varas F, Shah A, Aikens J, Nadkarni NP, Rozzell JD, Demirjian DC (1999) Visualization of enzyme-catalyzed reactions using pH indicators: rapid screening of hydrolase libraries and estimation of the enantioselectivity. Bioorg Med Chem 7:2183–2188
Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, Olson AJ (1998) Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J Comput Chem 19:1639–1662
Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ (2009) AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem 30:2785–2791
Norin M, Haeffner F, Achour A, Norin T, Hult K (1994) Computer modeling of substrate-binding to lipases from Rhizomucor miehei, Humicola lanuginosa, and Candida rugosa. Protein Sci 3:1493–1503
Peters GH, Svendsen A, Langberg H, Vind J, Patkar SA, Toxvaerd S, Kinnunen PKJ (1998) Active serine involved in the stabilization of the active site loop in the Humicola lanuginosa lipase. Biochemistry 37:12375–12383
Pleiss J, Fischer M, Peiker M, Thiele C, Schmid RD (2000) Lipase engineering database — understanding and exploiting sequence–structure–function relationships. J Mol Catal B-Enzym 10:491–508
Pollard DJ, Woodley JM (2007) Biocatalysis for pharmaceutical intermediates: the future is now. Trends Biotechnol 25:66–73
Rakels JLL, Straathof AJJ, Heijnen JJ (1993) A simple method to determine the enantiomeric ratio in enantioselective biocatalysis. Enzyme Microb Technol 15:1051–1056
Reetz MT (2012) Laboratory evolution of stereoselective enzymes as a means to expand the toolbox of organic chemists. Tetrahedron 68:7530–7548
Reetz MT, Wilensek S, Zha DX, Jaeger KE (2001) Directed evolution of an enantioselective enzyme through combinatorial multiple-cassette mutagenesis. Angew Chem Int Edit 40:3589–3591
Reetz MT, Prasad S, Carballeira JD, Gumulya Y, Bocola M (2010) Iterative saturation mutagenesis accelerates laboratory evolution of enzyme stereoselectivity: rigorous comparison with traditional methods. J Am Chem Soc 132:9144–9152
Romdhane IB, Frikha F, Maalej-Achouri I, Gargouri A, Belghith H (2012) Gene cloning and molecular characterization of the Talaromyces thermophilus lipase catalyzed efficient hydrolysis and synthesis of esters. Gene 494:112–118
Sambrook J, Russell DW (2001) Molecular cloning: a laboratory manual, 3rd edn. Cold Spring Harbor Laboratory, New York
Sanchis J, Fernandez L, Carballeira J, Drone J, Gumulya Y, Hobenreich H, Kahakeaw D, Kille S, Lohmer R, Peyralans J, Podtetenieff J, Prasad S, Soni P, Taglieber A, Wu S, Zilly F, Reetz M (2008) Improved PCR method for the creation of saturation mutagenesis libraries in directed evolution: application to difficult-to-amplify templates. Appl Microbiol Biotechnol 81:387–397
Schmid RD, Verger R (1998) Lipases: interfacial enzymes with attractive applications. Angew Chem Int Edit 37:1609–1633
Shu ZY, Duan MJ, Yang JK, Xu L, Yan YJ (2009) Aspergillus niger lipase: heterologous expression in Pichia pastoris, molecular modeling prediction and the importance of the hinge domains at both sides of the lid domain to interfacial activation. Biotechnol Prog 25:409–416
Silverman RB (2008) From basic science to blockbuster drug: the discovery of Lyrica. Angew Chem Int Edit 47:3500–3504
Simon L, Goodman JM (2010) Enzyme catalysis by hydrogen bonds: the balance between transition state binding and substrate binding in oxyanion holes. J Org Chem 75:1831–1840
Turner NJ (2009) Directed evolution drives the next generation of biocatalysts. Nat Chem Biol 5:568–574
Verger R (1997) ‘Interfacial activation’ of lipases: facts and artifacts. Nat Chem Biol 15:32–38
Wang J, Wang D, Wang B, Mei ZH, Liu J, Yu HW (2012a) Enhanced activity of Rhizomucor miehei lipase by directed evolution with simultaneous evolution of the propeptide. Appl Microbiol Biotechnol 96:443–450
Wang M, Si T, Zhao HM (2012b) Biocatalyst development by directed evolution. Bioresour Technol 115:117–125
Wenda S, Illner S, Mell A, Kragl U (2011) Industrial biotechnology—the future of green chemistry? Green Chem 13:3007–3047
Woodley JM (2008) New opportunities for biocatalysis: making pharmaceutical processes greener. Trends Biotechnol 26:321–327
Xie ZY, Feng JL, Garcia E, Bernett M, Yazbeck D, Tao JH (2006) Cloning and optimization of a nitrilase for the synthesis of (3S)-3-cyano-5-methyl hexanoic acid. J Mol Catal B-Enzym 41:75–80
Yapoudjian S, Ivanova MG, Brzozowski AM, Patkar SA, Vind J, Svendsen A, Verger R (2002) Binding of Thermomyces (Humicola) lanuginosa lipase to the mixed micelles of cis-parinaric acid/NaTDC — fluorescence resonance energy transfer and crystallographic study. Eur J Biochem 269:1613–1621
Zheng YY, Guo XH, Song NN, Li DC (2011) Thermophilic lipase from Thermomyces lanuginosus: gene cloning, expression and characterization. J Mol Catal B-Enzym 69:127–132
Zheng RC, Li AP, Wu ZM, Zheng YG (2012) Enzymatic production of (S)-3-cyano-5-methylhexanoic acid ethyl ester with high substrate loading by immobilized Pseudomonas cepacia lipase. Tetrahedron-Asymmetry 23:1517–1521
Zheng RC, Wang TZ, Fu DJ, Li AP, Li XJ, Zheng YG (2013) Biocatalytic synthesis of chiral intermediate of Pregabalin with high substrate loading by a newly isolated Morgarella morganii ZJB-09203. Appl Microbiol Biotechnol 97:4839–4847
Acknowledgements
This work was financially supported by the National High Technology Research and Development Program of China (no. 2012AA022201), the Key Scientific and Technology Programs of Zhejiang Province (no. 2012C03005-2) and the Natural Science Foundation of Zhejiang Province (no. Z4090612).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Li, XJ., Zheng, RC., Ma, HY. et al. Engineering of Thermomyces lanuginosus lipase Lip: creation of novel biocatalyst for efficient biosynthesis of chiral intermediate of Pregabalin. Appl Microbiol Biotechnol 98, 2473–2483 (2014). https://doi.org/10.1007/s00253-013-5136-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-013-5136-y