
Abstract
Biocatalyzed synthesis of nucleoside analogues was carried out using two thermostable nucleoside phosphorylases from the hyperthermophilic aerobic crenarchaeon Aeropyrum pernix K1. The synthesis of the 2,6-diaminopurine nucleoside and 5-methyluridine was used as a reaction model to test the process. Both the purine nucleoside phosphorylase (apPNP) and uridine phosphorylase (apUP) were functionally expressed in Escherichia coli. The recombinant enzymes were characterized after purification, and both enzymes showed high thermostability and broad substrate specificity. Both enzymes retained 100 % of their activity after 60 min at high temperature, and the optimum temperature for the enzymes was 90–100 °C. The nucleoside phosphorylases obtained from A. pernix are valuable industrial biocatalysts for high-temperature reactions that produce nucleoside drugs in high yields.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Nucleoside analogues are used extensively in antiviral therapies (Balfour 1999). Since 1985, when the anti-HIV activity of AZT was discovered, many nucleoside drugs have been designed. These compounds mainly act as inhibitors of viral replication or cellular DNA replication (Van Rompay et al. 2003). Purine nucleoside analogues such as abacavir (Carr et al. 2002; Foster and Faulds 1998), fludarabine (Rai et al. 2000), cladribine (Minton 2009), and amdoxovir (Thompson et al. 2005) as well as pyrimidine nucleoside analogues such as stavudine (Sacktor et al. 2009), zidovudine (Scruggs and Dirks Naylor 2008), and lamivudine (Xu et al. 2009) are widely used in the pharmaceutical industry.
Traditionally, nucleoside analogues are prepared by various chemical methods. There are many disadvantages of using chemical synthesis methods for nucleosides, which involve multiple procedures and require harsh reaction conditions, leading to pollution and increased time consumption (Hassan et al. 1994; Rao et al. 2007). In contrast, utilizing enzymatic methods for the synthesis of nucleoside analogues avoids the protection and deprotection steps that are required for chemical synthesis and circumvents the difficulties inherent to the formation of glycosidic bonds (Hori et al. 1989; Liang et al. 2010; Trelles et al. 2003). For example, Okuyama et al. heterologously expressed a thermostable pyrimidine nucleoside phosphorylase from Bacillus stearothermophilus and used it in the synthesis of 5-methyluridine (Kiyoshi et al. 1996). Tang et al. synthesized 5-fluorouridine by recombinant Escherichia coli pyrimidine nucleoside phosphorylase (Tang et al. 2010). Visser et al. developed a method for high-yield enzymatic synthesis of 5-methyluridine using a novel purine nucleoside phosphorylase from Bacillus halodurans ALK36 (Gordon et al. 2011; Visser et al. 2011, 2010b). Ge et al. synthesized 2,6-diaminopurine riboside by recombinant E. coli (Ge et al. 2009).
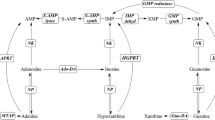
It is well-known that enzymes catalyzing the transglycosylation of ribonucleosides and deoxyribonucleosides are widely distributed in mammalian cells and bacteria, of which nucleoside phosphorylases are the main ones. Of all the nucleosides that can be selected as ribose donors in the synthesis of nucleoside drugs (including both purine and pyrimidine nucleoside analogues), inosine and guanosine are the least expensive. The main process for the synthesis of nucleoside drugs is shown in Fig. 1.

Process for synthesis of nucleoside analogues from inosine
Purine nucleoside phosphorylase (PNP, EC2.4.2.1) plays a key role in the salvage pathway, which provides an alternative to the de novo pathway of purine biosynthesis (Bzowska et al. 2000). Two main types of PNPs have been identified: high molecular mass hexameric PNPs (type I) showing broader substrate specificity and low molecular mass trimeric PNPs (type II) specific for 6-oxopurines and their nucleosides. Type I PNPs tend to be bacterial in origin, with the PNP from E. coli being the most typical. Type II PNPs tend to be eukaryotic in origin, with the PNP from humans being the most typical (Lewkowicz and Iribarren 2006).
Uridine phosphorylase (UP, EC2.4.2.3) is a key enzyme in the pyrimidine salvage pathway that catalyzes the reversible phosphorolysis of uridine to uracil and ribose 1-phosphate (Leer et al. 1977). UP belongs to the nucleoside phosphorylase superfamily 1, which includes enzymes that share a single subunit with either a trimeric or hexameric quaternary structure and accept both purine and pyrimidine as substrates (Lewkowicz and Iribarren 2006). UPs are widespread in prokaryotes, yeast, and higher organisms, and the amino acid sequences of UPs are highly conserved.
One bottleneck in the current process of synthesizing nucleoside analogues is that the sugar donors, inosine and guanosine, are relatively insoluble. Elevating reaction temperature by utilizing thermostable nucleoside phosphorylase is an efficient way to increase the solubility of the substrate in an aqueous solution and improve the reaction kinetics (Gordon et al. 2011); Higher reaction temperatures improve solubility, leading to improved productivity. Hori et al. used thermostable nucleoside phosphorylase from B. stearothermophilus to synthesize 5-methyluridine (Hori et al. 1991a, b, 1989), and Visser et al. found a thermostable purine phosphorylase from B. halodurans ALK36 (Gordon et al. 2011; Visser et al. 2010a, 2011).
Recently, extremophiles have attracted considerable attention, and research on extremely thermostable purine phosphorylase from Sulfolobus solfataricus has revealed some insight into the mechanism of thermostability (Appleby et al. 2001; Cacciapuoti et al. 2005, 1999, 1994). However, this thermostable nucleoside phosphorylase has not been employed to synthesize nucleoside analogues. In contrast to thermostable purine phosphorylases, only few thermostable uridine phosphorylases have been reported in previous studies.
Aeropyrum pernix was the first strictly aerobic hyperthermophilic Archaea to be discovered. It was originally isolated from heated marine sediments and venting water collected in 1996 (Mochizuki et al. 2010). This article describes the cloning, expression, characterization, and application of the thermostable nucleoside phosphorylases from A. pernix. Evidence for intersubunit disulfide bonds was found in uridine phosphorylase for the first time, and useful processes for the synthesis of both purine and pyrimidine nucleoside analogues via the two thermostable nucleoside phosphorylases were developed.
Materials and methods
Materials, microorganisms, plasmids, and culture conditions
Purine, purine nucleosides, pyrimidine, pyrimidine nucleosides, and isopropyl-thio-β-d-galactoside (IPTG) were purchased from Sigma (Germany) or J&K Scientific (China). Restriction enzymes, pEasy-T vector, and T4 DNA ligase were purchased from Takara Bio, Inc. (Japan). A TIANgel Midi Purification Kit was purchased from Tiangen Biotech (Beijing) Co., Ltd. and was used according to the manufacturer's recommendations. All organic solvents of high-performance liquid chromatography (HPLC)-grade were purchased from Acros Organics (USA). All other chemicals used were analytical-grade reagents and were purchased from Beijing Chemical Co., Ltd.
The genomic DNA of A. pernix was purchased from NITE Biological Resource Center in Japan (NBRC 100138). E. coli DH5α cells (Novagen, USA) were used for the cloning studies, and E. coli BL21 (DE3) (Novagen) strains were used as hosts for the expression of the gene. pET28a and pET30a (Invitrogen, USA) were used for standard cloning and gene expression studies. E. coli cells were routinely grown in Luria broth (LB) medium or on LB agar plates at 37 °C. In order to select bacteria carrying recombinant plasmids, 50 μg/ml of kanamycin was added into the medium. DNA was purified using the TIANgel Midi Purification Kit.
Cloning of the A. pernix apPNP and apUP
The genes encoding the purine nucleoside phosphorylase and uridine phosphorylase were amplified by polymerase chain reaction (PCR). The gene sequence encoding the purine nucleoside phosphorylase (apPNP) was amplified using the primers listed below based on the genomic DNA sequence annotation of A. pernix in GenBank (APE_0993.1). The sense primer apPNP-NdeI-fwd was 5′-gggaattccatatgaggaagccggttca-3′, and the antisense primer apPNP-HindIII-rwd was 5′-cccaagcttgactcctcctgtgaggac-3′; the NdeI and HindIII sites (underlined) were added to the forward and reverse primers, respectively. The gene sequence encoding the uridine phosphorylase (apUP) was also amplified based on the genomic DNA sequence in GenBank (APE_2105.1). The sense primer apUP-NdeI-fwd was 5′-gggaattccatatgggagacgagagtct-3′, and the NdeI site is underlined. The antisense primer apUP-XhoI-rwd was 5′-ccgctcgagtgtgcgtctgcacgccaggctc-3′. The antisense primer apUP-HindIII-rwd for the native enzyme was 5′-cccaagctttcatgtgcgtctgcacgccaggctc-3′.
PCR was performed with Red-Pfu DNA Polymerase (Biocolor BioScience & Technology Co., Beijing). The thermal cycling program involved an initial cycle of predenaturation at 94 °C for 5 min (first step), followed by 30 cycles of denaturation at 94 °C for 1 min (second step), annealing at 55 °C for 1 min (third step), and extension at 72 °C for 1 min. This was followed by a final extension at 72 °C for 10 min.
The PCR-amplified apPNP gene was purified and cloned into the NdeI and XhoI sites of the pET30a vector. The PCR-amplified apUP gene (primers apUP-NdeI-fwd and apUP-XhoI-rwd) was purified and cloned into the NdeI and XhoI sites of the pET28a vector and pET-30a vector, respectively, to construct double fusion and C-terminal fusion enzymes, respectively. Primers apUP-NdeI-fwd and apUP-HindIII-rwd were used to construct N-terminal fusion enzyme genes and native enzyme genes, respectively, into the pET28a vector and pET-30a vector, respectively.
Functional expression and purification of the apPNP and apUP
The constructs were transformed into E. coli BL21 (DE3) cells and grown overnight at 37 °C. Stock culture (5 ml) was transferred into 500 ml fresh culture medium and grown until the optical density at 580 nm (OD 580) was 0.8. IPTG was then added to a final concentration of 1.0 mM, and after incubation for another 4 h (OD 580 was about 1.2), the cells were harvested by centrifugation (4,000×g for 10 min).
Cells from the expression culture (500 ml total) were suspended in 40 ml binding buffer (50 mmol/l−1 NaH2PO4, 300 mmol/l−1 NaCl, and 10 mmol/l−1 imidazole; pH 8.0) and sonicated. The cell-free extract was then heated at 90 °C for 30 min and centrifuged at 12,000×g for 15 min. The clear supernatant was then applied to a 1-ml Novagen HisBand gravity flow column that had been equilibrated with 20 ml Ni-NTA binding buffer. The column was then washed with 20 ml wash buffer (50 mmol l−1 NaH2PO4, 300 mmol l−1 NaCl, and 20 mmol l−1 imidazole; pH 8.0). The His-tagged proteins were eluted with 10 ml elution buffer (50 mmol l−1 NaH2PO4, 300 mmol l−1 NaCl, and 200 mmol l−1 imidazole; pH 8.0). For the apUP, size exclusion chromatography was employed as a second purification step to obtain the protein in high purity. The column used was a 16/60 Superdex G75 prep grade (Amersham Biosciences) gel filtration column and was equilibrated with two column volumes of buffer (25 mM Hepes, 50 mM NaCl, pH 7.0). Subsequently, the protein solution was applied onto the column at a flow rate of 1 ml/min. Elution was performed with a flow rate of 1 ml/min and the wavelength chosen for detection of the protein was 280 nm. The fraction size was 2 ml and protein containing fractions were analyzed by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE).
To remove the imidazole present in the elution buffer, the eluate was applied to a 5-ml HiTrap desalting column connected to an AKTA FPLC system (GE Healthcare, USA), and the column was eluted with 50 mM Tris–HCl buffer (pH 7.5, 150 mM NaCl). The protein samples were collected and dialyzed against 5,000 ml of 20 mM phosphate buffer (pH 7.2). The buffer was changed three times over a period of 24 h, after which time the samples were ready for use. On-column transformation was carried out by re-equilibrating the protein-absorbed column with 20 ml Ni-NTA binding buffer.
Protein concentration was determined using the BCA Protein Assay Kit (Pierce Protein Biology Products, USA), with bovine serum albumin as the standard. SDS-PAGE was performed with a 6 % polyacrylamide stacking gel and a 12 % polyacrylamide separating gel. Aliquots of cell samples (5 μl, ~5.0 of OD 580) were loaded into the wells of the gel.
Enzyme activity assay
Enzyme activity was assayed by adding 30 μg of enzyme to 500 μl of the universal buffer (50 mM Tris, 50 mM boric acid, 33 mM citric acid, and 50 mM Na2HPO4; pH adjusted by NaOH to 7.0), which contained 5 mM substrate (inosine for the apPNP, 5-methyluridine for the apUP) (Visser et al. 2010a). The reaction solution was incubated at 90 °C for 60 min and ended in ice. The reaction solution was then centrifuged, and 5-μl aliquots of supernatant were analyzed by HPLC on a Shimadzu HPLC system interfaced with Shimadzu LC solution software, which was equipped with a detector at 260 nm and a Dikma Diamonsil C18 column (250 × 4.6 mm, 5 μ) at 25 °C. The mobile phase was 92 % water and 8 % methanol, with a flow rate of 1.0 ml/min. All purine and pyrimidine nucleoside analogues were analyzed separately by HPLC as a reference standard to identify the products. Conversion and production were calculated according to the peak area in the HPLC results.
Homology modeling and tertiary structures
Protein homology searches were conducted using BLASTX, and ClustalW was used for sequence alignments (Larkin et al. 2007). Homology modeling was performed using Accelrys Discovery Studio 2.0. The model of the apPNP was performed based on structures 1JDS of S. solfataricus (Appleby et al. 2001) and 1ODI of Thermus thermophilus (Tahirov et al. 2004). The model of the apUP was based on structures 3QPB of Streptococcus pyogenes (Tran et al. 2011), 3C74 of Salmonella typhimurium, and 1K3F of E. coli (Morgunova et al. 1995). Substrate binding was simulated by ligand docking with AutoDock Vina (Seeliger and de Groot 2010), and both the structures and docking were visualized with PyMOL.
SDS-PAGE was performed at room temperature using 12 % acrylamide resolving gel and 5 % acrylamide staking gel. The enzymes denatured in the presence and in the absence of dithiothreitol (DTT) were analyzed. Gel filtration was performed on an AKTA FPLC system equipped with a 23-ml Superdex HR 200 10/30 column (GE Healthcare). The column was calibrated with molecular mass standards (SERVA, Germany). After the column had been equilibrated with 0.5 M HEPES buffer containing 10 % (v/v) glycerol and 100 mM NaCl (pH 7.5), samples were eluted by the same buffer solution. For each chromatogram, a 2-mg sample was added to the column and 0.5-ml fractions were collected at 0.3 ml/min. The column was calibrated by using standard proteins of known molecular weight (phosphorylase B, 97.4 KDa; ovine serum albumin, 66.2 KDa, ovalbumin, 45.0 KDa; carbonic anhydrase, 31 KDa; trypsin inhibitor, 21.5 KDa; and lysozyme, 14.4 KDa).
Kinetic parameters and physical characteristics
The apparent kinetic parameters were determined by Lineweaver–Burk plot. Enzymatic reactions were carried out with substrate solutions of different concentrations (ranging from 0.05 to 2.5 mM). To ensure accuracy, substrate conversion was controlled so that it did not exceed 10 %. Inosine, guanosine, and adenosine were used as substrates for the apPNP assays, while uridine and 5-methyluridine were routinely used as substrates for the apUP assays. The activity temperature profile was determined at pH 7.5 to be 40–100 °C for the apUP (50–130 °C for the apPNP). Reactions over 100 °C were carried out in an autoclave using test tube. The thermal stability of the enzymes was determined by measuring their residual activity under standard conditions after incubation at a temperature ranging from 40 to 100 °C (apUP) or 50 to 130 °C (apPNP) for 60 min. The activity–pH profile was determined at 90 °C in buffers with pH values ranging from 3.0 to 12.0. To determine the stability at various pH values, the enzymes were incubated at 90 °C in various buffers for 60 min without substrate, and then, the residual activity was measured under standard conditions. The buffer used was universal buffer (50 mM Tris, 50 mM boric acid, 33 mM citric acid, and 50 mM Na2PO4). The buffers were adjusted by NaOH or HCl to obtain buffers with pH values ranging from 3.0 to 12.0.
Substrate specificity and enzymatic synthesis of nucleoside drugs
Substrate specificity of the apPNP toward various purine nucleosides and that of the apUP toward various pyrimidine nucleosides was determined using standard methods. The purified enzyme and different purine nucleosides (5 mM) were incubated in 50 mM universal buffer (pH 5.0) at 90 °C for 60 min; standard enzyme assays were then performed, and the highest activity which was set to 100 % 2,6-diaminopurine nucleoside was synthesized using inosine and 2,6-diaminopurine as the starting materials, and 5-methyluridine was synthesized using guanosine and thymine as the starting materials. The products were confirmed by standard sample. For the synthesis of 5-methyluridine, small scale screening experiments were conducted in order to establish the operating conditions of the biocatalytic reaction and identify the most important variables. Key reaction parameters such as pH, temperature, concentration of buffer, and substrates were investigated. Then, final reactions at a larger scale were conducted.
Results
Enzyme expression and purification
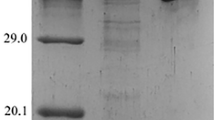
The gene encoding the apPNP was cloned by PCR amplification from chromosomal DNA into the cognate restriction sites of expression vector pET-30a (+); this vector adds a hexahistidine C-terminal tag in order to facilitate the purification of the protein. Heterologous expression of the apPNP was induced in E. coli BL21 (DE3) cells through the addition of IPTG (1 mmol/l−1). The resulting gene product was expressed efficiently in the host, and only the soluble fraction of the apPNP was used for further purification. The fact that the apPNP comes from an extreme thermophilic organism, with an optimum growth temperature of 100 °C, allows for the use of an easy purification method. The soluble extract was first heated to 90 °C for 30 min. This procedure allowed removing at least 80 % of the host protein by centrifugation. The His-tagged protein was purified efficiently by Ni2+ chelate affinity chromatography, from which a very pure recombinant protein was obtained (Fig. 2). Approximately 4.8 ± 0.1 mg of the recombinant protein was purified from 2,000 mg of crude extract.

SDS-polyacrylamide gel electrophoresis of PNP and UP. Lane M: marker. Lane 1: Nonreduced UP treated without DTT. Lane 2: Reduced UP treated with DTT. Lane 3: Nonreduced PNP treated without DTT. Lane 4: Reduced PNP treated with DTT
Three constructs of the apUP gene were prepared and expressed as His-tagged fusion proteins. Double fusion proteins (N- and C-terminal fusion proteins), N-terminal fusion proteins, and C-terminal fusion proteins were expressed in inactive form. According to the SDS-PAGE analyses, most of the expressed fusion proteins were expressed as inclusion bodies in cell pellets. The native apUP proteins (pET30a-apUP) without fusion tags were actively expressed, and about 50 % of the proteins were found in the soluble fraction of the cell lysate.
The native protein was purified to homogeneity by size exclusion chromatography after heat treatment (90 °C, 30 min, Fig. 2). Approximately 5.0 ± 0.1 mg of the recombinant protein was purified from 2,000 mg crude extract.
Homology modeling and tertiary structures
Sequence analysis showed that the closest related structure to the apPNP in the protein databases was the 5-methylthioadenosine phosphorylase (MTP) from S. solfataricus (44 % identity). According to the sequence identity, the apPNP belonged to the type I PNP family, and the phosphate-binding site, the ribose-binding site, and the purine-binding site were strictly conserved. According to the alignment results, the phosphate-binding site of the apPNP was composed of Gly22, Arg26, Arg88, and Thr91; the ribose-binding site consisted of His6, Ile66, Glu187, and Met187; and the purine-binding site was composed of Phe165, Glu168, and Asp210. Docking and modeling studies corroborated these results (Fig. 3a). The protein was denatured in the presence and in the absence of DTT and was analyzed on a 12 % SDS-PAGE gel. The results showed that in both the absence and the presence of the reducing agent, the predominant tertiary confirmation was a monomer. The estimation of molecular mass by gel filtration on a calibrated Sephacryl S-200 column also gave the same result, indicating that the apPNP is a monomer protein with a molecular weight of about 30 kDa that is different from both type I and type II PNPs. Even without the formation of disulfide bonds (which are present in the MTP from S. solfataricus), the apPNP still showed great thermostability. It is likely that several complex factors contribute to the thermostability of the enzyme.

Homology modeled three-dimensional structure of PNP (a), UP (b), and the enzyme–substrate interactions
The apUP displayed moderate homology (around 40 % identity) with the uridine phosphorylases from S. typhimurium and E. coli. For these proteins, the sequences for the phosphate-binding site, the ribose-binding site, and the uracil-binding site were strictly conserved, too. Docking and modeling studies were performed based on the alignment results (Fig. 3b). The phosphate-binding site of the apUP was composed of Arg41, Arg59, and Arg102; the ribose-binding site consisted of His21, Arg102, Glu208, and Met207; and the uracil-binding site was composed of Gln177, Arg179, and Arg233. The apUP structural gene was an 843-bp fragment. The native construct expressed a 32.0-kDa protein, which is accordance with the predicted weight of 30.2 kDa. The native protein was determined by gel filtration to be approximately 185.3 kDa. Judging from the results, the fusion enzyme would appear to be hexameric. The apUP was denatured in the presence and in the absence of DTT and was analyzed on a 12 % SDS-PAGE gel. The results showed that in the absence of the reducing agent, the apUP was a dimer, indicating the presence of three disulfide bonds that are probably intersubunit bonds, resulting in the organization of the enzyme into three dimers (Fig. 2).
Physical characteristics
As shown in Fig. 4a, the optimum temperature of the apPNP was found to be 100 °C, and the recombinant protein had nearly 100 % activity when incubated at 100 °C for 60 min. The apPNP showed an optimum pH of 5.0, retaining nearly 100 % activity between pH 3.0 and 11.0 when incubated at 90 °C for 60 min (Fig. 4b).

Effect of temperature and pH on the activity of the purified PNP (triangle). Effect of temperature and pH on the stability of the purified PNP (square). The error bars in the figure indicate the standard deviations from three independent samples. The highest activity was set to 100 %
The hydrolysis activity of the apUP reached a maximum at 90 °C. After incubation for 60 min at 90 °C, the enzyme remained 100 % active (Fig. 5a). Maximum activity of the apUP was found at pH 9.0 in universal buffers for uridine, and the apUP displayed extraordinary pH stability ranging from pH 3.0 to pH 9.0 (Fig. 5b). The apPNP and apUP did show good stability at different values of pH, with no change in activity over time and excellent stability under 90 °C. Therefore, they can be used as industrial biocatalysts in the synthesis of nucleoside drugs.

Effect of temperature and pH on the activity of the purified UP (triangle). Effect of temperature and pH on the stability of the purified UP (square). The error bars in the figure indicate the standard deviations from three independent samples. The highest activity was set to 100 %
Substrate specificity and kinetic properties
Linear transformation of velocity data obtained for varying initial substrate concentrations showed a good linear regression fit. Based on the values calculated from various plots (Lineweaver–Burk, Eadie–Hofstee, and Hanes–Woolf), K m and V max were determined. Subsequently, the turnover number (K cat) and the specificity constant were calculated using the monomer molecular weight; these values are listed in Table 1.
According to previous studies, type I PNPs show broad substrate specificity. The apPNP in this study belongs to the type I PNP family, so its broad substrate specificity was consistent with these previous findings. The enzyme can accept both 6-oxo- and 6-aminopurines, but it cannot accept 6-chloropurines. It also can accept 2′-deoxy purine nucleosides, illustrating its great potential for use as an industrial biocatalyst. Eight uridine derivatives were tested for enzyme specificity of the apUP. The results listed in Table 2 indicate that the optimal substrate for the apUP is uridine. The apUP showed no activity for 5-fluorouridine, cytidine, or 2-deoxycytidine (even when incubated with the substrate under optimal conditions for 24 h), indicating that it is a uridine phosphorylase rather than a pyrimidine nucleoside phosphorylase.
Enzymatic synthesis of nucleoside drugs
The target nucleoside analogue 2,6-diaminopurine nucleoside, which is an important intermediate, was successfully synthesized in a 60 % yield using inosine and 2,6-diaminopurine as the starting materials, and 5-methyluridine was synthesized in a high yield using guanosine and thymine as starting materials. In optimal conditions, 5-methyluridine was synthesized in an 85 % yield with a guanosine conversion of 96 % (Fig. 6). All processes can be performed at a high temperature, which will highly increase the solubility of the substrate; therefore, these processes are suitable for industrial applications.

HPLC analysis of the products from in vitro reactions. 5-methyluridine and 2,6-diaminopurine nucleoside were synthesized via thermostable phosphorylase
Discussion
Current advances in genome sequencing projects have revolutionized research in biotechnology, enabling not only a glimpse into the uncultured microbial population, but also the high-throughput discovery of new enzymes for industrial bioconversions (Ferrer et al. 2007). Recently, interest in extremely thermophilic microorganisms has increased considerably, resulting in the discovery of novel thermostable biocatalysts. The thermostable phosphorylases described in this paper originated from the hyperthermophilic aerobic crenarchaeon A. pernix, which was first isolated from heated marine sediments and venting water (Mochizuki et al. 2010).
Both the purine nucleoside phosphorylase and uridine phosphorylase were cloned, expressed, and purified, and their enzymatic properties were determined. Although some inclusion bodies were formed during expression, about 60 % of the apPNP was found in the soluble fraction of the cell homogenate. Using an easy two-step purification method comprising heat treatment and Ni2+ chelate affinity chromatography, the recombinant apPNP was quickly purified to homogeneity and functionally characterized. While the expressions of the fusion apUP gene in E. coli were unsuccessful, with most of the fusion proteins expressed in the form of inclusion bodies even after many attempts to optimize expression, the native apUP gene was expressed well. Because of its high thermostability, the protein was purified to 95 % homogeneity by heat treatment and gel filtration. In this study, His-tags resulted in the formation of inclusion bodies.
Both the apPNP and apUP showed high thermostability. The optimum temperature of the apPNP was obtained at 100 °C, and the recombinant protein had nearly 100 % activity when incubated at 100 °C for 60 min. The hydrolysis activity of the apUP reached a maximum at 90 °C. After incubation for 60 min at 90 °C, the enzyme remained 100 % active. Recent research on 5-methylthioadenosine phosphorylase from S. solfataricus has revealed some interesting results: the enzyme is hexameric, and there are six disulfide bonds formed between the subunits. Previous studies have shown that the involvement of disulfide bonds plays a key role in whether an enzyme is thermophilic and thermostable (Appleby et al. 2001). According to their sequence similarity, the apPNP in this study was likely to have a similar structure as the S. solfataricus MTP. Attempts were made to validate the hypothesis that some of the cysteine residues in the recombinant apPNP were in reduced form, but unfortunately, these efforts were not successful. Interestingly, the apUP was found to have this special property. The three disulfide bonds are probably intersubunit bonds, resulting in the organization of the enzyme into three dimers. This is the first known documentation of this characteristic in uridine phosphorylase.
The substrate specificity of the apPNP and that of the apUP was examined in detail. The apPNP showed a broader substrate specificity in that it can accept both 6-oxo- and 6-aminopurines, as well as 2′-deoxypurines. It cannot, however, accept 6-chloropurines. These results were similar to those for previously reported PNPs from E. coli (Luo et al. 2011). In addition to the high K m values of the apPNP, the K cat values were considerably higher than for other PNPs such as the PNP from B. halodurans (Visser et al. 2010b), indicating that it could serve as a useful biocatalyst in biochemistry and industrial biotechnology. The apUP was highly active on many uridine nucleoside analogues like uridine, 5-methyluridine, thymidine, and deoxyuridine, and the most closely related UPs, e.g., the enzymes from E. coli and S. typhimurium, also have similar specificity (Leer et al. 1977). However, the apUP was inactive on cytidine analogues and 5-fluoro analogues. 5-Methyluridine is an intermediate in the synthesis of thymidine and antiretroviral drugs such as zidovudine and stavudine, and the apPNP and apUP can be employed in the synthesis of 5-methyluridine using guanosine as the ribose donor.
As mentioned previously, in the synthesis of nucleoside analogues, inosine and guanosine are the least expensive ribose donors, with very low solubility in water. Elevating reaction temperature by utilizing thermostable nucleoside phosphorylase is an efficient way to increase the solubility of the substrate in an aqueous solution and to improve the reaction kinetics (Gordon et al. 2011); the higher the temperature at which the reaction is performed, the higher the productivity. Researchers have devoted a lot of attention toward screening new thermostable nucleoside phosphorylases. The PNP and UP from B. stearothermophilus JTS 859 were successfully used to synthesize 5-methyluridine (Hori et al. 1991b). Visser et al. found a novel thermostable purine nucleoside phosphorylase from B. halodurans ALK36, whose activity was highest at 70 °C and whose half-life was 20 h at 60 °C (Visser et al. 2010a). In combination with the uridine phosphorylase from E. coli and under optimal conditions, a guanosine conversion of >95 % and a >85 % yield of 5-methyluridine were achieved. An overall productivity of 10.6 g l−1 h−1 was obtained, with a final product concentration of 84 g l−1 (325 mM) (Gordon et al. 2011). Furthermore, replacement of the wild-type uridine phosphorylase with the mutant allowed the reaction temperature to increase to 65 °C. The elevated reaction temperature can increase the reaction productivity from 10 to 31 g l−1 h−1 (Visser et al. 2010a). Therefore, in this system, highly thermostable purine nucleoside phosphorylases and uridine phosphorylases are superior to their mesophilic counterparts.
While several thermostable purine nucleoside phosphorylases have been the subject of previous studies, there have been very few reports on thermostable uridine phosphorylases. To the best of our best knowledge, the uridine phosphorylase described in this paper is the most thermostable uridine phosphorylase reported to date. In this study, a highly thermostable purine nucleoside phosphorylase and a highly thermostable uridine phosphorylase from A. pernix were revealed and characterized. Both the enzymes maintained 100 % activity after being incubated at a high temperature. Using the apPNP, the 2,6-diaminopurine nucleoside was successfully synthesized in an approximately 60 % yield. Utilizing the two thermostable nucleoside phosphorylases, the synthetic process for 5-methyluridine was developed. Under optional conditions, a guanosine conversion of >96 % and a 5-methyluridine yield of >85 % were achieved. Higher reaction productivity (50 g l−1 h−1) can be obtained because the reaction can be performed at a higher temperature (Zhu et al. 2012).
This work has described the enzymatic synthesis of nucleoside analogues via thermostable nucleoside phosphorylase from A. pernix for the first time. These processes have an important impact on the biosynthesis of nucleoside analogues such as 5-methyluridine and 2,6-diaminopurine nucleoside, which are important drug intermediates. Thanks to the enhanced solubility of the substrate at an elevated reaction temperature, both of these processes provided high yields of nucleoside analogues. Therefore, these novel processes are suitable for large-scale industrial preparations.
References
Appleby TC, Mathews II, Porcelli M, Cacciapuoti G, Ealick SE (2001) Three-dimensional structure of a hyperthermophilic 5′-deoxy-5′-methylthioadenosine phosphorylase from Sulfolobus solfataricus. J Biol Chem 276(42):39232–39242. doi:10.1074/jbc.M105694200
Balfour HH (1999) Antiviral drugs. New Engl J Med 340(16):1255–1268. doi:10.1056/NEJM199904223401608
Bzowska A, Kulikowska E, Shugar D (2000) Purine nucleoside phosphorylases: properties, functions, and clinical aspects. Pharmacol & Therapeut 88(3):349–425
Cacciapuoti G, Porcelli M, Bertoldo C, De Rosa M, Zappia V (1994) Purification and characterization of extremely thermophilic and thermostable 5′-methylthioadenosine phosphorylase from the archaeon Sulfolobus solfataricus. Purine nucleoside phosphorylase activity and evidence for intersubunit disulfide bonds. J Biol Chem 269(40):24762–24769
Cacciapuoti G, Fusco S, Caiazzo N, Zappia V, Porcelli M (1999) Heterologous expression of 5-methylthioadenosine phosphorylase from the archaeon Sulfolobus solfataricus: characterization of the recombinant protein and involvement of disulfide bonds in thermophilicity and thermostability. Protein Expres Purif 16(1):125–135
Cacciapuoti G, Forte S, Moretti MA, Brio A, Zappia V, Porcelli M (2005) A novel hyperthermostable 5′-deoxy-5′-methylthioadenosine phosphorylase from the archaeon Sulfolobus solfataricus. FEBS J 272(8):1886–1899. doi:10.1111/j.1742-4658.2005.04619.x
Carr A, Workman C, Smith DE, Hoy J, Hudson J, Doong N, Martin A, Amin J, Freund J, Law M, Cooper DE, Group ftMTS (2002) Abacavir substitution for nucleoside analogs in patients with HIV lipoatrophy. JAMA-J Am Med Assoc 288(2):207–215. doi:10.1001/jama.288.2.207
Ferrer M, Golyshina O, Beloqui A, Golyshin PN (2007) Mining enzymes from extreme environments. Curr Opin Microbiol 10(3):207–214
Foster RH, Faulds D (1998) Abacavir. Drugs 55(5):729–736
Ge C, OuYang L, Ding Q, Ou L (2009) Co-expression of recombinant nucleoside phosphorylase from Escherichia coli and its application. Appl Biochem Biotech 159(1):168–177. doi:10.1007/s12010-008-8429-3
Gordon GER, Visser DF, Brady D, Raseroka N, Bode ML (2011) Defining a process operating window for the synthesis of 5-methyluridine by transglycosylation of guanosine and thymine. J Biotechnol 151(1):108–113
Hassan AEA, Shuto S, Matsuda A (1994) Nucleosides and nucleotides. 124. Chemical reactivity of the sugar moiety of 2′-deoxy-2′-methylidene pyrimidine nucleosides: synthesis of 3′-amino-2, 3′-dideoxy-2′-methylidene pyrimidine nucleosides via [2, 3]-sigmatropic rearrangement of allylic selenides as potential antitumor agents. Tetrahedron 50(3):689–700
Hori N, Watanabe M, Yamazaki Y, Mikami Y (1989) Synthesis of 5-methyluridine by a thermophile, Bacillus stearothermophilus JTS 859. Agr Biol Chem 53(1):197–202
Hori N, Watanabe M, Mikami Y (1991a) The effects of organic solvent on the ribosyl transfer reaction by thermostable purine nucleoside phosphorylase and pyrimidine nucleoside phosphorylase from Bacillus stearothermophilus jts 859. Biocatal Biotransfor 4(4):297–304. doi:10.3109/10242429109000693
Hori N, Watanabe M, Sunagawa K, Uehara K, Mikami Y (1991b) Production of 5-methyluridine by immobilized thermostable purine nucleoside phosphorylase and pyrimidine nucleoside phosphorylase from Bacillus stearothermophilus JTS 859. J Biotechnol 17(2):121–131
Kiyoshi O, Tomoki H, Toshitada N, Yuichiro M (1996) Molecular cloning and expression of the pyrimidine nucleoside phosphorylase gene from Bacillus stearothermophilus th 6–2. Biosci Biotech Bioch 60(10):1655–1659. doi:10.1271/bbb.60.1655
Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23(21):2947–2948. doi:10.1093/bioinformatics/btm404
Leer JC, Hammer-Jespersen K, Schwartz M (1977) Uridine phosphorylase from Escherichia coli. Eur J Biochem 75(1):217–224. doi:10.1111/j.1432-1033.1977.tb11520.x
Lewkowicz ES, Iribarren AM (2006) Nucleoside phosphorylases. Curr Org Chem 10(11):1197–1215
Liang S, Li W, Gao T, Zhu X, Yang G, Ren D (2010) Enzymatic synthesis of 2-deoxyadenosine and 6-methylpurine-2-deoxyriboside by Escherichia coli DH5a overexpressing nucleoside phosphorylases from Escherichia coli BL21. J Biosci Bioeng 110(2):165–168
Luo W, Liu Y, Zhu X, Zhao W, Huang L, Cai J, Xu Z, Cen P (2011) Cloning and characterization of purine nucleoside phosphorylase in Escherichia coli and subsequent ribavirin biosynthesis using immobilized recombinant cells. Enzyme Microb Tech 48(6–7):438–444
Minton K (2009) Cladribine hope for multiple sclerosis. Nat Rev Immunol 9(6):387
Mochizuki T, Yoshida T, Tanaka R, Forterre P, Sako Y, Prangishvili D (2010) Diversity of viruses of the hyperthermophilic archaeal genus Aeropyrum, and isolation of the Aeropyrum pernix bacilliform virus 1, APBV1, the first representative of the family Clavaviridae. Virology 402(2):347–354
Morgunova EY, Mikhailov AM, Popov AN, Blagova EV, Smirnova EA, Vainshtein BK, Mao C, Armstrong SR, Ealick SE, Komissarov AA, Linkova EV, Burlakova AA, Mironov AS, Debabov VG (1995) Atomic structure at 2.5 A resolution of uridine phosphorylase from E. coli as refined in the monoclinic crystal lattice. FEBS Lett 367(2):183–187
Rai KR, Peterson BL, Appelbaum FR, Kolitz J, Elias L, Shepherd L, Hines J, Threatte GA, Larson RA, Cheson BD, Schiffer CA (2000) Fludarabine compared with chlorambucil as primary therapy for chronic lymphocytic leukemia. New Engl J Med 343(24):1750–1757. doi:10.1056/NEJM200012143432402
Rao JR, Schinazi RF, Chu CK (2007) Enantioselective synthesis and antiviral activity of purine and pyrimidine cyclopentenyl C-nucleosides. Bioorgan Med Chem 15(2):839–846
Sacktor N, Nakasujja N, Skolasky RL, Robertson K, Musisi S, Ronald A, Katabira E, Clifford DB (2009) Benefits and risks of stavudine therapy for HIV-associated neurologic complications in Uganda. Neurology 72(2):165–170. doi:10.1212/01.wnl.0000339042.96109.86
Scruggs ER, Dirks Naylor AJ (2008) Mechanisms of zidovudine-induced mitochondrial toxicity and myopathy. Pharmacology 82(2):83–88
Seeliger D, de Groot B (2010) Ligand docking and binding site analysis with PyMOL and Autodock/Vina. J Comput Aid Mol Des 24(5):417–422. doi:10.1007/s10822-010-9352-6
Tahirov TH, Inagaki E, Ohshima N, Kitao T, Kuroishi C, Ukita Y, Takio K, Kobayashi M, Kuramitsu S, Yokoyama S, Miyano M (2004) Crystal structure of purine nucleoside phosphorylase from Thermus thermophilus. J Mol Biol 337(5):1149–1160
Tang Y, Ni M, Wu W, Sun J, Zhou C (2010) Biotransformation of the antitumor intermediate 5-fluorouridine by recombinant Escherichia coli with high pyrimidine nucleoside phosporylase activity. Biocatal Biotransfor 28(2):130–136. doi:10.3109/10242420903505842
Thompson MA, Kessler HA, Eron JJJ, Jacobson JM, Adda N, Shen G, Zong J, Harris J, Moxham C, Rousseau FS, Group D-S (2005) Short-term safety and pharmacodynamics of amdoxovir in HIV-infected patients. AIDS 19(15):1607–1615
Tran TH, Christoffersen S, Allan PW, Parker WB, Piskur J, Serra I, Terreni M, Ealick SE (2011) The crystal structure of Streptococcus pyogenes uridine phosphorylase reveals a distinct subfamily of nucleoside phosphorylases. Biochemistry 50(30):6549–6558. doi:10.1021/bi200707z
Trelles JA, Fernandez M, Lewkowicz ES, Iribarren AM, Sinisterra JV (2003) Purine nucleoside synthesis from uridine using immobilised Enterobacter gergoviae CECT 875 whole cells. Tetrahedron Lett 44(12):2605–2609
Van Rompay AR, Johansson M, Karlsson A (2003) Substrate specificity and phosphorylation of antiviral and anticancer nucleoside analogues by human deoxyribonucleoside kinases and ribonucleoside kinases. Pharmacol & Therapeut 100(2):119–139
Visser D, Hennessy F, Rashamuse K, Louw M, Brady D (2010a) Cloning, purification and characterisation of a recombinant purine nucleoside phosphorylase from Bacillus halodurans Alk36. Extremophiles 14(2):185–192. doi:10.1007/s00792-009-0297-4
Visser DF, Rashamuse KJ, Hennessy F, Gordon GER, Van Zyl PJ, Mathiba K, Bode ML, Brady D (2010b) High-yielding cascade enzymatic synthesis of 5-methyluridine using a novel combination of nucleoside phosphorylases. Biocatal Biotransfor 28(4):245–253. doi:10.3109/10242422.2010.493210
Visser DF, Hennessy F, Rashamuse J, Pletschke B, Brady D (2011) Stabilization of Escherichia coli uridine phosphorylase by evolution and immobilization. J Mol Catal B-Enzym 68(3–4):279–285
Xu WM, Cui YT, Wang L, Yang H, Liang ZQ, Li XM, Zhang SL, Qiao FY, Campbell F, Chang CN, Gardner S, Atkins M (2009) Lamivudine in late pregnancy to prevent perinatal transmission of hepatitis B virus infection: a multicentre, randomized, double-blind, placebo-controlled study. J Viral Hepatitis 16(2):94–103. doi:10.1111/j.1365-2893.2008.01056.x
Zhu S, Ren L, Wang J, Zheng G, Tang P (2012) Two-step efficient synthesis of 5-methyluridine via two thermostable nucleoside phosphorylase from Aeropyrum pernix. Bioorg Med Chem Lett 22(5):2102–2104
Acknowledgments
The authors express their gratitude to Prof. Chen Jinchun for inspiring discussions about the inclusion bodies. We also thank Min Cong for assistance in gene cloning.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Zhu, S., Song, D., Gong, C. et al. Biosynthesis of nucleoside analogues via thermostable nucleoside phosphorylase. Appl Microbiol Biotechnol 97, 6769–6778 (2013). https://doi.org/10.1007/s00253-012-4542-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-012-4542-x