
Abstract
Biological soil disinfestation (BSD) involves the anaerobic decomposition of plant biomass by microbial communities leading to control of plant pathogens. We analyzed bacterial communities in soil of a model experiment of BSD, as affected by biomass incorporation under various conditions, to find out the major anaerobic bacterial groups which emerged after BSD treatments. The soil was treated with Brassica juncea plants, wheat bran, or Avena strigosa plants, irrigated at 20 or 30 % moisture content and incubated at 25–30 °C for 17 days. The population of Fusarium oxysporum f. sp. spinaciae incorporated at the start of the experiment declined markedly for some BSD conditions and rather high concentrations of acetate and butyrate were detected from these BSD-treated soils. The polymerase chain reaction-denaturing gradient gel electrophoresis analysis based on the V3 region of 16S rRNA gene sequences from the soil DNA revealed that bacterial profiles greatly changed according to the treatment conditions. Based on the clone library analysis, phylogenetically diverse clostridial species appeared exceedingly dominant in the bacterial community of BSD soil incorporated with Brassica plants or wheat bran, in which the pathogen was suppressed completely. Species in the class Clostridia such as Clostridium saccharobutylicum, Clostridium acetobutylicum, Clostridium xylanovorans, Oxobacter pfennigii, Clostridium pasteurianum, Clostridium sufflavum, Clostridium cylindrosporum, etc. were commonly recognized as closely related species of the dominant clone groups from these soil samples.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Agricultural producers often apply some form of soil disinfestation before planting high-value cash crops, to reduce soil-borne crop pests including fungal, bacterial and nematode pathogens, weeds, and insects (Spadaro and Gullino 2005). For the last few decades, methyl bromide was the major fumigant used for pre-planting soil disinfestation, but it has been shown that gaseous methyl bromide may destroy stratospheric ozone (Prather and McElroy 1984). The search for alternatives to this effective soil fumigant has recently been emphasized in light of the phasing out of methyl bromide (Ristaino and Thomas 1997), giving preference to nonchemical fumigation methods. Instead of the chemical disinfestants, steam disinfestation (Katan 2000), soil solarization (Katan 1981), or flooding (Stover 1979) has been mentioned but may have some practical limitations (Goud et al. 2004).
Biological soil disinfestation (BSD) is one of the alternative methods of soil disinfestation using anaerobic decomposition of organic matter in soil, which has been developed in the Netherlands (Blok et al. 2000) and Japan (Shinmura 2004; Shinmura et al. 1999) as an environmentally friendly approach. BSD can be a promising alternative for all other fumigation methods due to its broad spectrum for suppression of soil-borne pathogens at various incubation duration, temperature, and location (Goud et al. 2004; Shinmura 2004; Messiha et al. 2007). The principles of BSD conducted in Japan include three steps: (1) incorporating the plant biomass into soil, (2) flooding the soil by irrigation, and (3) covering the soil surface with a plastic film to induce reducing soil conditions (Shinmura 2000, 2004). The process lasts for about 3 weeks and becomes suitable for crop cultivation soon after removing the plastic film followed by plowing the field. A number of plant biomass sources such as Brassica spp., wheat bran, rice bran, grasses, or other organic substances have been used successfully as BSD materials for controlling soil-borne pests and diseases (Shinmura 2004; Goud et al. 2004; Momma 2008).
It has been reported that survival of many plant pathogens is significantly decreased under anaerobic soil conditions (Blok et al. 2000; Messiha et al. 2007), and the bacterial communities developed due to anaerobiosis may enhance the pathogen elimination efficacy. In a previous report, we analyzed the bacterial communities in soil of a BSD experiment using Brassica juncea plants or wheat bran (Mowlick et al. 2012). The BSD treatment with these plant biomass successfully decreased the population of the pathogen (Fusarium oxysporum f. sp. lycopersici, fusarium wilt pathogen of tomato) incorporated in the soil before the treatment, and it was found by clone library analysis based on the 16S rRNA gene sequences that anaerobic bacteria including especially the members of the class Clostridia in the phylum Firmicutes became the major and dominant bacterial groups in the soil. The members in the class Clostridia are known to produce some harmful compounds to pathogens, such as skatole, indole, cresol, or some phenolic compounds, by the fermentation of amino acids such as tryptophan and tyrosine under anaerobic conditions (Macfarlane and Macfarlane 1995). Besides, fatty acids like acetate or butyrate produced by these bacterial groups were supposed to contribute to pathogen inactivation (Momma et al. 2006). These acids were actually detected in the BSD soil (Mowlick et al. 2012), and it was strongly suggested that acetate, butyrate, or other substances could be the products of the clostridial groups responsible for the suppression of some soil-borne pathogens. Thus, it seems that proliferation of clostridial species in BSD-treated soil may be important to make the treatment more effective.
Considering the facts stated, the aim of this study was to confirm the effects of various plant biomass sources and incubation conditions on the suppression of pathogens and to identify common bacterial groups that proliferated during the BSD treatment. We analyzed the bacterial communities of BSD-treated soils amended with three types of biomasses, mustard (B. juncea L.) plants, wheat (Triticum aestivum L.) bran, and lopsided oat (Avena strigosa L.) plants under different incubation conditions (temperature and moisture content (MC)) of a model experiment using closed vessels. Molecular techniques such as polymerase chain reaction-denaturing gradient gel electrophoresis (PCR-DGGE) (Muyzer et al. 1993) and clone library analysis (Maidak et al. 1999) were mainly used to determine the changes in the major bacterial populations and detailed community composition in the BSD soil. To compare with our previous results (Mowlick et al. 2012), soil was obtained from a different district in Japan and a different pathogen was used in the experiment.
Materials and methods
BSD experiments using closed vessels
The model experiment of BSD was carried out by using 640-ml plastic pots. Soil obtained from a field (gray lowland soil at pH 5.18) in the NARO Western Region Agricultural Research Center, Fukuyama, Hiroshima, Japan (34.5° N, 133.4° E) was air-dried and passed through a sieve with 2 mm opening. The nitrate-nonutilizing (nit) mutant of F. oxysporum f. sp. spinaciae cultivated in potato sucrose broth medium (Takehara and Kuniyasu 1994) was incorporated into the bulk soil (5×105 CFU/g) and mixed thoroughly to make uniformly pre-infested soil. The nit mutant strain (M2-1) was generated from the parent strain Spin-2 (wild type, the NIAS GenBank accession No. MAFF103060) by incubating mycelial plugs of the wild-type strain on a chlorate-containing medium for 12 days according to the method described by Puhalla (1985). Strain M2-1 was comparable to strain Spin-2 in growth and pathogenicity. The pots were filled with the F. oxysporum-infested soil (450 g dry soil/pot) and treated with three types of plant biomass, namely, mustard (B. juncea) plants, wheat bran, and lopsided oat (A. strigosa) plants. The incorporation amounts of plant biomass were determined based on the standard methods for BSD treatment (Goud et al. 2004; Shinmura 2004). Leaves of the B. juncea plants were taken from the field before the flowering stage, dried at 40 °C for 2 days, and incorporated into pot soil at the rate of 4.0 g of dry matter/kg of soil (1.8 g/pot). Wheat bran was applied at the rate of 10.9 g/kg of soil (4.9 g/pot). Grass plants of A. strigosa were cut above the ground at the flowering stage, dried at 40 °C for 4 days, and incorporated into pot soil at a rate of 7.3 g of dry matter/kg of soil (3.3 g/pot). For the control treatment, none of the plant material or any other substances was incorporated into the soil.
The field capacity for MC measured for the original soil was 24.5 % (w/w of wet soil). Soil in each pot was irrigated at 20 or 30 % MC, and the pots were covered with lids tightly to avoid penetration of oxygen. The pots containing 30 % MC soil were incubated at three different temperatures (25, 27.5, and 30 °C) for each biomass condition as well as the control, while all pots with 20 % MC soil were incubated at 30 °C. Each treatment condition was applied in duplicate resulting to 32 pots in total. The names of the treatments were designated considering the biomass (or control), incubation temperature, and MC, e.g., Br30-20 for Brassica-treated soil at 30 °C with 20 % MC (Table 1). Soil samples were collected from all the pots using sterile spatulas after 17 days of incubation. In addition to these treated soil samples, the original field soil sample without any treatment was also collected in duplicates (Y1 and Y2). The oxidation-reduction potential (ORP) of soil was measured for all treatments by electrodes (Ag/AgCl)) inserting into the soil directly. The number of nit mutant of the pathogen incorporated into the soil was determined by the dilution plate technique using a selective medium for the mutant (CMP medium) (Takehara et al. 2003) for all soil samples.
Determination of concentrations of volatile fatty acids in soil samples
A five-gram soil sample of each pot soil collected was suspended in 5 ml deionized water and shaken for 20 min with a reciprocating shaker. The slurry samples were centrifuged at 2,500 rpm for 10 min and the concentrations of volatile fatty acids (VFAs) and alcohols in the supernatants were analyzed by gas chromatography (Hitachi G-3500 and G-5000) as described previously (Ueki et al. 1986). Concentrations of VFAs and alcohols are expressed in the text as those determined in the supernatant of slurry samples.
DNA extraction and PCR amplification
About 1 g soil from each sample was taken, and DNA was extracted using “Ultra Clean™ Soil DNA Isolation kit” (MO BIO Laboratories, Inc., Carlsbad, CA) according to the manufacturer’s instructions. Finally, 50-μl volume was extracted for each sample in which the DNA was eluted from the spin column at a concentration of 48–72 ng/μl. For PCR-DGGE, the V3 region of 16S rRNA gene from DNA samples was PCR amplified using a primer set B341fGC (5′-CGC CCG CCG CGC GCG GCG GGC GGG GCG GGG GCA CGG GGG GCC TAC GGG AGG CAG CAG-3′, with underlined GC clamps) and 534r (5′-ATT ACC GCG GCT GCT GG-3′) (Muyzer et al. 1993). For clone library analysis, bacterial 16S rRNA genes were amplified using a primer set B27f (5′-AGA GTT TGA TYM TGG CTC AG-3′) and U1492r (5′-GGY TAC CTT GTT ACG ACT T-3′). The PCR mixture (50 μl) contained 1.25 U of Taq DNA polymerase (Amplitaq Gold; Applied Biosystems, Foster, CA), 15 mM tris(hydroxymethyl)aminomethane-HCl (pH 8.0), 50 mM KCl, 1.5 mM MgCl2, 0.1 % bovine serum albumin, each deoxynucleotide triphosphate mixtures at a concentration of 200 μM, 0.25 μM of each primer, and 60–100 ng of template DNA. The amplification conditions for the primer set B341fGC and 534r or B27f and U1492r were followed as described in the previous study (Mowlick et al. 2012). Amplified DNA fragments were confirmed after agarose gel electrophoresis staining with ethidium bromide.
DGGE analysis
For the DGGE analysis, PCR products of the 16S rRNA gene were separated by DGGE using a DCode™ system (Bio-Rad Laboratories, Hercules, CA). A total of 10 μg DNA sample was applied in each lane representing the soil samples. The DNA samples were loaded to 10 % acrylamide gels with a urea–formamide denaturing gradient of 30–60 % (100 % denaturant was defined as 7 M urea and 40 % formamide) at an electrophoretic movement for 3.5 h and 200 V. The gels were stained in SYBR Gold solution and viewed by a UV transilluminator. The photographic images were transformed into digital data, and the positions of major DNA bands in the DGGE profiles were numerically designated for performing cluster analysis.
Clone library and nucleotide sequencing
The following ten soil samples were selected based on the various data obtained (soil parameters, DGGE, etc.) as representatives for the clone library analysis of the bacterial community in the soil samples: Br30-20 (pot no. A1), Br25-30 (B2), Br30-30 (E1), Wh25-30 (H1), Wh30-30 (K1), Av25-30 (M2), Av30-30 (P2), Co25-30 (R1), Co30-30 (U1), and original (Y2) (Table 1). The PCR products of DNA from these samples were purified using the QIAquick Gel Extraction Kit (Qiagen, Valencia, CA) and cloned into Escherichia coli JM109-competent cells following the instructions of pGEM-T Easy Vector Systems (Promega, Madison, WI). The vector-harboring clones containing an insert of appropriate sizes (about 1500 bp) were obtained in the Luria-Bertani plates by the standard methods (Kaku et al. 2005). Sequence analysis (about 600 bp) was done for a total of 96 clones from each soil sample using a sequence primer U515f (5′-GTG YCA GCM GCC GCG GTAA-3′) according to the Dye Terminator method using a capillary sequencer at Takara Co. Ltd.
Statistical analysis
The major DGGE bands detected on the profiles were numbered and classified into four categories for the cluster analysis depending on their intensities (0 = no band; 1 = weak; 2 = moderate; 3 = strong) (Watanabe et al. 2009). The cluster analysis with normalized data was carried out using the PAST program and Ward method (Hammer et al. 2001). Database searches for related 16S rRNA gene sequences were conducted using BLAST program and GenBank database (Altschul et al. 1997). The profile alignment function of ClustalW program was used to align the sequences. The phylogenetic trees were constructed by the neighbor-joining method (Saitou and Nei 1987) with Njplot program in ClustalW package (Thompson et al. 1994). Bootstrap resampling analysis for 1000 replicates was performed to determine the confidence of the tree topology. An OTU (operational taxonomic unit) was designated as a phylogenetic group or unit consisted of the resulting clones showing almost 97 % similarity of nucleotide sequences. All the representative clones of the OTUs were analyzed to determine the taxonomic affiliations of the clones of all libraries. All 16S rRNA gene sequences obtained in the present study were checked for possible chimeras using the chimera check online analysis program (http://comp-bio.anu.edu.au/bellerophon/bellerophon.pl) of the Bellerophon server. Finally, nonchimeric clonal sequences were validly used for the analysis of the bacterial community for each soil sample. A rarefaction analysis of the 16S rRNA gene sequences in the clone libraries was carried out with the software aRarefactWin (http://www.uga.edu/strata/software/Software.html). The coverage of the clone libraries (C) was calculated (Good 1953) from the equation \( C=1-\left( {{n_1 \left/ {N} \right.}} \right) \); where n 1 is the number of clones that occurred only once (frequency 1), and N is the total number of clones examined. The richness (chao 1) was determined for all the clone libraries using an online grouping method “Fastgroup” (http://biome.sdsu.edu/fastgroup/fg_tools.htm). Bacterial diversity was calculated using the Simpson's diversity index (D) by the function, \( D=1-\sum n{{{\left( {n-1} \right)}} \left/ {{\left( {N\left( {N-1} \right)} \right)}} \right.} \), where n is the total number of clones of a particular OTU and N is the total number of clones of all OTUs. Besides, Shannon–Wiener diversity index (H′) was determined to compare the changes in diversity of bacterial communities within the libraries by the function: H′ = −∑P i log P i, where the proportion of OTU i relative to the total number of OTU (p i) was calculated. All those diversity indexes were calculated using online biodiversity calculator (http://www.alyoung.com/labs/biodiversity_calculator.html).
The nucleotide sequences obtained from the clone library analyses have been reported in DDBJ/GenBank under the accession numbers AB642681-AB643454 (774 entries).
Results
Status of the treated soil samples
The incorporated pathogen (the nit mutant of F. oxysporum) was enumerated at 5 × 105 CFU/g of dry soil at the start of the incubation. Densities of the pathogen in all treated soils after 17 days of incubation were determined along with the measurements of ORP and concentrations of VFAs and alcohols (Table 1). A high density of the fungal pathogen (higher than 1 × 103 CFU/g) was detected from all the control soil samples (without plant biomass), indicating inability to kill the pathogen effectively without biomass incorporation. In contrast, the Fusarium population was eliminated completely from all wheat bran-treated soil irrespective of the soil conditions. For Brassica and Avena treatments, the pathogen was not detected at all from soil treated at 30 °C for both 20 and 30 % MC, while it was still alive in soil treated at 25 and 27.5 °C.
The ORP values for all biomass-treated (BSD) soil dropped considerably as compared with the values of the original or the control soils. The results indicated the development of a much reduced condition for all BSD-treated soils compared with the control soil. No VFAs was detected from the control as well as the original soil samples, while considerable amounts of acetate were detected from all conditions of BSD-treated soils followed by butyrate (for wheat bran-treated soils mainly) together with traces of propionate. In case of wheat bran-treated soil, when compared with other BSD soils, the concentrations of acetate were almost double and those of butyrate were much higher. Small amounts of ethanol (3.2–4.0 mmol/l) were detected from some BSD conditions.
PCR-DGGE analysis
The PCR-DGGE analysis was carried out to know the differences in the bacterial community profiles of these differently treated soil samples. The DGGE banding patterns and cluster analysis of the Brassica-treated soils showed almost similar community profiles between duplicate samples with an exception (B1 and B2 for Br25-30), indicating that similar bacterial communities usually developed in soil treated with the same condition (Fig. 1). The differences of incubation temperature or MC, however, clearly resulted in distinct profiles.

a DGGE patterns of bacterial populations in soil treated with B. juncea plants under different conditions. b Cluster analysis of DGGE band pattern for similarity index. V3 region of bacterial 16S rRNA gene was PCR amplified with a primer set of B341fGC/B534r. Amplified products were separated on a gradient gel of 30–60 % denaturant. The lanes A1 and A2 are from soil samples at 30 °C and 20 % MC, respectively; whereas others are from samples treated at 30 % MC under different temperatures—25 (lanes B1 and B2), 27.5 (lanes C1 and C2), and 30 °C (lanes E1 and E2). The numbers appearing aside of bands indicate the band numbers and positions for performing cluster analysis. The numbering was done chronologically starting from the upper position of the left-sided DGGE lane
The DGGE profiles for other soil samples (wheat bran-treated, Avena-treated, and control soils) with the cluster analysis (Fig. 2) revealed that banding patterns for soil samples treated at 25 and 27.5 °C were rather similar for each treatment while those were distinctive from soil treated at 30 °C for all cases irrespective of MC. This indicates that the rise in the incubation temperature from 27.5 to 30 °C affected the bacterial communities more strongly as compared with the rise from 25.0 to 27.5 °C.

a DGGE patterns for control (lanes U1, S2, R1, and Q2), Avena-treated (lanes P2, N2, M2, and L1), and wheat bran-treated (lanes K1, J1, H1, and G2) soil samples at different conditions. b Cluster analysis of DGGE band pattern for similarity index. The lanes Q2, L1, and G2 represent samples treated at 30 °C with 20 % MC, whereas others were treated at 30 % MC under different temperatures—30 (lanes U1, P2, and K1), 27.5 (lanes S2, N2, and J1), 25 °C (lanes R1, M2, and H1). Other notifications of DGGE were similar as described in Fig. 1
The DGGE analysis was carried out for selected soil samples including the original soil to know the effects of different plant biomass sources and MC on the bacterial community compositions (Fig. 1 in the ESM). Clear changes were shown in the profiles for all treated soil as compared with the original soil. Amendment with Brassica plants or wheat bran resulted in greater differences from the control soil than the amendment with Avena plants. Although the variation in plant biomass sources resulted in rather different DGGE profiles, the difference of MC (20 % or 30 %) for each biomass condition did not bring about such clear differences in the profiles (Fig. 1b in the ESM).
Clone library analysis
Soil samples treated at 25 and 30 °C under 30 % MC as well as the original soil were selected for the clone library analysis. In addition, for Brassica-treated soil, the soil sample treated at 30 °C under 20 % MC (Br30-20) was also selected, because it effectively eliminated the pathogen. One of the soil samples from the duplicate pots was selected for each treatment based on all results shown above.
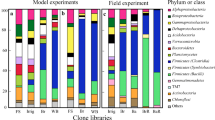
The clones from all libraries appeared to be assigned to at least six major phyla of the domain Bacteria, namely Firmicutes, Proteobacteria, Acidobacteria, Actinobacteria, Bacteroidetes, and Chloroflexi. However, the phylogenetic compositions and proportions of the major phyla or classes varied markedly depending upon the type of biomass used and the treatment conditions applied. The affiliations of clone sequences from each soil sample are shown in Fig. 3 in relation to the percentages of the number of clones belonging to each phylum or class. Out of the phylogenetic trees constructed for all ten libraries, those for Br30-30 (E1) and Wh25-30 (H1) were selected as representative trees from the soil samples that eliminated the pathogen completely (Fig. 4). In addition, major OTUs (consisting of more than two clones) and their closest relatives are presented in Table 2 for each library.

Phylogenetic composition (phylum or class) of bacteria based on 16S rRNA gene sequences from different clone libraries. The compositions are represented as relative abundances in relation to the percentages of number of clones belonging to each phylum or class. Clone libraries: A1, Br30-20; B2, Br25-30; E1, Br30-30; H1, Wh25-30; K1, Wh30-30; M2, Av25-30; P2, Av30-30; R1, Co25-30; U1, Co30-30; Y2, original. Symbols— , Alphaproteobacteria;
, Alphaproteobacteria;  , Betaproteobacteria;
, Betaproteobacteria;  , Gammaproteobacteria;
, Gammaproteobacteria;  , Deltaproteobacteria;
, Deltaproteobacteria;  , Acidobacteria;
, Acidobacteria;  , Bacteroidetes;
, Bacteroidetes;  , Planctomycetes;
, Planctomycetes;  , Firmicutes (Clostridia);
, Firmicutes (Clostridia);  , Firmicutes (Symbiobacterium);
, Firmicutes (Symbiobacterium);  , Firmicutes (Bacilli);
, Firmicutes (Bacilli);  , Actinobacteria;
, Actinobacteria;  , Chloroflexi; and
, Chloroflexi; and  , others
, others

Neighbor-joining trees showing the phylogenetic relationships of all OTUs derived from Brassica- (30 °C–30 % MC) and wheat bran-treated (25 °C–30 % MC) soil samples based on 16S rRNA gene sequences. Bootstrap values (n = 1,000) above 70 % are indicated at branch nodes. The scale bar represents 2 % estimated difference in nucleotide sequence position. The abbreviation C. indicates the genus Clostridium. The name of each clone starts with the pot number for each treatment (E1 for Br30-30 and H1 for Wh25-30). As the outgroup, Sulfolobus acidocaldarius (D14053; the domain Archaea) 16S rRNA gene sequence was used. Accession numbers of the species are shown in the parentheses. Numbers in the parentheses aside each clone name denote the number of clones assigned to the OTU. Each clone name without parenthesis represents one OTU with one clone. Abbreviations: α-Prot Alphaproteobacteria, γ-Prot Gammaproteobacteria, Actino Actinobacteria, Acido Acidobacteria
Phylogenetic groups in the original and the control soil
The clone library for the original soil (Y2) showed much diversified populations of different phylogenetic groups. The most abundant taxonomic group of the original soil was allocated to the phylum Proteobacteria (about 48 % of the total number of clones; Alpha- and Gammaproteobacteria classes mainly). Other clone sequences belonged to the phyla Firmicutes (7 % from the Clostridia and 8 % from the class Bacilli), Actinobacteria, Acidobacteria, Planctomycetes, etc.
In case of the control library Co25-30 (R1), diversified bacterial populations were detected. An OTU relating to the unique clade “Symbiobacterium,” a facultatively anaerobic group in the Firmicutes, at about 90–91 % of sequence similarity was the most dominant (24 %), and a few clones were retrieved from the Bacilli and Clostridia (Fig. 3). Other phylogenetic groups detected were almost similar with those from the Original clone library. For another control library Co30-30 (U1), the Symbiobacterium-related bacterial groups were also dominant with a few number of clostridial clones. Besides, Rhodanobacter terrae in the Gammaproteobacteria was assigned to the closest relative of the major OTUs commonly appeared in both control libraries as well as in the original soil (Table 2).
Phylogenetic groups in the BSD-treated soil
Clone libraries for all BSD-treated soil samples showed substantial differences as compared with those of the original and control libraries (Fig. 3). The BSD libraries irrespective of the treatment conditions showed the presence of exclusively dominant bacterial taxonomic groups in the communities. In case of the Brassica-treated libraries, about 80 % clones were assigned to the phylum Firmicutes, of which the majority belonged to the class Clostridia (strictly anaerobic spore-formers), with relatively minor clones in the class Bacilli (aerobic or facultatively anaerobic spore-formers). The clone libraries for the Brassica-treated soils, that is, Br30-20 (A1), Br25-30 (B2), and Br30-30 (E1), contained 54, 50, and 75 % clostridial clones, whereas, 41, 27, and 6 % clones from the class Bacilli, respectively (Fig. 3). The other clones were mainly affiliated with the different classes of the phylum Proteobacteria.
The compositions of the major Firmicutes groups for the Brassica-treated three clone libraries were compared (Table 2). The clones closely related at high similarities (about 99 %) to Clostridium saccharobutylicum occupied major portions of those clone library, especially for the Br30-20 library. A number of clones related to diverse species in clostridial groups such as Oxobacter pfennigii, Zymophilus raffinosivorans, Clostridium xylanovorans, Clostridium cylindrosporum, Clostridium sufflavum, etc. increased with the rise of MC and temperature (Fig. 4). The clones closely related to Bacillus azotoformans and Bacillus niacini in the class Bacilli were also the dominating OTUs in both Br30-20 and Br25-30 libraries.
For the wheat bran-treated two clone libraries Wh25-30 (H1) and Wh30-30 (K1), about 76 and 58 % of clones were detected from the class Clostridia with a few number of clones from the class Bacilli (Fig. 3). Other phylogenetic groups retrieved from the two libraries were placed in the phyla Proteobacteria and Actinobacteria mainly. The closely related species of the major bacterial OTUs in the wheat bran-treated clone libraries showed that the lower temperature (25 °C) stimulated the clostridial ratios as well as diversification consisting of Clostridium pasteurianum, Clostridium acetobutylicum, C. xylanovorans, C. saccharobutylicum, Clostridium tetanomorphum etc. (Table 2, Fig. 4) in the community. Instead, higher temperature (30 °C) induced two dominant groups closely related to B. niacini and Azotobacter chroococcum as well as diversified clostridial clones (Table 2).
For the Avena-treated clone libraries, members of the Firmicutes also occupied the dominant position. From the library Av25-30 (M2), 51 % clones belonged to the Firmicutes phylum (43 % from the Clostridia and 8 % from the Bacilli) (Fig. 3). Clones relating to C. saccharobutylicum, C. cylindrosporum, C. xylanovorans, and B. niacini, detected in other BSD libraries, were also the most dominant groups in the library. A number of clones from the phyla Proteobacteria and Bacteroidetes were also detected. On the other hand, the Av30-30 (P2) library contained the classes Clostridia and Bacilli in the Firmicutes at almost the same percentage. Clones closely related to Veillonella rogosae in the Clostridia and Streptococcus parasanguinis in the Bacilli occupied extraordinarily dominant positions in the library, respectively (Table 2).
Bacterial diversity in the clone libraries
Rarefaction analysis based on the OTU clustering (97 % sequence similarity cut-off level) (Fig. 5) suggests that the curve for the Original library was far from the saturation. Among the rarefaction curves for the BSD-treated libraries, the curves for Br30-20 and Av30-30 seemed to almost reach the plateau, whereas others were far from it and showed similar curves as those for the Co25-30 and Co30-30 libraries. The results suggest that bacterial richness in these samples was not fully recovered in these clone libraries with some exceptions.

Rarefaction curves for the bacterial communities from all clone libraries. A1, Br30-20; B2, Br25-30; E1, Br30-30; H1, Wh25-30; K1, Wh30-30; M2, Av25-30; P2, Av30-30; R1, Co25-30; U1, Co30-30; Y2, original
Table 3 shows estimated bacterial richness and diversity for each clone library. A total of 49 OTUs were recognized in the original library, whereas the numbers decreased for all the treated soil libraries. The estimates of richness and diversity in the communities suggest that the original soil harbors the most diverse communities. All treatments reduced the diversity as compared with the original soil, and especially, the bacterial diversities in the libraries Br30-20 and Av30-30 were decreased considerably. However, high bacterial diversities were still maintained for other BSD treatments, especially in case of Br25-30, Br30-30, and Wh25-30 communities. The result suggests that although the BSD treatment increased the percentages of clostridial clones considerably, bacterial diversity at the species (97 % sequence similarity) level was not usually reduced so much.
Phylogenetic diversity of clostridial groups after BSD
Based on the suppression of F. oxysporum population incorporated, compositions of clostridial groups retrieved from the four libraries, Br30-20, Br30-30, Wh25-30, and Wh30-30, were further analyzed phylogenetically to identify the major common clostridial species and to compare the clostridial diversities among the BSD samples.
A phylogenetic tree consisting of all OTUs assigned to the class Clostridia from these BSD samples was generated (Fig. 6). All OTUs related to the clostridial groups were divided into six clusters (Collins et al. 1994) including Oxobacter, Caloramator, and Pelotomaculum groups, indicating occurrence of extraordinarily diverse species in the class under these BSD conditions. Majority (63 %) of the clostridial clones from the four libraries were classified into the cluster I (Clostridium sensu stricto) including the Oxobacter and Caloramator groups, and the remaining clones (37 %) were affiliated with other miscellaneous clusters (clusters III, IV, VI, IX, and XIVa). For the wheat bran-treated libraries, almost two thirds (66 %) of the clostridial clones were assigned to the cluster 1 group. On the other hand, clostridial groups in the Brassica-treated soil, especially for the Br30-30 library, were distributed to cluster 1 and other clusters almost in an equal proportion. The result indicates that the phylogenetic composition of clostridial communities changed with the influence of biomass and treatment conditions.

Neighbor-joining tree showing the phylogenetic relationships of clostridial clones from some effective BSD samples based on 16S rRNA gene sequences (according to the clostridial cluster analysis by Collins et al. 1994). As the outgroup, Bacillus subtilis DSM10 16S rRNA gene sequence was used. The abbreviation C. indicates the genus Clostridium. The name of each clone starts with the pot number for each treatment: A1, Br30-20; E1, Br30-30; H1, Wh25-30; K1, Wh30-30. Accession numbers of the species are shown in the parentheses. Numbers in parentheses aside each clone name denote the number of clones in the OTU of each library. Tree construction and other notifications are similar as described in Fig. 4
From the combined clostridial phylogenetic tree, it was revealed that OTUs closely related to C. saccharobutylicum (about 99 % sequence similarity) or C. xylanovorans (about 95–96 %) were recognized in all four libraries as the dominant groups, and the clones relating to C. cylindrosporum, O. pfennigii, or Pelotomaculum shinkii were also detected commonly as relatively minor groups. The clostridial clones assigned to the cluster III (e.g., C. sufflavum) appeared only in the Brassica-treated libraries (Table 2).
The rarefaction analysis for the clostridial clones (Fig. 2 in the ESM) almost corresponded with it for all communities (Fig. 5) and also did not reach the plateau except for the Br30-20 library. The result of richness and diversity estimates of OTUs (Table 1 in the ESM) suggests that diversities of clostridial groups were higher in the Br30-30 and Wh25-30 BSD samples, and the diversity was much reduced in case of Brassica-treated soil of lower MC (Br30-20) and wheat bran-treated soil at higher temperature (Wh30-30).
Discussion
Achieving a reducing state or a decrease in ORP in soil is the most important aspect during BSD (Shinmura et al. 1999; Blok et al. 2000). Microbial activities can affect soil ORP by influencing redox couples including NO3 –/NO2 –, Fe3+/Fe2+, H+/H2, SO4 2–/HS–, and CO2/CH4 along with the partial or complete consumption of oxygen by soil microbes under the higher MC (Peters and Conrad 1996; Savant and Ellis 1964). In this study, the ORP decreased for most of BSD treatments as compared with the soil without biomass incorporation (control) and it should stimulate growth and multiplication of anaerobic bacteria in the BSD soil. In fact, a large number of bacterial species relating to the strictly anaerobic clostridial groups were detected as the closest relatives for the clone libraries of the BSD-treated soil samples.
The antagonistic activity of some soil microorganisms may be involved during BSD against soil-borne pathogens (Bailey and Lazarovits 2003; Kubo et al. 2005; Larkin and Griffin 2007). It has been pointed out that accumulation of VFAs such as acetate and butyrate would be the important factors for killing the plant pathogens during the process of disinfestation (Momma et al. 2006). In our study, rather high amounts of acetate were detected from all BSD soil samples together with butyrate especially for the wheat bran-treated soils, while no VFAs was detected from the control soils. Since the incorporation of plant biomasses to the soil strongly enhanced growth of clostridial species as shown by the clone library analysis, VFAs might be produced by these anaerobic bacteria through decomposition of the biomass. These results coincided with the results of our previous study (Mowlick et al. 2012). As mentioned below, we have already isolated many strains of anaerobic bacteria from both B. juncea- and wheat-bran-treated soil used in this study and confirmed formation of acetate and butyrate as fermentation products for many of the isolates (data not shown).
It is reported that BSD using Brassica plants, wheat bran, grasses or molasses effectively killed a wide range of soil-borne pathogens including Botrytis cinerea, Fusarium spp., Rhizoctonia solani, Phomopsis scleroides, Verticillium dahliae, Ralstonia solanacearum, Meloidogyne incognita, etc. (Shinmura 2000, 2004; Takeuchi 2004; Messiha et al. 2007). Although the incorporated pathogen seemed to be completely eliminated from the wheat bran-treated soils for all conditions in this study, the effects of BSD on the pathogen were different depending on the incubation conditions for Brassica and Avena treatments. For these Brassica- and Avena-treated soils, the variations in the incubation temperature greatly influenced the suppression of the pathogenic population during BSD. The result indicates that temperature control is very important to conduct the BSD treatment successfully. However, the temperature effect should be verified in further studies by considering other factors including the amount of plant material incorporated.
Soil harbors highly diverse bacterial communities with up to 50,000 (Sandaa et al. 1999) or even up to millions (Gans et al. 2005) of different 16S rRNA gene sequences or of different phylogenetic groups at the phylum or class level (Janssen 2006). The clone library for the original soil showed that the soil contained diversified bacterial groups of different phyla or classes. The bacterial compositions of both control libraries (Co25-30 and Co30-30) were somewhat similar with that from the original soil in containing clones relating to the Proteobacteria as major groups. The similarities of DGGE banding patterns between the original and the two control soils coincided with the results of the clone library analysis. We tried to know the differences in the recovery of the bacterial diversities among treatments analyzing the rarefaction curves. Based on the results, we think that, in the future studies, we need to analyze more number of clones for most of the soil samples to know the saturation point of the bacterial diversities.
In both control soils, a group relating to Symbiobacterium thermophilum, a very deep group within the Firmicutes or, possibly, a novel phylum (Beppu and Ueda 2009), proliferated, however, the strict anaerobes relating to the typical clostridial groups did not occupy a major position. In the BSD-treated libraries, clones affiliated with the general Firmicutes were exceedingly dominant and the ratios of the clostridial clones (50–75 %) and Bacillus group (6–50 %) varied considerably. Soil microbial communities can be influenced by many factors like temperature (Pietikänen et al. 2005), water availability (Fierer et al. 2003), ORP status, and others. In this study, it appeared that both temperature and MC contributed to the changes in bacterial communities in the BSD-treated soils. As shown in Fig. 3, increase in the incubation temperature stimulated the clostridial proliferation, but decreased the Bacillus group for Brassica-treated soil, while the opposite results were obtained for wheat bran- and Avena-treated soils. The effect of MC was very distinct in case of Brassica-treated soil, where the ratio of clones related to the class Bacilli was declined markedly with an increase in MC. Most of the species in the Bacilli produce spores and they should be alive as spores even in dry soil. Although they might start to grow under the humid condition immediately after the start of the treatment, earlier exhaustion of oxygen under the higher MC should suppress aerobic growth.
It was found that the members related to the Clostridia class appeared as major and important taxonomic groups in the bacterial communities in the BSD soils. The results agreed well with our previous results (Mowlick et al. 2012). Among the plant biomass incorporated in this study, Brassica or wheat bran was considered to stimulate the development of clostridial community during BSD. The clones assigned to the Clostridia class actually contained phylogenetically diverse members and it was indicated that the growth of abundant clostridial species might contribute to the total diversity of the bacterial communities in the BSD soil. Diverse species related to the class Clostridia should be also present as spores even in the aerobic field soil, and start to grow using plant biomass as growth substrates soon after the development of reduced condition. The result indicates that field soil, even when the soil has been maintained under the aerobic condition for a long time, harbors exceedingly diverse, strictly anaerobic, clostridial species, and the introduction of reduced condition to the soil induces drastic changes in the bacterial community composition in the soil along with their active growth.
In case of the five clone libraries (Br30-20, Br30-30, Wh25-30, Wh30-30, and Av30-30), where the pathogen was effectively eliminated, the abundance of the members of the Firmicutes was almost 70–90 % of the total clones, of which the majorities were related to the strictly anaerobic, clostridial species. Therefore, it seems likely that the dominant clostridial groups in these treatment conditions might have an important role to suppress the pathogen during BSD. Bacterial groups related to C. acetobutylicum, C. saccharobutylicum, C. xylanovorans, C. sufflavum, C. pasteurianum, O. pfennigii, Z. raffinosivorans, C. cylindrosporum, Caloramator mitchellensis, V. rogosae, etc. seemed to be commonly proliferated anaerobes by the BSD treatment.
It is known that most of the closely related clostridial species of the clones detected in this study produce various products including acetates, butyrate, alcohol, and H2 gas as well as indole or skatole (Krumholz and Bryant 1985; Rainey et al. 2009; Wiegel 2009; Schleifer 2009). Since the similarity levels of 16S rRNA gene sequences between the major clone groups and their closest relatives are different depending on the clones, physiological properties and fermentation products of the species in the BSD soil represented by the clone sequences should be different from those of the related species. However, some species such as C. acetobutylicum, C. saccharobutylicum, C. pasteurianum, and C. xylanovorans were closely related with the major OTUs at considerably high similarity values (95–99 %), respectively, suggesting that bacterial groups represented by the clones have similar properties as these recognized species.
Besides the clostridial population, the clones of the classes Bacilli and Gammaproteobacteria also proliferated in the BSD soils. The major Bacillus species included B. azotoformans (nitrogen-forming bacterium) and B. niacini (nicotinic acid-metabolizing) (Logan and Vos 2009; Priest 2009), and the Gammaproteobacteria group contained A. chroococcum (nitrogen fixer) (Kumar and Singh 2001). B. niacini and A. chroococcum were also detected as the closest relatives for the clones in our previous BSD soils (Mowlick et al. 2012). Thus, it is possible that species relating to these species might contribute partly for changing the soil status during BSD. In addition, in our previous study, nitrogen-fixing bacteria such as C. acetobutylicum, C. pasteurianum, and A. chroococcum were also appeared as the closely related species of the major groups in the BSD communities. It is of interest to know their roles in nitrogen supply to the soil microbial communities.
We have isolated many clostridial strains relating to the clone sequences shown above from the BSD-treated soil samples examined in this study. We are now investigating the physiological properties of these isolates to confirm the effects of clostridial species on the pathogens during the BSD treatment. Especially, it should be important to clarify the products to suppress the pathogens or to find out some other effective functions during the BSD treatment for these anaerobes. We have used F. oxysporum (fusarium wilt pathogen of tomato or spinach) as pathogenic agents in the present study as well as in our previous study (Mowlick et al. 2012). Other plant pathogens including fungi, bacteria, etc. should be also considered in the future studies. Moreover, BSD experiments should be applied in field conditions to confirm the suppression of plant pathogens and proliferation of clostridial communities.
References
Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402
Bailey KL, Lazarovits G (2003) Suppressing soil borne diseases with residue management and organic amendments. Soil Till Res 72:169–180
Beppu T, Ueda K (2009) Genus I. Symbiobacterium Ohno, Shitatori, Park, Saitoh, Kumon, Yamshita, Hirata, Nishida, Ueda and Beppu 2000, 1832VP. In: Whitman WB, Parte AC (eds) Bergey’s manual of systematic bacteriology, vol 3. Springer, New York, pp 1188–1190
Blok WJ, Lamers JG, Termorshuizen AJ, Bollen GJ (2000) Control of soilborne plant pathogens by incorporating fresh organic amendments followed by tarping. Phytopathology 90:253–259
Collins MD, Lawson PA, Willems A, Cordoba JJ, Fernandez-Garayzabal J, Garcia P, Cai J, Hippe H, Farrow JAE (1994) The phylogeny of the genus Clostridium: proposal of five new genera and eleven new species combinations. Int J Syst Bacteriol 44:812–826
Fierer N, Schimel JP, Holden PA (2003) Influence of drying rewetting frequency on soil bacterial community structure. Microb Ecol 45:63–71
Gans J, Wolinsky M, Dunbar J (2005) Computational improvements reveal great bacterial diversity and high metal toxicity in soil. Science 309:1387–1390
Good IJ (1953) The population frequencies of species and the estimation of population parameters. Biometrika 40:237–262
Goud JKC, Termorshuizen AJ, Blok WJ, van Bruggen AHC (2004) Long-term effect of biological soil disinfestation on Verticillium wilt. Plant Dis 88:688–694
Hammer Ø, Harper DQAT, Ryan PD (2001) PAST: Paleontological statistics software package for education and data analysis. Palaeontologia Electronica 4:4. http://palaeo-electronica.org/2001_1/past/issue1_01.htm
Janssen PH (2006) Identifying the dominant soil bacterial taxa in libraries of 16S rRNA and 16S rRNA genes. Appl Environ Microbiol 72:1719–1728
Kaku N, Ueki A, Ueki K, Watanabe K (2005) Methanogenesis as an important terminal electron accepting process in estuarine sediment at the mouth of Orikasa River. Microb Environ 20:41–52
Katan J (1981) Solar heating (solarization) of soil for control of soil borne pests. Annu Rev Phytopathol 19:211–236
Katan J (2000) Physical and cultural methods for the management of soil borne pathogens. Crop Prot 19:25–31
Krumholz LR, Bryant MP (1985) Clostridium pfennigii sp. nov. uses methoxyl groups of monobenzenoids and produces butyrates. Int J Syst Bacteriol 35:454–456
Kubo C, Ushio S, Katase M, Takeuchi T (2005) Analysis of factors involved in sterilization effect by soil reduction. Jpn J Phytopathol 71:281–282, abstract in Japanese
Kumar V, Singh KP (2001) Enriching vermicompost by nitrogen fixing and phosphate solubilizing bacteria. Biores Technol 76:173–175
Larkin RP, Griffin TS (2007) Control of soil borne potato diseases using Brassica green manures. Crop Prot 26:1067–1077
Logan NA, Vos PD (2009) Genus I. Bacillus Cohn 1872, 174AL. In: Whitman WB, Parte AC (eds) Bergey’s manual of systematic bacteriology, vol 3. Springer, New York, pp 21–111
Macfarlane J, Macfarlane GT (1995) Proteolysis and amino acid fermentation. In: Gibson GR, Macfarlane GT (eds) Human colonic bacteria. CRC Press, New York, pp 75–100
Maidak BL, Cole JR, Jr Parker CT, Garrity GM, Larsen N, Li B, Lilbum TG, McCaughey MJ, Olsen GJ, Overbeek R, Pramanik S, Schmidt TM, Tiedje JM, Woese CR (1999) A new version of the RDP (Ribosomal Database Project). Nucleic Acids Res 27:171–173
Messiha NAS, van Diepeningen AD, Wenneker M, van Beuningen AR, Janse JD, Coenen TGC, Termorshuizen AJ, van Bruggen AHC, Blok WJ (2007) Biological soil disinfestation (BSD), a new control method for potato brown rot, caused by Ralstonia solanacearum race 3 biovar 2. Eur J Plant Pathol 117:403–415
Momma N (2008) Biological soil disinfestation (BSD) of soil-borne pathogens and its possible mechanisms. JARQ 42:7–12
Momma N, Yamamoto K, Simandi P, Shishido M (2006) Role of organic acids in the mechanisms of biological soil disinfestation (BSD). J Gen Plant Pathol 72:247–252
Mowlick S, Hirota K, Takehara T, Kaku N, Ueki K, Ueki A (2012) Development of anaerobic bacterial community consisted of diverse clostridial species during biological soil disinfestation amended with plant biomass. Soil Sci Plant Nutr 58:273–287
Muyzer G, De Waal EC, Uitterlinden AG (1993) Profiling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction-amplified genes coding for 16S rRNA. Appl Environ Microbiol 59:695–700
Peters V, Conrad R (1996) Sequential reduction processes and initiation of CH4 production upon flooding of oxic upland soils. Soil Biol Biochem 28:371–382
Pietikäinen J, Pettersson M, Bååth E (2005) Comparison of temperature effects on soil respiration and bacterial and fungal growth rates. FEMS Microbiol Ecol 52:49–58
Prather MJ, McElroy MB, Wofsy SC (1984) Reductions in ozone at high concentrations of stratospheric halogens. Nature 312:227–231
Priest FG (2009) Genus I. Paenibacillus Ash, Priest and Collins 1994, 852VP. In: Whitman WB, Parte AC (eds) Bergey’s manual of systematic bacteriology, vol 3. Springer, New York, p 280
Puhalla JE (1985) Classification of strains of Fusarium oxysporum on the basis of vegetative compatibility. Can J Bot 63:179–183
Rainey FA, Hollen BJ, Small A (2009) Genus I. Clostridium Prazmowski 1880, 23AL. In: Whitman WB, Parte AC (eds) Bergey’s manual of systematic bacteriology, vol 3. Springer, New York, pp 738–828
Ristaino JB, Thomas W (1997) Agriculture, methyl bromide, and the ozone hole: can we fill the gaps. Plant Dis 81:954–975
Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425
Sandaa RA, Torsvik V, Enger O, Daae FL, Castberg T, Hahn D (1999) Analysis of bacterial communities in heavy metal-contaminated soils at different levels of resolution. FEMS Microbiol Ecol 30:237–251
Savant NK, Ellis R (1964) Changes in RP and phosphorus availability in submerged soil. Soil Sci 98:388–394
Schleifer KH (2009) Genus XXVI. Zymophilus Schleifer, Leuteritz, Weiss, Ludwig, Kirchhof and Seidel-Rüfer 1990, 26VP. In: Whitman WB, Parte AC (eds) Bergey’s manual of systematic bacteriology, vol 3. Springer, New York, p 1119
Shinmura A (2000) Causal agent and control of root rot of welsh onion. PSJ Soilborne Disease Workshop Report 20:133–143 (in Japanese with English Summary)
Shinmura A (2004) Principle and effect of soil sterilization method by reducing redox potential of soil. PSJ Soilborne Disease Workshop Report 22:2–12 (in Japanese with English Summary)
Shinmura A, Sakamoto N, Abe H (1999) Control of Fusarium root rot of welsh onion by soil reduction. Jpn J Phytopathol 65:352–353
Spadaro D, Gullino ML (2005) Improving the efficacy of biocontrol agents against soil borne pathogens. Crop Prot 24:601–613
Stover RH (1979) Flooding of soil for disease control. In: Mulder D (ed) Soil disinfestation. Elsevier, Amsterdam, pp 19–28
Takehara T, Kuniyasu K (1994) Use of nitrate-nonutilizing mutants in ecological studies of Fusarium diseases. II. Isolation of nitrate non-utilizing mutants of Fusarium oxysporum on selective media. Ann Phytopathol Soc Jpn 60:705–710 (In Japanese with English summary)
Takehara T, Kuniyasu K, Mori M, Hagiwara H (2003) Use of a nitrate-nonutilizing mutant and selective media to examine population dynamics of Fusarium oxysporum f. sp. spinaciae in soil. Phytopathology 93:1173–1181
Takeuchi T (2004) Effect of sterilization by soil reduction on soil borne diseases in Chiba Prefecture. PSJ Soilborne Disease Workshop Report, 22:13–21 (In Japanese with English summary)
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680
Ueki A, Matsuda K, Ohtsuki C (1986) Sulfate reduction in the anaerobic digestion of animal waste. J Gen Appl Microbiol 32:111–123
Watanabe T, Cahyani VR, Murase J, Ishibasi E, Kimura M, Asakawa S (2009) Methanogenic archaeal communities developed in paddy fields in the Kojima Bay polder, estimated by denaturing gradient gel electrophoresis, real time PCR and sequence analyses. Soil Sci Plant Nutr 55:73–79
Wiegel J (2009) Family I. Clostridiaceae Pribram 1933, 90AL. In: Whitman WB, Parte AC (eds) Bergey’s manual of systematic bacteriology, vol 3. Springer, New York, p 737
Acknowledgments
This work was partly supported by a grant from the Ministry of Agriculture, Forestry and Fisheries of Japan (development of mitigation and adaptation techniques to global warming in the sectors of agriculture, forestry, and fisheries). We also greatly appreciate the technical assistance of Y. Ohtaki.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PPT 334 kb)
Rights and permissions
About this article
Cite this article
Mowlick, S., Takehara, T., Kaku, N. et al. Proliferation of diversified clostridial species during biological soil disinfestation incorporated with plant biomass under various conditions. Appl Microbiol Biotechnol 97, 8365–8379 (2013). https://doi.org/10.1007/s00253-012-4532-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-012-4532-z