
Abstract
Factors related to ethanol production from xylose in engineered Saccharomyces cerevisiae that contain an exogenous initial metabolic pathway are still to be elucidated. In the present study, a strain that expresses the xylose isomerase gene of Piromyces sp. Pi-xylA and overexpresses XKS1, RPE1, RKI1, TAL1, and TKL1, with deleted GRE3 and COX4 genes was constructed. The xylose utilization capacity of the respiratory deficiency strain was poor but improved via adaptive evolution in xylose. The μ max of the evolved strain in 20 g l−1 xylose is 0.11 ± 0.00 h−1, and the evolved strain consumed 17.83 g l−1 xylose within 72 h, with an ethanol yield of 0.43 g g−1 total consumed sugars during glucose–xylose cofermentation. Global transcriptional changes and effect of several specific genes were studied. The result revealed that the increased xylose isomerase acivity, the upregulation of enzymes involved in glycolysis and glutamate synthesis, and the downregulation of trehalose and glycogen synthesis, may have contributed to the improved xylose utilization of the strain. Furthermore, the deletion of PHO13 decreased the xylose growth in the respiration deficiency strain although deleting PHO13 can improve the xylose metabolism in other strains.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Saccharomyces cerevisiae is a promising organism for lignocellulosic ethanol fermentation. It effectively ferments glucose, fructose, and mannose through glycolysis (van Maris et al. 2006). Xylose is the second predominant sugar in plant biomass after glucose. Converting xylose into ethanol using S. cerevisiae presents an economic interest (Hahn-Hägerdal et al. 2007; Liu et al. 2010). However, S. cerevisiae cannot naturally utilize xylose, although genes that encode the required metabolic enzymes are included in its genome (Hahn-Hägerdal et al. 2007; Jeffries and Jin 2004; Wang et al. 2004). Metabolic engineering was performed to construct the xylose-utilizing strain by introducing the heterogeneous xylose metabolic pathway, which was xylose reductase-xylitol dehydrogenase (XR-XDH) or xylose isomerase (XI). The xylose-utilizing capacity of recombinant strain then further optimized by enhancing the downstream metabolic pathway such as xylulokinase (XK) and the pentose phosphate pathway (PPP). Moreover, to decrease the by-production of xylitol, the aldose reductase (encoded by the GRE3 gene) was destroyed and the redox state of XR-XDH-expressing strain was balanced via various cofactor engineering (Eliasson et al. 2000b; Hahn-Hägerdal et al. 2007; Jeffries and Jin 2004; Karhumaa et al. 2007; Kuyper et al. 2005a; Peng et al. 2012). However, the xylose-fermenting efficiency of strain, which was obtained using only these rational manipulations, is less than expected.
Evolutionary engineering can develop strain with expected phenotypes based on the principles of mutation and selection (Sauer 2001). This strategy has been applied successfully by various groups in improving xylose fermentation. Sonderegger and Sauer (2003) developed a Ps-XR-XDH-expressing strain using EMS mutation and adaptive cultivation. The mutant, TMB3001C5, achieved a ∼50 % higher specific xylose utilization rate and produced 19 % more ethanol. Kuyper et al. (2005b, 2004) and Wisselink et al. (2009) reported an evolved strain that is capable of cofermenting glucose, xylose, and arabinose faster and more effectively through cycles of adaptive batch and chemostat cultivation.
To understand the molecular mechanism of enhanced xylose metabolism and provide new guidelines for further strain modification, the transcriptional profile of the evolved strain and its response to xylose were investigated in XR-XDH expressing strains (Jin et al. 2004; Salusjärvi et al. 2006; Sonderegger et al. 2004). Meanwhile, the transcriptomic data for the XR-XDH evolved strain exhibited a respiratory response (Jin et al. 2004). The conversion of xylose into ethanol in the XI-expressing strain does not require cofactors for redox metabolic balance. Therefore, it can be a good model for studying the mechanism underlying the metabolic regulation of xylose fermentation in S. cerevisiae.
In the present work, an effective xylose-fermenting strain was obtained by adaptive evolution based on the strain using techniques, such as the expression of the xylose isomerase of Piromyces sp. (Pi-xylA) and enhancement of the downstream metabolic pathway of xylose. Then, the global transcriptional profile of the evolved strain was compared with the reference strain. Several significantly regulated genes were tested for their effect on xylose metabolism.
Materials and methods
The construction of plasmids and strains
The yeast expressional plasmid pJFE3 (Fig. 1) was constructed by introducing the TEF1 promoter (TEF1p) and the PGK1 terminator (PGK1t) in the YEplac195 vector (Gietz and Akio 1988). Then, the xylose isomerase gene from Piromyces sp. (Pi-xylA, GenBank accession no. CAB76571.1) was artificially synthesized and cloned into pJFE3 under the control of TEF1p and PGK1t to generate the pJX5 plasmid (Fig. 1). The plasmid pJX5 was transformed into the respiratory-deficient strain BSPC042 (Peng et al. 2012), to produce the strain BSPC095 (Pi-XI, XK, gre3::PPP, and cox4Δ; Table 1).

Plasmids pJFE3 and pJX5
Gene deletion was preformed as previously described (Peng et al. 2012) and was confirmed by PCR. The selection marker, but not the deleted gene sequence, was replicated from the genome of strains. The integrating vector pYMIK with the PGK1 promoter (Wang et al. 2004) was employed for the overexpression procedure, and the transformants were confirmed by reverse transcription polymerase chain reaction (RT-qPCR). The plasmids and the S. cerevisiae strains used in this work are listed in Table 1.The primers used for PCR are listed in Table S1 in the Electronic supplementary material (ESM).
Adaptive evolution of strain on xylose
The adaptive evolution process was performed according to the methods of Peng et al. (2012) until the biomass doubling time for xylose did not show significant shortening. Several single clones were isolated and evaluated. One of the fastest growing clones was selected and designated as BSPX013 (Pi-XI, XK, gre3::PPP, cox4Δ, and AE). The abbreviation AE was used to indicate the strain after adaptive evolution.
Batch fermentation
Minimal medium (6.7 g l−1 YNB, pH 6.0) with different glucose and/or xylose concentrations was used for batch fermentation. The cells were grown at 30 °C and 200 rpm. A cotton plug and a rubber stopper were used in the shake flasks for aerobic and air-limited cultivation conditions, respectively.
Measurement of enzymatic activity
The cells were culture in minimal medium and harvested when the OD600 was 4.0 and were washed with sterile water. Cell-free extracts were prepared as the crude enzyme in 100 mmol l−1 Tris–HCl buffer (pH 7.5) with a proteinase inhibitor cocktail (for fungal/yeast cells; Sangon Biotech Co., Ltd., Shanghai, China) using a FastPrep cell homogenizer (Thermo Savant, Germany) as previously described (Peng et al. 2012). Protein concentration was measured using a BCA protein assay reagent kit SK3051 (Sangon Biotech Co., Ltd.). A spectrophotometer (Helios Gamma, Thermo Fisher Scientific, Waltham, MA) was employed to determine enzymatic activity by measuring the concentration change of reduced and oxidized forms of the coenzymes at 340 nm.
The xylose isomerase activity of the cell extracts was determined at 30 °C. The 1-ml reaction mixture contained 100 mmol l−1 Tris–HCl buffer (pH 7.5), 10 mmol l−1 MgCl2, 500 mmol l−1 xylose, 1 U of sorbitol dehydrogenase (Roche, Boulder, CO), 0.15 mmol l−1 NADH, and 0.05 ml of the cell extract (Kersters-Hilderson et al. 1987). One unit of enzyme activity was defined as the amount of enzyme required to oxidize 1 μmol of coenzyme/min, and the specific activity was expressed in units per milligram of protein.
The activity of xylose reductase and xylitol dehydrogenase in the cell extracts were measured at 30 °C using a previously described method (Eliasson et al. 2000b). One unit of enzyme activity was defined as the amount of enzyme required to reduce or oxidize 1 μmol of coenzyme/min, and the specific activity was expressed in units per milligram of protein.
Extracellular metabolite analysis and calculation
The concentrations of glucose, xylose, xylitol, glycerol, acetate, and ethanol were determined using HPLC using an Aminex HPX-87H ion exchange column (Bio-Rad, Hercules, CA) as previously described (Peng et al. 2012). The biomass concentration, the biomass yield, and the specific consumption or production rates for xylose, xylitol, glycerol, acetate, and ethanol, as well as carbon recovery, were calculated as previously described (Peng et al. 2012).
The biomass was determined by measuring the optical density of the culture at 600 nm using a spectrophotometer (Eppendorf AG, 22331 Hamburg, Germany) and the dry weight. Nitrocellulose filter membranes (0.45 nm; Sartorius, Göttingen, Germany) were pre-dried at 105 °C until a constant weight was achieved. Then, 5 mL of the cell culture was filtered and washed with three volumes of distilled water and dried at 105 °C. The weight differences of the filter membranes with and without cells were determined as the dry weight of the biomass.
Gene expression profile analysis by microarray
The cells were cultured in minimal medium with 10 g l−1 glucose and 20 g l−1 xylose and were then collected during the exponential phase (OD600 = 0.8) for RNA extraction. The total RNA was isolated using Trizol reagent (Takara, Tokyo, Japan) and purified using a NucleoSpin® Extract II Kit (Machery-Nagel Corp., Dueren, Germany). JINGXIN cDNA amplification and labeling kits (CapitalBio Corp., Beijing, China) and a two-channel 6K yeast genome array (CapitalBio Corp.) were employed for cy3 or cy5 labeling of the cDNA and microarray hybridization (Zhang et al. 2002). The array images were scanned using a LuxScan10KA Microarray Scanner (CapitalBio Corp.) and analyzed with LuxScan 3.0 software (CapitalBio Corp.). Space- and intensity-dependent normalization was employed based on a locally weighted scatter plot smoothing regression program (Yang et al. 2002). Fold changes in gene transcription ≥2 were considered significant, whereas changes ≥1.5 were also considered in the analysis. KegArray (http://www.genome.jp/kegg/expression/) was used to map the gene expression data to KEGG BRITE database for primary functional classification. The gene annotation information was based on the Saccharomyces Genome Database (http://www.yeastgenome.org/). For gene ontology (GO) analysis and pathway analysis, genes with changes in transcription level ≥2 were chosen, and the Molecule Annotation System (Küfer et al. 2001) (CapitalBio Corp.) was used.
Quantitative real-time PCR
The cDNA was reverse-transcribed from the mRNA using random oligonucleotides and a Reverse Transcription System kit (CapitalBio Corp.). qPCR analyses were performed using a SYBR Green PCR Master Mix (Toyobo, Osaka, Japan) following the manufacturer’s operation manual. ACT1 was used as the normalization standard. All data were the average of triplicate values.
Plasmid copy number determination
Cells were cultured in minimal medium and harvested at OD600 2.5. The TE buffer (10 mM Tris–Cl, pH 7.5, 1 mM EDTA) resuspended cells were broken by the FastPrep cell homogenizer (Thermo Savant, USA) as described above and the protein was removed by phenol chloroform method. The total DNA was precipitated by ethanol and resuspend by water. The total DNA was used to analyze the plasmid copy number by qPCR as described before (Lee et al. 2006). The specific primers (Table S1 in the ESM) for gene Pi-xylA exist in the plasmid only and the reference gene ACT1 exist in yeast chromosome only were employed for the analysis. The amplification efficiency ratio of Pi-xylA and ACT1 is close proximity to 1. The plasmid copy number was calculated according to the equation of plasmid copy number = 2CT(act1)/2CT(xylA).
Fluorescence microscopy
The cells were cultured until the late exponential phase in minimal medium containing 20 g l−1glucose and 20 g l−1 xylose at 30 °C. The cells were then fixed and stained with Calcofluor (Fluorescent Brightener 28, Sigma, St. Louis, MO) as previously described (Pringle 1991).
Results
Optimization of xylose metabolism via adaptive evolution
The recombinant plasmid pJX5 containing the Pi-xylA gene was introduced into the xylose downstream metabolic pathway-enhanced strain BSPC042 (XK, gre3::PPP, and cox4Δ), wherein xylulokinase and four enzymes (RPE1, RKI1, TAL1, and TKL1) of the nonoxidative pentose phosphate pathway were overexpressed, the aldose reductase gene GRE3 was deleted and the electron transfer chain was disrupted by deleting the COX4 gene (Peng et al. 2012). The resulting strain BSPC095 (Pi-XI, XK, gre3::PPP, and cox4Δ) showed (0.47 ± 0.00) U mg−1 protein of XI activity, but no growth on xylose plates. However, weak growth was observed in liquid xylose minimal medium after 10 days of incubation.
Then, the adaptive evolution of BSPC095 was performed by sequentially transferring the culture into fresh xylose minimal medium during the early stationary phase. The cells showed gradually improved growth after more than 1,000 h of adaptive incubation on xylose. The doubling time was shortened from ∼80 to 5 h (Fig. S1 in the ESM). The fastest growing colony of BSPX013 (Pi-XI, XK, gre3::PPP, cox4Δ, and AE) in xylose, with a μ max of 0.11 ± 0.00 h−1, was selected. Although no base changes were found in the whole isomerase expression cassette (TEF1p-Pi-xylA-PGK1t) of the pJX5 plasmid recovered from the evolved BSPX013 strain, the XI activity in BSPX013 was surprisingly increased to 1.02 U ± 0.27 U mg−1, which is approximately 2-fold higher than that of its parental strain BSPC095 (0.47 U ± 0.00 U mg−1).
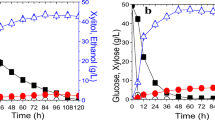
The results of the glucose–xylose cofermentation (Fig. 2; Table 2) revealed that the adaptive evolution process significantly increases the xylose fermentation capacity. The xylose-specific consumption rate of the evolved strain BSPX013 was 2.5-fold that of PC095 during glucose–xylose cofermentation phase and 8.5-fold that of BSPC095 during the xylose fermentation phase. The reference strain only utilized 3.68 g l−1 xylose within 120 h, whereas BSPX013 consumed 17.83 g l−1 xylose within 72 h. The consumed xylose was mainly converted into ethanol, and the ethanol yield was 0.43 g g−1 of consumed total sugar. However, the evolved strain required an additional 12 h to consume glucose completely (Fig. 2b).

Oxygen-limited shake flask fermentation of a glucose–xylose mixture by a BSPC095 (Pi-XI, XK, gre3::PPP, and cox4Δ) and b the evolved strain BSPX013 (Pi-XI, XK, gre3::PPP, cox4Δ, and AE). Black square, glucose; black diamond, xylose; black up-pointing triangle, xylitol; black circle, ethanol; error marks, glycerol; and solid line, OD600. Fermentation was performed at 30 °C. Then, 20 g l−1 glucose and 20 g l−1 xylose were used as the carbon source. One unit at an OD600 was equal to 0.23 g l−1 of BSPC095 biomass and 0.26 g l−1 of BSPX013 biomass. The glucose–xylose cofermentation phase of BSPC095 and of BSPX013 lasted 12 and 24 h, respectively
The potential effect from plasmid change was studied. The plasmid copy numbers relative to the chromosomal gene ACT1 in BSPC095 and BSPX013 are similar, which are 7.2 ± 0.1 and 6.5 ± 0.4, respectively. Moreover, the strain obtained by transforming the plasmid re-isolated from the evolved strain BSPX013 into strain BSPC042 did not show better growth on xylose than strain BSPC095, and no obvious difference on xylose metabolism were observed between the strain BSPX013 and its derivate strain, which were obtained by transforming the plasmid removed BSPX013 by original plasmid pJX5.
Transcriptional profile of adaptive evolution strain
To investigate the possible genetic factors that improved yeast xylose fermentation, the global transcription profile of the cells cultivated with glucose and xylose was analyzed. The data from the microarray were confirmed using qPCR (Table 3).
Compared with BSPC095 (Pi-XI, XK, gre3::PPP, and cox4Δ), the evolved strain BSPX013 (Pi-XI, XK, gre3::PPP, cox4Δ, and AE) showed that 172 genes were upregulated by >1.5-fold and 59 genes by >2-fold, whereas 107 genes were downregulated by >1.5-fold and 45 genes by >2-fold. The GO analysis enriched proteins mainly involved in response to stress (GO:0006950), glycogen biosynthesis (GO:0005978), trehalose biosynthesis (GO:0005992), cellular response to nitrogen starvation (GO:0006995), nitrogen compound metabolism (GO:0006807), cell wall organization and biogenesis (GO:0007047), amino acid transport (GO:0006865), etc.
The transcription levels of most genes in the glycolysis and pentose phosphate pathway involved in xylose fermentation did not change significantly, except for genes encoding 6-phosphofructo-2-kinase (PFK27) and pyruvate decarboxylase isozyme (PDC6), which increased by 1.69- and 1.75-fold, respectively (Fig. 3). The expression of glucokinase genes, HXK1 and EMI1, decreased by >2-fold. GDH2 (which encodes NAD+-dependent glutamate dehydrogenase) and GLN1 (glutamine synthetase), which encoded proteins involved in glutamate synthesis, were upregulated by 12.5- and 1.89-fold, respectively. The trehalose synthetic genes TSL1, TPS2, and TPS3 were downregulated (2.6-, 1.9-, and 1.9-fold, respectively) similar to the glycogen synthesis genes GLG1, GSY1, GSY2, and GLC3, which were downregulated 2.2-, 5.9-, 1.6-, and 2.2-fold, respectively. The gene encoding transcription factor Yak1p (serine-threonine protein kinase), which regulates trehalose and glycogen synthesis by activating Msn2p/Msn4p (Busti et al. 2010; Lee et al. 2008), was reduced by 1.96-fold. Additional information regarding the changes in gene expression is listed in Table S2 in the ESM.

Transcriptional changes in the genes involved in metabolic pathways were compared between the evolved strain BSPX013 (Pi-XI, XK, gre3::PPP, cox4Δ, and AE) and BSPC095 (Pi-XI, XK, gre3::PPP, and cox4Δ). The cells were cultured in minimal medium with 10 g l−1 glucose and 20 g l−1 xylose and then collected during the exponential growth phase (OD600 = 0.8)
The effect of several genes involved in xylose metabolism in different engineered strains
Seven interesting genes were selected basing transcriptional analysis to study their effect on xylose metabolism. The central carbon metabolism genes PFK27 and PDC6, the oligopeptide transporter gene OPT2, and a cell wall-localized protein gene YLR040C were overexpressed in serial strains that contained various modified pathways. They are BSPC016 (XR, XDH, and XK), BSPC112 (Pi-XI and XK), BSPC212 (XR, XDH, XK, and gre3::PPP), BSPC312 (Pi-XI, XK, and gre3::PPP), BSPX095 (Pi-XI, XK, gre3::PPP, and cox4Δ), and BSPX013 (Pi-XI, XK, gre3::PPP, and cox4Δ, AE). The overexpression of these genes was confirmed by RT-qPCR (Table 3). In addition, the alkaline phosphatase gene PHO13, the high-affinity inorganic phosphate transporter gene PHO84, and the cell wall mannoprotein gene CWP1, which relative transcriptional levels in the evolved strain BSPX013 were 1.25-, 0.27-, and 0.15-fold of those in BSPX095, were disrupted in the same strains mentioned above.
The growth characteristic of the engineered strains on the xylose plates is shown in Fig. 4. The deletion or overexpression of the genes did not restore the growth of the respiratory-deficient strain BSPC095 (Pi-XI, XK, gre3::PPP, and cox4Δ) on xylose. The new genetic operation in the xylose metabolic pathways of the well-modified strains BSPC212 (XR, XDH, XK, and gre3::PPP), BSPC312 (Pi-XI, XK, and gre3::PPP), and BSPX013 (Pi-XI, XK, gre3::PPP, cox4Δ, AE) did not further improve the growth of the strains on the xylose plates. The deletion of the PHO84 gene arrested the growth of BSPC016 (XR, XDH, and XK) and BSPC112 (Pi-XI and XK) on xylose. This result reveals that a functional Pho84p is necessary for xylose utilization of the strains contained only the xylose metabolic upstream pathways and metabolizes xylose inefficiently. In BSPX013 (Pi-XI, XK, gre3::PPP, cox4Δ, and AE), the expression of PHO84 was downregulated by ∼3.7-fold, and the deletion of PHO84 gene did not affect the growth of strain on xylose. This suggested that the some unknown alteration compensated the function of Pho84p in BSPX013. PHO13 deletion enhanced the growth of BSPC016 and BSPC112 on xylose plate, especially the XR-XDH background strain BSPC016. This result confirmed the earlier conclusions that deletion of PHO13 gene can enhance the xylose utilizing of strains (Fujitomi et al. 2012; Van Vleet et al. 2008). However, deletion of PHO13 gene in BSPX013, who can efficiently metabolize xylose, weakened the growth of that strain. Deletion of CWP1 improved the growth of the XR-XDH background strain BSPC016. However, the opposite effect was observed in the Pi-XI background strain BSPC112. The other tested genes showed less effect on the cell growth. Hence, the effect of the tested genes on the different xylose-utilizing engineered strains was multifarious. Similar to the findings of Parachin et al. (2010), the effect was more significant in the strains that grew poorly on xylose. Therefore, the additional effects of these genes were further studied in the inefficient xylose metabolism strains. However, the results (Table S3 in the ESM) show that the BSPC112-derived (Pi-XI and XK) strains consumed too little xylose to evaluate. Hence, further discussion focused mainly on the BSPC016-derived (XR, XDH, and XK) strains.

The growth characteristics of the engineered strains on xylose plates. Samples of the serial 10-fold dilutions (4 μL) with an initial OD600 of 1 were spotted onto minimal medium plates with 20 g l−1 xylose as the sole carbon source and then cultured at 30 °C for 3 days
Effect on the specific activity of XR and XDH
The specific activity of XR (with both cofactors NADPH and NADH) and XDH in BSPC016 (XR, XDH, and XK)-derived strains show that PHO13 deletion and PFK27 and PDC6 overexpression slightly changes the enzymatic activity. The CWP1 deletion decreased the enzymatic activity (Table 4).
Xylose aerobic fermentation of the strains
The fermentation data using xylose as sole carbon source under aerobic condition are shown in Table 5. Based on the BSPC016 (XR, XDH, and XK) strain, PHO13 deletion positively affected the xylose utilization of the strain. The maximum specific growth rate (μ max) of the strain increased from 0.039 to 0.168 h−1. The xylose specific consumption rate increased from 0.156 to 0.203 g g−1 biomass h−1. Then, 80 % xylose was consumed within 48 h by the pho13Δ strain, which produced ∼1.6 g l−1 ethanol and less xylitol and glycerol accumulated compared with its reference strain BSPC016. These results demonstrate that inactivation of Pho13p improves ethanol production from xylose. Deletion of CWP1 negatively affected the xylose utilization of the strain in the liquid culture, although the cells grew better on the xylose plate (Fig. 4). The μ max of the cwp1Δ strain was 0.024 h−1, which is lower than that of the control strain BSPC016 (0.039 h−1). The cwp1Δ strain consumed only 3.69 g l−1 xylose during fermentation, and no metabolite was detected. The overexpression of PFK27 and PDC6 did not affect the μ max of the strain in xylose. However, more biomass and less glycerol were produced by the PDC6-overexpressing strains. The glycerol production of the PDC6-overexpressing strain was only 24 % that of BSPC016.
Glucose–xylose cofermentation of the strains
The data for the glucose–xylose cofermentation under oxygen-limited conditions are listed in Table 6. Deletion of the PHO13 gene did not affect the growth and consumption ability of the strains on glucose. However, during the xylose consumption phase (after glucose was exhausted), the pho13Δ strain showed higher xylitol and acetate production rates but lower glycerol and ethanol production rates, compared with the reference strain BSPC016 (XR, XDH, and XK). Deletion of CWP1 decreased the xylose-metabolizing ability of the strain but increased their growth rate on glucose. The μ max of the cwp1Δ strain was 35 % higher than that of the reference strain BSPC016 (0.383 vs. 0.284 h−1) during glucose–xylose cofermentation. Therefore, the biomass and ethanol yield of the cwp1Δ strain were higher than those of BSPC016 (0.116 vs. 0.080 and 0.384 vs. 0.370 g g−1 total sugar). However, the lower specific consumption and production rate of the cwp1Δ strain during the xylose consumption phase indicates that deletion of CWP1 decreases the xylose-metabolizing ability of the strain even though sufficient biomass was obtained from glucose. The overexpression of PFK27 increased the specific production rate of acetate, which suggests that the acetaldehyde flux was increased. The PDC6-overexpressing strain consumed 14 % more xylose, both its ethanol and xylitol production rates increased and higher ethanol and xylitol yields were observed.
Deletion of CWP1 affects the cell wall structure
Mannoproteins contribute to form the external cell wall layer, which determines wall porosity (Klis et al. 2006). One of the two major mannoproteins that localizes specifically to the birth scars of daughter cells (Smits et al. 2006) is encoded by CWP1 (Zhang et al. 2008).
In the present work, deletion of CWP1 led to the expansion of cells; the cells of the cwp1Δ strain were thus larger than its reference strain BSPC016 (XR, XDH, and XK) (Fig. S2A, B in the ESM). The cell dry weight of the cwp1Δ strain increased to 0.212 g l−1 OD−1, whereas that of BSPC016 was 0.190 g l−1 OD−1. Furthermore, the cwp1Δ strain was more readily stained with Calcofluor white (Fig. S2C, D in the ESM), which suggests that the cell wall structure of the strain was changed by the CWP1 deletion.
At low xylose concentrations, such as at 4 g l−1, xylose uptake is controlled at the transport level (Gardonyi et al. 2003; Runquist et al. 2009). The μ max of the cwp1Δ strain in 4 g l−1 xylose was 0.008 h−1, which is only 57 % that of BSPC016 (Fig. S3 in the ESM). The xylose uptake of the strain might have decreased after the deletion of CWP1.
Discussion
Glucose and xylose co-utilization is one of the most important economic strategies in the production of second-generation bioethanol using S. cerevisiae. However, the xylose metabolic regulatory mechanism needs to be understood further to enhance the xylose-fermenting efficiency of recombinant yeast strain (Hahn-Hägerdal et al. 2007; Jeffries and Jin 2004; van Maris et al. 2006).
In the present work, a strain was engineered via the expression of the xylose isomerase gene of Piromyces sp. (Pi-xylA), the overexpression of xylulokinase (XKS1) and the nonoxidative PPP genes RPE1, RKI1, TAL1, and TKL1, and deletion of the nonspecific aldose reductase gene (GRE3). However, the strain with the aforementioned genotypes did not exhibit sufficiently fast and effective xylose fermentation, similar to that previously reported in another study (Kuyper et al. 2005b). Then, the COX4 gene that encodes subunit IV of cytochrome c oxidase was deleted to induce respiratory deficiency. The resulting strain BSPC095 (Pi-XI, XK, gre3::PPP, and cox4Δ) showed weakened growth on xylose. However, after prolonged adaptive evolution in xylose, which is an effective nonrational method of microorganism breeding, the evolved strain BSPX013 (Pi-XI, XK, gre3::PPP, cox4Δ, and AE) was obtained. BSPX013 showed a significantly improved capacity for fermenting xylose, and the fermentation was an anaerobic one since the respiratory was destroyed. The strain consumed 17.83 g l−1 xylose within 72 h; however, the glucose consumption rate was slightly decreased (Fig. 2). The increased enzymatic activity of xylose isomerase may have contributed to this improvement. However, no base change was found in the whole isomerase expression cassette (TEF1p-Pi-xylA-PGK1t), and the plasmid copy number was not increased after evolution. The mechanism underlying this unexplained increase needs to be elucidated. Then, the global transcriptional profile of the evolved strain was compared with the corresponding reference. The effect of several interesting genes was analyzed to investigate the mechanism for effective xylose fermentation.
The transport of xylose, which is regulated by hexose transporters in S. cerevisiae, is the first challenge in xylose utilization. In contrast to several previously reported evolved strain (Garcia Sanchez et al. 2010; Kuyper et al. 2005b), the expression of the hexose transporters (SNF3, HXT1-7, HXT9, HXT11, HXT14, and HXT16) was decreased at the transcriptional level after evolution. Except for the metabolic competition during increased xylose flux, the decreased transport capacity could account for the decrease in the glucose consumption rate.
Deletion of the cell wall mannoprotein gene CWP1 reportedly produces cell wall abnormalities and enhances cell permeability in yeast (Zhang et al. 2008), which reduces the resistance of the yeast cells to metals and chemicals (dos Santos and Sá-Correia 2011; van Bakel et al. 2005). In the present work, after adaptive evolution, CWP1 expression was downregulated by ∼6.7-fold. The cells of the CWP1-deleted strain were bigger and heavier than those of the reference strain because of the changes in the structure and permeability of the cell wall. However, deletion of CWP1 did not improve xylose utilization. It weakened the uptake of xylose, and decreased XR and XDH activity, which may account for the lower xylose utilization.
The PFK27 and PDC6 genes were upregulated by 1.69- and 1.75-fold, respectively, unlike most of the genes involved in glycolysis and the pentose phosphate pathway, which did not change significantly at the transcriptional level. PFK27 encodes the isozyme of 6-phosphofructo-2-kinase, which catalyzes the synthesis of fructose-2,6-bisphosphate (F2,6bP) from fructose-6-phosphate and ATP (Boles et al. 1996; Kretschmer and Fraenkel 1991). In yeast, F2,6bP is the activator of 6-phosphofructo-l-kinase and inhibitor of fructose-1,6-bisphosphatase in vitro. It does not increase the glycolytic flux but rather is concerned with the homeostasis of metabolite concentrations (Müller et al. 1997). In this study, there was no obvious effect of PFK27 overexpression to the ethanol production from glucose and xylose. PDC1, PDC5, and PDC6 encode three different isozymes of pyruvate decarboxylase (Hohmann 1991). Kim et al. (2012) confirmed that reduced mRNA levels of pyruvate decarboxylase (PDC1) may cause low ethanol production from xylose. The specific rate of glycerol formation in the pdcΔ mutant was double that of the wild-type strain (Nevoigt and Stahl 1996). Pdc6p is not expressed during glucose fermentation but is induced during growth on nonfermentable carbon sources (Hohmann 1991). This finding indicates that increased PDC6 expression may improve the metabolism of xylose, a nonfermentable carbon source (Jin et al. 2004). In this study, the positive effect of PDC6 overexpression was demonstrated, and the PDC6-overexpressing strain consumed more xylose and produced less glycerol compared with the reference strain.
The expression of the genes GDH2 and GLN1, which are involved in glutamate synthesis, were significantly upregulated in the evolved strain BSPX013. This result is consistent with that of a previous study that reported that GDH2 and GLN1 overexpression improves xylose utilization and ethanol production (Roca et al. 2003; Xiong et al. 2011). The increased GDH2 and GLN1 expression might contribute to the increased xylose fermentation in BSPX013.
Several “omics” studies have suggested a relationship between the stress response and xylose metabolism (Wenger et al. 2010; Wohlbach et al. 2011). In the evolved strain, the YAK1 transcription level was reduced by 1.96-fold. YAK1 encodes serine–threonine protein kinase, which phosphorylates and activates the transcriptional activators Msn2p/Msn4p (Lee et al. 2008). After adaptive evolution, lower Msn2p/Msn4p (∼70 % of reference strain) expression levels were detected. Stress response genes, such as the trehalose and glycogen synthetic genes, were consequently downregulated. Trehalose and glycogen accumulate under carbon starvation, nitrogen limitation, and certain toxic stress conditions (Parrou et al. 1999). The downregulation of the trehalose and glycogen synthetic genes TSL1, TPS2, TPS3, and GLC3 was also observed in xylose-cultured strain (Salusjärvi et al. 2006). In addition, the ethanol yield of TPS1 and TPS2 (trehalose 6-phosphate synthase genes) knockout S. cerevisiae strains from xylulose increased by 20 % (Eliasson et al. 2000a). The decrease in trehalose and glycogen synthesis might be another reason for the increased xylose utilization in the evolved strain.
Deletion of the PHO13 gene, which encodes a protein that has both alkaline phosphatase activity specific for p-nitrophenyl phosphate and protein phosphatase activity, enhances the growth of S. cerevisiae on xylose and its ethanol production from xylose (Fujitomi et al. 2012; Van Vleet et al. 2008). Deletion of PHO13 may alter the redox levels because increased acetate levels and decreased glycerol levels were observed during xylose fermentation (Van Vleet et al. 2008). This phenomenon was also confirmed by the results of the glucose and xylose fermentation (Tables 5 and 6) in this work. However, the transcription of PHO13 was upregulated by 1.25-fold in the evolved strain BSPX013 and the deletion of PHO13 in the evolved strain sharply weakened the growth of BSPX013 on xylose plate (Fig. 4). The most important differences of BSPX013 to other strains is the destroyed respiration. These results suggest that the positive effect of PHO13 deletion to xylose fermentation might be depended on cellular respiration.
Using the adapted evolution strategy, the xylose fermentation ability of an engineered Pi-xylA-containing strain was significantly improved. The increased xylose isomerase activity, the upregulated expression of genes involved in glycolysis and glutamate synthesis, as well as the downregulated expression of trehalose and glycogen synthetic genes might have contributed to the improvement.
References
Boles E, Gohlmann HW, Zimmermann FK (1996) Cloning of a second gene encoding 5-phosphofructo-2-kinase in yeast, and characterization of mutant strains without fructose-2,6-bisphosphate. Mol Microbiol 20(1):65–76
Busti S, Coccetti P, Alberghina L, Vanoni M (2010) Glucose signaling-mediated coordination of cell growth and cell cycle in Saccharomyces cerevisiae. Sensors 10(6):6195–6240
dos Santos SC, Sá-Correia I (2011) A genome-wide screen identifies yeast genes required for protection against or enhanced cytotoxicity of the antimalarial drug quinine. Mol Genet Genomics 286(5–6):333–346
Eliasson A, Boles E, Johansson B, Osterberg M, Thevelein JM, Spencer-Martins I, Juhnke H, Hahn-Hägerdal B (2000a) Xylulose fermentation by mutant and wild-type strains of Zygosaccharomyces and Saccharomyces cerevisiae. Appl Microbiol Biotechnol 53(4):376–382
Eliasson A, Christensson C, Wahlbom CF, Hahn-Hägerdal B (2000b) Anaerobic xylose fermentation by recombinant Saccharomyces cerevisiae carrying XYL1, XYL2, and XKS1 in mineral medium chemostat cultures. Appl Environ Microbiol 66(8):3381–3386
Entian KD, Kötter P (1998) Yeast mutant and plasmid collections. In: Brown AJP, Tuite MF (eds) Methods in microbiology, volume 26. Academic, San Diego, pp 431–449
Fujitomi K, Sanda T, Hasunuma T, Kondo A (2012) Deletion of the PHO13 gene in Saccharomyces cerevisiae improves ethanol production from lignocellulosic hydrolysate in the presence of acetic and formic acids, and furfural. Bioresour Technol 111:161–166
Garcia Sanchez R, Karhumaa K, Fonseca C, Sanchez Nogue V, Almeida JR, Larsson CU, Bengtsson O, Bettiga M, Hahn-Hägerdal B, Gorwa-Grauslund MF (2010) Improved xylose and arabinose utilization by an industrial recombinant Saccharomyces cerevisiae strain using evolutionary engineering. Biotechnol Biofuels 3:13
Gardonyi M, Jeppsson M, Lidén G, Gorwa-Grauslund MF, Hahn-Hägerdal B (2003) Control of xylose consumption by xylose transport in recombinant Saccharomyces cerevisiae. Biotechnol Bioeng 82(7):818–824
Gietz RD, Akio S (1988) New yeast-Escherichia coli shuttle vectors constructed with in vitro mutagenized yeast genes lacking six-base pair restriction sites. Gene 74(2):527–534
Hahn-Hägerdal B, Karhumaa K, Fonseca C, Spencer-Martins I, Gorwa-Grauslund MF (2007) Towards industrial pentose-fermenting yeast strains. Appl Microbiol Biotechnol 74(5):937–953
Hohmann S (1991) Characterization of PDC6, a third structural gene for pyruvate decarboxylase in Saccharomyces cerevisiae. J Bacteriol 173(24):7963–7969
Jeffries TW, Jin YS (2004) Metabolic engineering for improved fermentation of pentoses by yeasts. Appl Microbiol Biotechnol 63(5):495–509
Jin YS, Laplaza JM, Jeffries TW (2004) Saccharomyces cerevisiae engineered for xylose metabolism exhibits a respiratory response. Appl Environ Microbiol 70(11):6816–6825
Karhumaa K, Garcia Sanchez R, Hahn-Hägerdal B, Gorwa-Grauslund MF (2007) Comparison of the xylose reductase-xylitol dehydrogenase and the xylose isomerase pathways for xylose fermentation by recombinant Saccharomyces cerevisiae. Microb Cell Factories 6:5
Kersters-Hilderson H, Callens M, Van Opstal O, Vangrysperre W, De Bruyne CK (1987) Kinetic characterization of d-xylose isomerases by enzymatic assays using d-sorbitol dehydrogenase. Enzyme Microb Technol 9(3):145–148
Kim DM, Choi SH, Ko BS, Jeong GY, Jang HB, Han JG, Jeong KH, Lee HY, Won Y, Kim IC (2012) Reduction of PDC1 expression in S. cerevisiae with xylose isomerase on xylose medium. Bioprocess Biosyst Eng 35(1–2):183–189
Klis FM, Boorsma A, De Groot PW (2006) Cell wall construction in Saccharomyces cerevisiae. Yeast 23(3):185–202
Kretschmer M, Fraenkel DG (1991) Yeast 6-phosphofructo-2-kinase: sequence and mutant. Biochemistry 30(44):10663–10672
Küfer R, Thamasett S, Volkmer B, Hautmann RE, Gschwend JE (2001) New-generation lithotripters for treatment of patients with implantable cardioverter defibrillator: experimental approach and review of literature. J Endourol 15(5):479–484
Kuyper M, Winkler AA, van Dijken JP, Pronk JT (2004) Minimal metabolic engineering of Saccharomyces cerevisiae for efficient anaerobic xylose fermentation: a proof of principle. FEMS Yeast Res 4(6):655–664
Kuyper M, Hartog MM, Toirkens MJ, Almering MJ, Winkler AA, van Dijken JP, Pronk JT (2005a) Metabolic engineering of a xylose-isomerase-expressing Saccharomyces cerevisiae strain for rapid anaerobic xylose fermentation. FEMS Yeast Res 5(4–5):399–409
Kuyper M, Toirkens MJ, Diderich JA, Winkler AA, van Dijken JP, Pronk JT (2005b) Evolutionary engineering of mixed-sugar utilization by a xylose-fermenting Saccharomyces cerevisiae strain. FEMS Yeast Res 5(10):925–934
Lee C, Kim J, Shin SG, Hwang S (2006) Absolute and relative QPCR quantification of plasmid copy number in Escherichia coli. J Biotechnol 123:273–280
Lee P, Cho BR, Joo HS, Hahn JS (2008) Yeast Yak1 kinase, a bridge between PKA and stress-responsive transcription factors, Hsf1 and Msn2/Msn4. Mol Microbiol 70(4):882–895
Liu K, Lin X, Yue J, Li X, Fang X, Zhu M, Lin J, Qu Y, Xiao L (2010) High concentration ethanol production from corncob residues by fed-batch strategy. Bioresour Technol 101(13):4952–4958
Müller S, Zimmermann FK, Boles E (1997) Mutant studies of phosphofructo-2-kinases do not reveal an essential role of fructose-2,6-bisphosphate in the regulation of carbon fluxes in yeast cells. Microbiology 143(Pt 9):3055–3061
Nevoigt E, Stahl U (1996) Reduced pyruvate decarboxylase and increased glycerol-3-phosphate dehydrogenase NAD+ levels enhance glycerol production in Saccharomyces cerevisiae. Yeast 12(13):1331–1337
Parachin NS, Bengtsson O, Hahn-Hägerdal B, Gorwa-Grauslund MF (2010) The deletion of YLR042c improves ethanolic xylose fermentation by recombinant Saccharomyces cerevisiae. Yeast 27(9):741–751
Parrou JL, Enjalbert B, Plourde L, Bauche A, Gonzalez B, Francois J (1999) Dynamic responses of reserve carbohydrate metabolism under carbon and nitrogen limitations in Saccharomyces cerevisiae. Yeast 15(3):191–203
Peng B, Chen X, Shen Y, Bao X (2011) Effect of controlled overexpression of xylulokinase by different promoters on xylose metabolism in Saccharomyces cerevisiae. Wei Sheng Wu Xue Bao 51(7):914–922
Peng B, Shen Y, Li X, Chen X, Hou J, Bao X (2012) Improvement of xylose fermentation in respiratory-deficient xylose-fermenting Saccharomyces cerevisiae. Metab Eng 14(1):9–18
Pringle JR (1991) Staining of bud scars and other cell wall chitin with calcofluor. Methods Enzymol 194:732–735
Roca C, Nielsen J, Olsson L (2003) Metabolic engineering of ammonium assimilation in xylose-fermenting Saccharomyces cerevisiae improves ethanol production. Appl Environ Microbiol 69(8):4732–4736
Runquist D, Fonseca C, Rådström P, Spencer-Martins I, Hahn-Hägerdal B (2009) Expression of the Gxf1 transporter from Candida intermedia improves fermentation performance in recombinant xylose-utilizing Saccharomyces cerevisiae. Appl Microbiol Biotechnol 82(1):123–130
Salusjärvi L, Pitkänen JP, Aristidou A, Ruohonen L, Penttilä M (2006) Transcription analysis of recombinant Saccharomyces cerevisiae reveals novel responses to xylose. Appl Biochem Biotechnol 128(3):237–261
Sauer U (2001) Evolutionary engineering of industrially important microbial phenotypes. Adv Biochem Eng Biotechnol 73:129–169
Smits GJ, Schenkman LR, Brul S, Pringle JR, Klis FM (2006) Role of cell cycle-regulated expression in the localized incorporation of cell wall proteins in yeast. Mol Biol Cell 17(7):3267–3280
Sonderegger M, Sauer U (2003) Evolutionary engineering of Saccharomyces cerevisiae for anaerobic growth on xylose. Appl Environ Microbiol 69(4):1990–1998
Sonderegger M, Jeppsson M, Hahn-Hägerdal B, Sauer U (2004) Molecular basis for anaerobic growth of Saccharomyces cerevisiae on xylose, investigated by global gene expression and metabolic flux analysis. Appl Environ Microbiol 70(4):2307–2317
van Bakel H, Strengman E, Wijmenga C, Holstege FC (2005) Gene expression profiling and phenotype analyses of S. cerevisiae in response to changing copper reveals six genes with new roles in copper and iron metabolism. Physiol Genomics 22(3):356–367
van Maris AJ, Abbott DA, Bellissimi E, van den Brink J, Kuyper M, Luttik MA, Wisselink HW, Scheffers WA, van Dijken JP, Pronk JT (2006) Alcoholic fermentation of carbon sources in biomass hydrolysates by Saccharomyces cerevisiae: current status. Antonie Van Leeuwenhoek 90(4):391–418
van Vleet JH, Jeffries TW, Olsson L (2008) Deleting the para-nitrophenyl phosphatase (pNPPase), PHO13, in recombinant Saccharomyces cerevisiae improves growth and ethanol production on d-xylose. Metab Eng 10(6):360–369
Walfridsson M, Anderlund M, Bao X, Hahn-Hägerdal B (1997) Expression of different levels of enzymes from the Pichia stipitis XYL1 and XYL2 genes in Saccharomyces cerevisiae and its effects on product formation during xylose utilisation. Appl Microbiol Biotechnol 48(2):218–224
Wang Y, Shi WL, Liu XY, Shen Y, Bao XM, Bai FW, Qu YB (2004) Establishment of a xylose metabolic pathway in an industrial strain of Saccharomyces cerevisiae. Biotechnol Lett 26(11):885–890
Wenger JW, Schwartz K, Sherlock G (2010) Bulk segregant analysis by high-throughput sequencing reveals a novel xylose utilization gene from Saccharomyces cerevisiae. PLoS Genet 6(5):e1000942
Wisselink HW, Toirkens MJ, Wu Q, Pronk JT, van Maris AJ (2009) Novel evolutionary engineering approach for accelerated utilization of glucose, xylose, and arabinose mixtures by engineered Saccharomyces cerevisiae strains. Appl Environ Microbiol 75(4):907–914
Wohlbach DJ, Kuo A, Sato TK, Potts KM, Salamov AA, Labutti KM, Sun H, Clum A, Pangilinan JL, Lindquist EA, Lucas S, Lapidus A, Jin M, Gunawan C, Balan V, Dale BE, Jeffries TW, Zinkel R, Barry KW, Grigoriev IV, Gasch AP (2011) Comparative genomics of xylose-fermenting fungi for enhanced biofuel production. Proc Natl Acad Sci U S A 108(32):13212–13217
Xiong M, Chen G, Barford J (2011) Alteration of xylose reductase coenzyme preference to improve ethanol production by Saccharomyces cerevisiae from high xylose concentrations. Bioresour Technol 102(19):9206–9215
Yang YH, Dudoit S, Luu P, Lin DM, Peng V, Ngai J, Speed TP (2002) Normalization for cDNA microarray data: a robust composite method addressing single and multiple slide systematic variation. Nucleic Acids Res 30(4):e15
Zhang L, Zhang Y, Zhou Y, An S, Cheng J (2002) Response of gene expression in Saccharomyces cerevisiae to amphotericin B and nystatin measured by microarrays. J Antimicrob Chemother 49(6):905–915
Zhang M, Liang Y, Zhang X, Xu Y, Dai H, Xiao W (2008) Deletion of yeast CWP genes enhances cell permeability to genotoxic agents. Toxicol Sci 103(1):68–76
Acknowledgments
This work was supported by the National Key Basic Research Program (2011CB707405), the International S&T Cooperation Program of China (2010DFA32560), and the National Natural Science Foundation of China (30970091 and 31070096).We would like to thank Dr. Peter Kötter of the Johann Wolfgang Goethe-University in Frankfurt for supplying the CEN.PK strains.
Author information
Authors and Affiliations
Corresponding author
Additional information
Yu Shen and Xiao Chen are co-first authors.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOCX 314 kb)
Rights and permissions
About this article
Cite this article
Shen, Y., Chen, X., Peng, B. et al. An efficient xylose-fermenting recombinant Saccharomyces cerevisiae strain obtained through adaptive evolution and its global transcription profile. Appl Microbiol Biotechnol 96, 1079–1091 (2012). https://doi.org/10.1007/s00253-012-4418-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-012-4418-0