
Abstract
Selenium nanoparticles (Se NPs) have been recognized as promising materials for biomedical applications. To prepare Se NPs which contained cancer targeting methods and to clarify the cellular localization and cytotoxicity mechanisms of these Se NPs against cancer cells, folic acid protected/modified selenium nanoparticles (FA–Se NPs) were first prepared by a one-step method. Some morphologic and spectroscopic methods were obtained to prove the successfully formation of FA–Se NPs while free folate competitive inhibition assay, microscope, and several biological methods were used to determine the in vitro uptake, subcellular localization, and cytotoxicity mechanism of FA–Se NPs in MCF-7 cells. The results indicated that the 70-nm FA–Se NPs were internalized by MCF-7 cells through folate receptor-mediated endocytosis and targeted to mitochondria located regions through endocytic vesicles transporting. Then, the FA–Se NPs entered into mitochondria; triggered the mitochondria-dependent apoptosis of MCF-7 cells which involved oxidative stress, Ca2+ stress changes, and mitochondrial dysfunction; and finally caused the damage of mitochondria. FA–Se NPs released from broken mitochondria were transported into nucleus and further into nucleolus which then induced MCF-7 cell cycle arrest. In addition, FA–Se NPs could induce cytoskeleton disorganization and induce MCF-7 cell membrane morphology alterations. These results collectively suggested that FA–Se NPs could be served as potential therapeutic agents and organelle-targeted drug carriers in cancer therapy.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Cancer is a great threat to public health, with more than 3.2 million new cases being diagnosed and more than 1.7 million patients lose their lives each year just in Europe (Milosavljevic et al. 2002). Most of cancer patients are suffering from the toxicity and side effect of chemotherapy (Stéphanie et al. 2011). The development of new drugs with high efficacy and high selectivity to cancer cells is more and more important.
The trace mineral selenium (Se) is an essential nutrient of fundamental importance to human biology and involves a lot physiological functions. It has many important health effects particularly in relation to the immune response and cancer prevention activity, which are almost certainly linked to Se-dependent enzyme (Rayman 2000) or selenoprotein functions (Rock and Moos 2009). The risk of many cancers such as prostate, lung, and colon cancers is decreased with Se supplementation (Fang et al. 2010; Reid et al. 2002). Simultaneously, the side effect of radiotherapy can also be reduced by Se supplement (Tabassum et al. 2010) which both indicate the potential application of Se compounds in cancer treatment. But Se is also toxic at a level not much higher than the beneficial requirement (Koller and Exon 1986). The dose and form of Se also remarkably affect their toxicity and anticancer activity (Abdulah et al. 2005; El-Bayoumy and Sinha 2004). Nano-Se, a red nanomaterial, is an excellent antioxidant with reduced risk of Se toxicity and has many applications in life science (Wang et al. 2007). As an antioxidant, Nano-Se can be used as a protective agent against chemotherapy such as the protection effect in preventing the renal injury induced by cisplatin (Li et al. 2011a). Moreover, Nano-Se also has wonderful anti-cancer activity to various cancers (Huang et al. 2010; Kong et al. 2011; Zheng et al. 2011). The synthetic methods of Se nanoparticles (Se NPs) are also extensively developed in recent years (Li et al. 2010; Xia 2007). But it is unfortunate that there is still no cancer targeting method. In addition, the interaction of Se NPs with tumor cells, their uptake mechanism, and the relationship of subcellular localization and cytotoxicity are also poorly understood.
Folic acid (FA), a kind of vitamin which solves the expensive and short life issues of antibody and oligopeptide as a target ligand, is studied extensively as an optimal targeting ligand for cell imaging and cancer treatment (Wang et al. 2010). Folate receptor (FR), known as a high affinity membrane folate-binding protein, is overexpressed in many tumors (Elnakat and Ratnam 2006). FA modified nanomaterials for specific targeting FR overexpressed cancers are also widely developed (Yoo and Park 2004; Zhang et al. 2007). Those methods can improve the uptake efficiency of nanomaterials in FR-positive cancer cells effectively and imply a wonderful prospect of FA linked drugs in cancer therapy (Sudimack and Lee 2000).
In the present work, we have synthesized folate molecules protected nanoparticles (FA–Se NP) by one-step method and characterized it using transmission electron microscope (TEM), dynamic light scattering (DLS), ultraviolet and visible (UV/VIS), and Fourier transform infrared spectra (FTIR). Free FA competitive inhibition assay was used to study the intracellular uptake of FA–Se NP by inductively coupled plasma mass spectrometry (ICP-MS). Cellular uptake and intracellular localization of FA–Se NP in MCF-7 cells was detected by TEM experiments. In the present study, we also tried to examine the cytotoxic mechanism of FA–Se NP in MCF7 cells. Our results suggest that the internalization and subcellular localization of FA–Se NPs play a critical role in their cytotoxicity to MCF-7 cells.
Materials and methods
Reagents, cell line, and cell culture
Sodium selenite, ascorbic acid, and FA/folate were both purchased from Sigma (USA). Fetal bovine serum, Dulbecco’s modified Eagle medium (DMEM), trypsin, and 3-(4, 5)-dimethylthiazo(-z-y1)-3,5-di-phenytetrazoliumromide (MTT) kit were obtained from Gibco (USA). Annexin V-FITC/PI apoptosis detection kit, cell cycle phase determination kit, rhodamine 123 kit, DCFH-DA kit, 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethyl-imidacarbocyanine iodide (JC-1) kit, 2-(4-amidinophenyl)-6-indolecarb-amidine dihydrochloride (DAPI) kit, and fluo-3 AM kit were purchased from Beyotime (China). All reagents used in the experiments were analytical grade. Ultrapure water used for all experiments was supplied by cascade RO water purification system from Pall (USA). The cord blood mesenchymal stem cell (CBMSC) and MCF-7 cell lines were purchased from Life Science Research Institute of the Cell Resource Center, Shanghai, China. Cells were cultured at 37 °C and 5 % CO2 in DMEM supplemented with 10 % fetal bovine serum, 100 U/mL penicillin, and 100 g/mL streptomycin.
Preparation of FA–Se NPs
A 2.6-mL folic acid solution (1 g/L) was mixed with 300 μL sodium selenite solution (0.05 M) in a 10-mL beaker. A 2.4-mL aqueous ascorbic acid solution (0.05 M) was slowly added into the mixtures under sonication condition. After 15 min reaction under sonication, some sodium hydroxide (0.5 M) was added into the above solutions until it came to be clear. At last, the solutions were dialyzed (MWCO 8,000–14,000) against ultrapure water at 4 °C until no Se was detected in the outer solutions as determined by inductively coupled plasma-atomic emission spectrometry (ICP-AES) analysis. The dialyzed FA–Se NPs was serially passed through 0.22-μm pore-size filters to get rid of bacteria. The Se concentration of the obtained FA–Se NPs solutions was also determined by ICP-AES analysis and was restored at 4 °C for subsequent experiments. The control Se NPs were prepared the same way as FA–Se NPs but without FA molecules as protective agent.
Characterization of FA–Se NPs
UV/VIS absorbance of as-prepared products was characterized by a scanning spectrophotometer (Varian, USA) with a 1-cm path length. All the spectra were subtracted background with baseline which is obtained from the absorption of purified water. The FTIR spectra of FA, FA–Se NPs, and control Se NPs were recorded with KBr pellets using a spectrometer (Bruker, Germany) at room temperature.
The FA–Se NPs solution was dropped onto the copper net and dried at room temperature for 12 h. TEM images were taken by using a transmission electron microscope (Philips Co, Holland) operating at an accelerating voltage of 200 kV. DLS was used to test size distribution and zeta potential of FA–Se NPs, which was performed at 25 °C (Malvern, UK), equipped with a 532-nm laser at a fixed scattering angle of 90°(n > 3).
ICP-MS analysis
Human MCF-7 breast cancer cells, which overexpress FR (Pan and Feng 2008), were chosen for the following experiments. The intracellular uptake of FA–Se NPs in MCF-7 cells was analyzed by inductively coupled plasma mass spectroscopy (Bruker, Germany) using a free FA competitive inhibition assay as described previously (Li et al. 2009). When free FA molecules were added into the cell culture medium together with FA-modified nanoparticles, there would be a competition between free FA molecules and FA molecules on the nanoparticles when binding to FR on the FR-positive cell surface. This competition would inhibit the intracellular uptake of FA modified nanoparticles through inhibiting the FR-mediated endocytosis. MCF-7 cells were plated at a density of 2 × 105 cells per well in six-well plates. After 24 h incubation, the medium was replaced with medium contained 2.5 μg/mL FA–Se NPs alone or with medium contained 2.5 μg/mL FA–Se NPs and 0.01, 0.05, 0.1, 0.2, 0.4, and 0.5 mM free FA molecules to incubate for 3 h, respectively. The treatment of samples for ICP-MS was also performed as reported (Ke et al. 2010). The medium was removed and the cells were then washed three times with ice-cold phosphate buffer solution (PBS) to remove the media residues. After lysed by trypsin and washed three times with PBS, the centrifuged cell pellets were dissolved in 37%HCl and were incubated for 1 h at 70 °C. The samples were diluted to 4 mL before the selenium concentration was determined by ICP-MS. Uptake of FA–Se NPs is expressed as the content of selenium per cell. Measurement for each treatment was repeated in triplicate.
TEM analysis for cell samples
The uptake process of FA–Se NPs into MCF-7 cells and the distribution of FA–Se NPs absorbed into MCF-7 cells were imaged by TEM (Philips Co, Holland). The cells were seeded at a density of 1 × 106 cells each well in a six-well plate and treated with 2.5 μg/mL FA–Se NPs 3, 6, 24, and 48 h, respectively. After harvesting, the cells were washed with PBS, fixed with 2.5 % glutaraldehyde, embedded in resin, and obtained thin section through microtome which were stained with osmic acids and imaged with TEM at last.
MTT assay
MTT assays were used to test the cell viability changes caused by FA–Se NPs. MCF-7 cells and CBMSC were seeded into 96-well plate with a density of 3 × 103 for 24 h and incubated with different concentration of FA–Se NPs for 48 h. MTT reagents (10 μL, 5 mg/mL PBS) were then added into each well for 4 h incubation, the medium was removed, and the cells were suspended in 150 μL DMSO for 15 min. A spectrophotometer (TECAN, Switzerland) was used to test absorbance with wavelength at 570 nm.
Determination of apoptosis rate and cell cycle distribution
To detect the apoptotic rate of FA–Se NPs treated MCF-7 cells, Annexin V-FITC/PI apoptosis detection kit was used according to the manufacturer’s instructions. After incubation with different concentrations of FA–Se NPs for 48 h, cells were harvested, washed with PBS, suspended in Annexin V binding buffer, and incubated with FITC-labeled Annexin V and PI for 15 min at room temperature in dark. Then, the samples were immediately analyzed by a flow cytometer (BD Inc, USA) with excitation wavelength at 488 nm.
After 48 h treatment with FA–Se NPs, the cells were harvested, washed with PBS, and fixed with 70 % ethanol overnight at 4 °C. The fixed cells were washed twice with PBS, treated with RNase A, stained with PI (50 μg/mL), and analyzed by a flow cytometer excitated by 488 nm.
Measurements of ROS and free Ca2+ levels
After incubation with different concentrations of FA–Se NPs for 24 h, cells were harvested, washed twice with PBS, and suspended in PBS. To detect the changes in ROS and free Ca2+ levels, the suspended MCF-7 cells were incubated with DCFH-DA and fluo-3 AM for 30 min in dark at room temperature, respectively. After collected and washed twice with PBS, the samples were measured by flow cytometer with excitation wavelengths both at 488 nm for DCFH-DA and fluo-3 AM.
Measurements of mitochondrial membrane potential disruption
The mitochondrial specific probe JC-1 was used following the instructions to confirm the disruption of mitochondrial membrane potential (MMP). After incubated with FA–Se NPs and washed with PBS, 1 mL DMEM medium and 1 mL JC-1 staining solution were added to incubate for 20 min. After incubation, the medium was removed, washed twice with JC-1 cleaning solution, and immediately examined under a confocal microscope with excitation wavelength set at 514 nm.
Rhodamine 123 was also used to detect the disruption of MMP and detected by flow cytometer. After treated with FA–Se NPs, the harvested and washed cells were incubated with rhodamine 123 for 30 min in dark at room temperature. Flow cytometer with excitation wavelengths at 488 nm was used to detect the fluorescence intensity of rhodamine 123 after the cells were collected and washed twice with PBS.
Caspase-3 and caspase-9 activity assay
Caspase-3 and caspase-9 activities were both determined using a similar fluorescence quantitative method as described previously (Patil et al. 2010). After incubation, cells were harvested by scraping and washed twice with PBS. The cell pellets were then suspended in cell lysis buffer and incubated for 15 min on ice. After centrifugation at 16,000×g for 15 min, the collected supernatants were immediately measured to obtain the protein concentration and caspase activity. The cell lysates were added into 96-well plates and then incubated with specific caspase-3 substrates (Ac-DEVD-pNA) and specific caspase-9 substrates (Ac-LEHD-pNA) for 2 h at 37 °C, respectively. Caspase-3 and caspase-9 activities were determined by fluorescence intensity with the excitation wavelength both set at 405 nm using a spectrophotometer.
Measurements of the organization and the amounts of F-actin
The untreated and FA–Se NPs treated MCF-7 cells were fixed by 4 % paraformaldehyde for 15 min and incubated with 1 μM rhodamine–phalloidin for 60 min in the dark at room temperature. After washed triple with PBS, cells were then incubated with 50 μM DAPI for 4 min and washed triple with PBS. A laser scanning confocal microscope (Carl Zeiss, Germany) was used to image the nuclear morphology and the organization of F-actin cytoskeleton, respectively.
The amounts of F-actin in FA–Se NPs untreated or treated MCF-7 cells were determined by flow cytometer with special staining of actin-tracker green. The harvested and washed cells were fixed by 4 % paraformaldehyde for 15 min, washed triple with PBS, incubated with actin-tracker green for 30 min, washed triple with PBS, and then suspended in PBS. At last, samples were immediately analyzed by a flow cytometer (BD Inc, USA) and excitated by 496 nm.
Atomic force microscope measurements
Atomic force microscope (AFM) (Veeco, USA) was used to obtain topographic images of FA–Se NPs treated or untreated MCF-7 cells in the contact mode. Organic contaminates of the silicon nitride tips used in all measurements were removed by ultraviolet irradiation. The curvature radius of the tips is less than 10 nm, with a force constant set at 10 mN/m and an oscillation frequency set at 255 kHz. Samples were fixed by paraformaldehyde solution and detected in air after dried at room temperature. Single-cell imaging was obtained from more than five cells, and each cell was scanned for three times.
Statistical analysis
Each experimental group was repeated at least three times. Statistical analysis was performed using Student’s t test. P < 0.05 was regarded as statistically significant.
Results
Characterization of FA–Se NPs
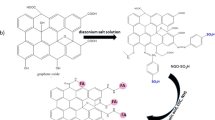
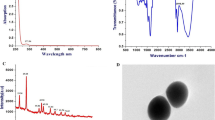
In this study, we introduced a new method to prepare FA–Se NPs by a redox system. Figure 1a showed the size distribution of FA–Se NPs obtained by TEM which indicated that the uniform spherical FA–Se NPs were obtained in an average diameter of 70 nm and was similar with the results obtained by DLS in Fig. 1b. The zeta potential of FA–Se NPs in aqueous solution was obtained by DLS experiments and showed a mean zeta potential of −45 mV (Fig. 1b).

Characterization of FA–Se NPs. a TEM images of FA–Se NPs with different ranges, the given bar in a and b are 500 and 200 nm, respectively. b DLS characterization of FA–Se NPs. a Size distribution and b zeta potential of FA–Se NPs in aqueous solution obtained by DLS. c Spectrum information of FA–Se NPs, a UV/VIS absorbance spectra of FA–Se NPs, FA, and Se NPs, respectively. b FIRT absorbance spectra of FA–Se NPs, free FA, and Se NPs, respectively. d The scheme for the formation process of FA–Se NPs
UV/VIS absorbance spectra and FIRT absorbance spectra were used to confirm the successful modification of Se NPs with FA molecules. Figure 1c showed the UV/VIS absorbance spectra of FA–Se NPs solution, free FA solution, and Se NPs solution, respectively. There was a clear peak at 279 nm in the spectra of free FA solution, and a similar peak was also found in the spectra of FA–Se NPs solution at 276 nm. But in the spectra of Se NPs solution, there was just one clear peak at 261 nm which was distinct from 279 nm. Similarly, there was a clear peak at 340 nm in the spectra of free FA solution which also expressed in the spectra of FA–Se NPs, while nearly no absorbance was observed at 340 nm in the spectra of Se NPs solution.
The FTIR spectra of FA–Se NPs we prepared showed two clear absorbance peaks at 1,694.2 and 1,604.1 cm−1 when the IR spectra of FA showed a similar absorbance peaks at 1,693.8 and 1,605.7 cm−1 (Fig. 1d). But there were no similar special absorbance peaks in the IR spectra of Se NPs which had just one clear peak at 1,623.8 cm−1 in this region. The disappearance of O–H broad absorbance peak between 2,500and 3,000 cm−1 in the FTIR spectra of FA–Se NPs indicated that the successful modification of FA molecules onto Se NPs was mostly due to carboxyl functionalities of FA molecules. These results indicated that FA–Se NPs were prepared successfully to obtain an average diameter of 70 nm and protected by FA molecules by carboxylation (shown as Fig. 1e).
Cellular uptake
The above characterizations demonstrated the successful modification of FA onto the surface of Se NPs. To further investigate the cellular uptake mechanism of FA–Se NPs in FR overexpressed cancer cells, we performed a free FA competitive inhibition experiments in MCF-7 cells. The results determined by ICP-MS were expressed as Se content per cell and given in Fig. 2a a. As expected, when additional free FA molecules were introduced, the uptake of FA–Se NPs decreased even at low FA concentrations. When more FA molecules were added, the cellular uptake of FA–Se NPs decreased again in a dose-dependent manner which suggested that additional FA molecules could inhibit the intracellular uptake of FA–Se NPs.

FR-mediated endocytosis and intracellular localization of FA–Se NPs in MCF-7 cells. a FR-mediated endocytosis of FA–Se NPs in MCF-7 cells. a Free FA competitive inhibition assay detected by ICP-MS. The cellular uptake of FA–Se NPs by MCF-7 cells was expressed here as Se content per cell. Data were the average of three independent experiments ± SD. b, c TEM images showed the endocytosis process of FA–Se NPs in MCF-7 cells. b The process of FA–Se NPs were released into cytoplasm from endocytic vesicles and then transported into the mitochondria. c The mitochondria damage induced by FA–Se NPs. d The FA–Se NPs released from broken mitochondria translocated into nucleus or nucleolus and induced the nucleolus structural damage. Scale bars in all these images were 1 μm. FA–Se NPs in MCF-7 cells were indicated by white thick arrow. White arrows in these images showed the locations of FA–Se NPs in MCF-7 cells. Letters N, NE, V, and M are abbreviations of nucleus, nucleolus, vesicle, and mitochondria, respectively
Recently, TEM has always been used to test the endocytosis process and intracellular distribution of nanoparticles in cells (Evans et al. 2011). It could give some significant internal information about the relationship between nanoparticles and cellular organelles such as mitochondria, lysosomes, and nucleus (Qiu et al. 2010). In order to corroborate the endocytosis process of FA–Se NPs, we checked the endocytosis process of the MCF-7 cells upon FA–Se NPs treatments using TEM. From Fig. 2a (b, c), we found that the FA–Se NPs in medium were bigger than 70 nm and had no definite shape or volume. This indicated that FA–Se NPs could form some amorphous aggregates with different sizes before entering into cells. Those aggregates can be enclosed by the plasma membrane of MCF-7 cells to form endocytic vesicles for intracellular transport.
Accumulation of FA–Se NPs into mitochondria and nucleus in MCF-7 cells
The cytotoxicity of nanoparticles is closely related to the cellular ultrastructure and intracellular trafficking (Wang et al. 2011a). TEM images can provide spatial and temporal information about the endocytosis process, trafficking process, and subcellular localization of FA–Se NPs in MCF-7 cells. The results in Fig. 2a had showed that FA–Se NPs were internalized by MCF-7 cells through FR-mediated endocytosis way. The endocytic vesicles formed during the endocytic process were used to transport FA–Se NPs into cells. After FA–Se NPs were transport further into the cell by endocytic vesicles, they were released into cytoplasm from endocytic vesicles after 3 h treatment (Fig. 2b a). After continuous incubation for 6 h, most FA–Se NPs were transported to mitochondria located region in cytoplasm (Fig. 2b b) and then began to enter into the mitochondria (Fig. 2b c). This implied that mitochondria were the main targeted organelle of FA–Se NPs in MCF-7 cells in the early period of time after been internalized. Before the entry of FA–Se NPs, there showed regular and clear mitochondrial crista structures in cross section of mitochondria (indicated by black thick arrows in Fig. 2b c) or intensive and intact structures in longitudinal sections of mitochondria (Indicated by black thick arrows in Fig. 2b a, b). This indicated that these mitochondria were under health physiological status with normal mitochondrial structures before FA–Se NPs were trafficked into them.
After treatment with FA–Se NPs for 24 h, the longitudinal sections of mitochondria presented characteristic vacuolation (indicated by black thick arrows in Fig. 2c c). The mitochondrial crista in cross section of mitochondria nearly all disappeared and even appeared breakages with fogged mitochondrial membrane after the entry of FA–Se NPs (indicated by black thick arrows in Fig. 2c b). These changes of mitochondrial structure indicated that the entry of FA–Se NPs into mitochondria could affect the structure and physiological status of mitochondria directly. Due to the damage of mitochondria, FA–Se NPs were released from the broken mitochondria into cytoplasm again. These released FA–Se NPs did not transport and distribute randomly in cells or get out of cells, but been trafficked into nucleus (Fig. 2d a). After incubated with FA–Se NPs for 48 h, FA–Se NPs were transported into cell nucleus and some FA–Se NPs even got into nucleolus. It is worth noting that there were some voids appeared in nucleolus after the entry of FA–Se NPs into nucleolus (indicated by black thick arrow in Fig. 2d b, c).
Anti-proliferative effect of FA–Se NPs
In order to determine if FA–Se NPs have selective killing effect on FR over expressed cancer cells, the viability to MCF-7 and CBMSC was assessed by MTT assays. The results shown in Fig. 3a revealed that FA–Se NPs strongly suppressed MCF-7 cell growth after 48 h treatment with a dose-dependent killing effect. The half maximal inhibitory concentration (IC50) of FA–Se NPs for MCF-7 cells was 2.47 μg/mL. As our expected, FA–Se NPs showed lower cytotoxicity toward normal CBMSC whose IC50 was 22.52 μg/mL, much higher than that of MCF-7 cells. These data suggested that FA–Se NPs could suppress the growth of MCF-7 cells effectively and selectively.

FA–Se NPs inhibit the growth of MCF-7 cells. a Viability of FA–Se NPs to MCF-7 and CBMSC for 48 h determined by MTT assay. b Cell cycle distribution of MCF-7 cells untreated or treated with FA–Se NPs for 48 h detected by flow cytometric analysis. Mean values ± standard deviation, N = 3. c Early apoptosis rate of MCF-7cells treated with different concentrations of FA–Se NPs for 48 h. Lower right (LR) quadrant represents percentage of early apoptotic cells, upper right (UR) quadrant represents percentage of late apoptotic cells
Apoptosis effects and cell cycle arrest induced by FA–Se NPs
Flow cytometric analysis was performed to further assess the apoptotic MCF-7 cells induced by FA–Se NPs. As seen in Fig. 3c, when treated with 5 μg/mL FA–Se NPs, the number of early apoptotic cells (12.74 %) was significant higher than that of control group (4.27 %). Additionally, the number of necrosis or late apoptotic MCF-7 cells was also much higher than that of control group. These results indicated that FA–Se NPs could induce apoptosis of MCF-7 cells.
Besides, the effect of FA–Se NPs on cell cycle distribution was also detected with PI staining and determined by flow cytometric. The average percentage of cells in S phase increased from 20.2 to 33.5, 39.1, 36.8, and 35.3 % after 0.5, 2.5, 5, and 10 μg/mL FA–Se NPs treatment, respectively(Fig. 3b), implying that FA–Se NPs treatment arrested the MCF-7 cell cycle in S phase.
Mitochondria-dependent apoptosis induced by FA–Se NPs
ROS were highly reactive molecules which generated from cellular metabolism as a byproduct. To determine if ROS were involved in FA–Se NPs induced apoptosis, we examined the induction of cellular ROS production in response to FA–Se NPs exposure by flow cytometry. The results (Fig. 4a, c) implied that mean fluorescence intensity (MFI) of FA–Se NPs treated cells ranged from 46.60 to 85.95 while the MFI of control group was just 30.63, showing the potential role of ROS in FA–Se NPs induced MCF-7 cell apoptosis.

Flow cytometric analysis of ROS and Ca2+ levels in designated concentrations FA–Se NPs treated MCF-7 cells. a Changes of ROS levels in FA–Se NPs treated MCF-7 cells. b Changes of Ca2+ concentration in FA–Se NPs treated MCF-7 cells. c ROS levels changes in MCF-7 cells, ROS levels in each FA–Se NPs treatment group treatment are significantly different with control. d Ca2+ concentration changes in MCF-7 cells, ROS levels in each FA–Se NPs treatment group are significantly different with control. The data in c, d both represent three separated experiments
Increasing of intracellular Ca2+ concentrations was also recognized to be an important factor for cell death and injury. We found that the MFI of intracellular free Ca2+ was 29.98 for control group but increased remarkably after the FA–Se NPs treatment from 41.00 to 79.41 (Fig. 4b, d). The results indicated that the increase of intracellular Ca2+ was also closely related to FA–Se NPs induced MCF-7 cell apoptosis.
In this work, JC-1 staining was used to confirm the disruption of MMP as a qualitative method (Li et al. 2011b), and rhodamine 123 was used to detect MMP as a quantitative method (Huang et al. 2012). The JC-1specfic staining could provide a distinct visualized result because red fluorescence was the indicator of higher MMP while green fluorescence was the indicator of lower MMP. FA–Se NPs induced a dose-dependent shift of fluorescence from red to green which indicated that FA–Se NPs induced the disruption of MMP in MCF-7 cells (Fig. 5a). Moreover, the MFI of rhodamine 123 also decreased in a dose-dependent manner after the treatment of FA–Se NPs against MCF-7 cells (shown in Fig. 5b). These information both demonstrated that FA–Se NPs could induce MMP disruption in MCF-7 cells due to the FA–Se NPs entered into mitochondria.

The mitochondria-dependent apoptosis induced by FA–Se NPs in MCF-7 cells. a Changes of MMP in FA–Se NPs treated MCF-7 cells tested by confocal microscopy (JC-1 staining), the scale bars were all 30 μm. b Changes of MMP in MCF-7 cells treated with FA–Se NPs tested by flow cytometry. c Caspase-9 and Caspase-3 activity were measured with whole cell extracts by a fluorometric method. All data were expressed as mean values ± standard deviation from three independent experiments
In order to confirm the apoptosis pathways, we also tested the activity of caspases which were central initiators and executioners of the apoptotic process (Danial and Korsmeyer 2004). As shown in Fig. 5c, the activity of caspase-9 significantly increased to 442 % after treated with 5 μg/mL FA–Se NPs for 24 h. Similarly, the activity of caspase-3 increased to 243 % with 5 μg/mL FA–Se NPs treatment.
Cell morphological changes induced by FA–Se NPs
The morphological changes of F-actin and nucleus induced by FA–Se NPs in MCF-7 cells were tested by specific staining of the F-actin cytoskeleton with rhodamine-labeled phalloidin and staining of the nucleus with DAPI. As shown in Fig. 6a, the staining of control MCF-7 cells showed a mainly homogeneous cytoplasmic distribution of F-actin with intact, plump nucleus, and the cells contact with each other intimately. After 24 h treatment with FA–Se NPs, the F-actin aggregated and formed dot-like structures(shown as white arrows), with some shrinked and broken nuclear bodies (shown as gray arrows); furthermore, the cells appeared more spread out on the substratum. The amounts of F-actin before or after FA–Se NPs treatment in MCF-7 cells was tested by staining F-actin with actin-tracker green and shown in Fig. 6b. The fluorescent signal of actin-tracker green in MCF-7 cells decreased with FA–Se NPs treatment indicated that FA–Se NPs could induce the depolymerization of F-actin.

Changes of F-actin cytoskeletons and nucleus after FA–Se NPs treatment. a The changes in morphology of F-actin in the cytoskeleton and the changes of nucleus before and after FA–Se NPs treatment. Scale bar 30 μm. b The F-actin expression before and after FA–Se NPs treatment. All results were obtained in three independent experiments. The white arrows indicated the dot-like structure of F-actin, and the gray arrows indicated the shrinking nucleus
AFM was used to observe a variety of changes in surface morphology and membrane ultrastructure of MCF-7 cells before or after FA–Se NPs treatment. It was demonstrated that control MCF-7 cells had oval shape and the cell membrane was relatively smooth and intact, but the FA–Se NPs treated MCF-7 cells were deformed and the cell membrane became more roughness with shrunk cell tails (Fig. 7a). The distribution of nanoparticles on cell membrane implied that the nanoparticles (indicated by white arrows in Fig. 7b) on cell membrane became bigger from 120 to 360 nm with FA–Se NPs treatment (shown in Fig. 7c). Additionally, the height of these nanoparticles was also increased, from 41 nm for the control cells to 120 and 228 nm for 0.5 and 2.5 μg/mL FA–Se NPs treated group, respectively (Fig. 7d).

Cell morphology of FA–Se NPs treated and control MCF-7 cells imaged by AFM. a Topography images and their corresponding 3-D mode of control, 0.5 μg/mL FA–Se NPs treated and 2.5 μg/mL FA–Se NPs treated MCF-7 cells. Scale bar 10 μm. The black arrows in these images indicated the shrunk cell tails. b Topography of MCF-7 cell membrane ultrastructure on corresponding cell region indicated by black frame in a. Scale bar 1 μm. c Histograms of the particle size extracted from images in b. d Height profiles of the particle in c. Cell imaging was obtained more than five cells per group, and each cell was scanned for three times
Discussion
This study provides a new method to prepare folate-modified selenium nanoparticles and provides some important information to understand the subcellular localization and cytotoxicity mechanism of selenium nanoparticles in MCF-7 cells. It is reported that the sol system was stable when its absolute value of average zeta potential was higher than 30 mV (Muller and Jacobs 2002). This demonstrated that FA–Se NPs were electronegative and stable in aqueous solution. This stable property would contribute to its further medical and nutritional applications. The special peak at 276 and 340 nm in the spectra of FA–Se NPs but not found in the spectra of Se NPs could be ascribed to the FA molecules conjugated on Se NPs. Additionally, the characteristic IR absorption peaks of folic acid at 1,605 and 1,693 cm−1 (Zhang et al. 2002) were also observed in the spectrum of FA–Se NPs, but not observed in the IR absorption of Se NPs. These results demonstrated that the FA molecules had been successfully conjugated onto Se NPs.
Free FA molecules can inhibit the binding capacity of FA molecules linked drugs to FR on cell membrane (Kalber et al. 2011). When free FA molecules were added, there would be a competition between free FA molecules and FA molecules on the Se NPs when binding to FR on the MCF-7 cell surface. This competition would inhibit the intracellular uptake of FA–Se NPs through inhibiting FR-mediated endocytosis of FA–Se NPs. This inhibition effect demonstrated that the FA molecules on the surface of Se NPs facilitated the uptake efficiency of FA–Se NPs into MCF-7 cells through FR-mediated endocytosis way. Based on the cell cycle distribution results, we could conclude that that FA–Se NPs arrested the cell cycle of MCF-7 cells at S phase, whose main aim was to synthetize DNA for cell division (Laskey et al. 1989). The reason why FA–Se NPs arrested MCF-7 cell cycle at S phase was mostly due to the influence of FA–Se NPs on the DNA synthesis process in S phase after the entry of FA–Se NPs into nucleolus. Before the entry of FA–Se NPs into nucleus, MCF-7 cells had intact, plump nucleolus with no voids and the chromatin is homogeneously distributed in the nucleolus, without chromatin condensation or breakage (labeled by NE in Fig. 2b b). The voids appeared in nucleolus also demonstrated the damage of chromatin and the broken of nucleolar structures.
High levels of ROS could cause cellular damage depending on the duration of ROS stress to mediate apoptosis by regulating the expression of various apoptosis regulatory proteins (Trachootham et al. 2006) and could therefore function as anti-tumor species (Lu et al. 2007). There even highlighted a novel strategy for cancer therapy that preferentially eradicates cancer cells by targeting the ROS stress-response pathway (Raj et al. 2011). ROS generation also acted as an important role in selenium compounds, especially in Se NP-induced cancer cell apoptosis (Chen et al. 2008). The increasing of ROS induced by FA–Se NPs demonstrated that FA–Se NPs could disturb the balance of ROS in MCF-7 cells to induce cell apoptosis. The loss of MMP would lead to the release of cytochrome c, the decrease of adenosine triphosphate generation (Waterhouse et al. 2001), the activation of caspases, and the initiation of apoptotic cascades (Hu and Kavanagh 2003). Caspase-9 has been identified as the indicator of mitochondria-dependent apoptosis pathways and always activated by Apaf-1 or cytochrome c, which present only in mitochondria while caspase-3 has been regarded as the downstream effector caspase of apoptosis (Adams 2003). The loss of MMP, the increasing of caspase-9 and caspase-3 activity, indicated the activation of mitochondria-dependent apoptosis pathways. These results demonstrated that FA–Se NPs induced MCF-7 cell apoptosis through mitochondria -dependent pathways involved the increasing of intracellular ROS level and Ca2+ level as critical mediators. These changes led to the damage of mitochondria membrane, induced the release of FA–Se NPs from the broken mitochondria, and been further transported into nucleus.
F-actin had been viewed as a “good thing” that is necessary for cell motility and sustained cell signaling because it could provide the capability of cells to move organelles like mitochondria within the cytoplasm (Dustin 2006). More significantly, F-actin might be critical for delivering proapoptotic molecules to mitochondria (Tang et al. 2006). Our study showed that FA–Se NPs were not only able to alter the organization of cytoskeleton architecture by disrupting F-actin distribution but also could decrease the expression of F-actin to change the cytoskeleton composition of MCF-7 cells. As a nondestructive cell surface imaging tool, AFM could provide us a lot of information about ultrastructural changes in cell-surface topography at nanoscale (Jin et al. 2010) so that AFM was always used in cell imaging, especially in cancer detection (Lekka and Laidler 2009). The nanoparticles on cell surface observed by AFM were reported as clusters of membrane proteins (Grimellec et al. 1998). The changes of cell membrane morphology and membrane proteins implied that some physiological changes could occur in membrane proteins, such as opening/closing of ion channels, disrupting of structure, or changing of chemical composition of the outer membrane proteins (Wang et al. 2011b).
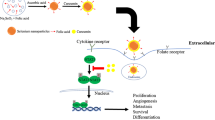
Taking all results obtained here into account, we proposed a mechanism of intracellular localization and cytotoxicity for FA–Se NPs in MCF-7 cells (Fig. 8). At first, when FA–Se NPs were added into the culture medium, some irregular aggregates formed before FA–Se NPs were internalized by cell membrane. The FA molecules on FA–Se NPs aggregates were attracted by the FR on MCF-7 cell membrane. After binding with FR, the plasma membrane surrounding the FA–Se NPs began to invaginate until an endocytic vesicle was formed to transport FA–Se NPs into MCF-7 cells. The endocytic vesicles which were transported further into the cell would rapidly be acidified due to the colocalized proton pumps action in the vesicle membrane (Lee et al. 1996). This acidification environment presumably protonated the carboxyl moieties on the FR protein, thus promoting a conformational change that would enable the escaping of FA–Se NPs from FR (Leamon and Reddy 2004). The nonbinding FA–Se NPs would then be released from endocytic vesicles into cytoplasm. The unconstrained FA–Se NPs would not deliver randomly in cytoplasm but accumulate to the region around mitochondria spontaneously and then entered into mitochondria. The entering of FA–Se NPs into mitochondria induced the generating of ROS, releasing of free Ca2+ and finally led to the damage of mitochondria. Those changes then induced the activation of caspase-9, the activation of downstream apoptosis executor caspase-3, and further induced mitochondria-dependent apoptosis in MCF-7 cells. The FA–Se NPs broke out from the disrupted mitochondria and then translocated to the nucleus. The FA–Se NPs entered nucleus kept moving into nucleolus and induced the disruption of chromatin in nucleolus which made the intact and plump nucleolus damage. As results, the FA–Se NPs in nucleolus would induce MCF-7 cell cycle arrested in S phase. Additionally, the FA–Se NPs in cytoplasm induced depolymerization of F-actin and disorganization of F-actin cytoskeleton which directly caused the morphology changes of MCF-7 cells.

The proposed mechanism for cellular uptake, subcellular localization, and cytotoxicity of MCF-7 cells by FA–Se NPs
Based on the above experiments, we have proposed a mechanism to understand the subcellular localization and cytotoxicity of FA–Se NPs in MCF-7 cells. We believe that understanding the role of cell internalization, intracellular transportation, and subcellular distribution of FA–Se NPs in cancer cells will shed new light in delineating the cytotoxicity mechanisms of Se NPs. The mitochondria-targeted and nucleus-targeted properties of FA–Se NPs in FR overexpressed cancer cells make it a potential carrier for releasing mitochondria-specific or nucleus-specific drugs to kill FR overexpressed tumor selectively. The similar manner would work for other Se NPs and contribute to the design of organelle-targeted drug carriers.
In summary, we first reported that FA can function as both protecting and modifying agent in the production of Se NPs. The spherical FA–Se NPs were successfully modified and protected by FA molecules and obtained in an average diameter of 70 nm. FA–Se NPs were internalized by MCF-7 cells through a FR-mediated endocytosis way and formed endocytic vesicles to transport FA–Se NPs into cytoplasm. The FA–Se NPs in endocytic vesicles were then released into cytoplasm and accumulated to mitochondria. The entry of FA–Se NPs into mitochondria induced mitochondria-dependent apoptosis and further induced the disruption of mitochondria. FA–Se NPs in cytoplasm then transported into nucleus and nucleolus which induced the damage of nucleolus and the arrest of cell cycle. Our findings suggested that FA–Se NPs can be targeted to tumor cells in a site-dependent manner and thus exhibit potential application in targeted therapy of tumors.
References
Abdulah R, Miyazaki K, Nakazawa M, Koyama H (2005) Chemical forms of selenium for cancer prevention. J Trace Elem Med Biol 19:141–150
Adams JM (2003) Ways of dying: multiple pathways to apoptosis. Genes Dev 17:2481–2495
Chen TF, Wong YS, Zheng WJ, Bai Y, Huang L (2008) Selenium nanoparticles fabricated in Undaria pinnatifida polysaccharide solutions induce mitochondria-mediated apoptosis in A375 human melanoma cells. Colloid Surf B 67:26–31
Danial NN, Korsmeyer SJ (2004) Cell death: critical control points. Cell 116:205–219
Dustin ML (2006) When F-actin becomes too much of a good thing. Science 313:767–768
El-Bayoumy K, Sinha R (2004) Mechanisms of mammary cancer chemoprevention by organoselenium compounds. Mutat Res 551:181–197
Elnakat H, Ratnam M (2006) Role of folate receptor genes in reproduction and related cancers. Front Biosci 11:506–519
Evans CW, Fitzgerald M, Clemons TD, House MJ, Padman BS, Shaw JA, Saunders M, Harvey AR, Zdyrko B, Luzinov I, Silva GA, Dunlop SA, Iyer KS (2011) Multimodal analysis of PEI-mediated endocytosis of nanoparticles in neural cells. ACS Nano 5:8640–8648
Fang WF, Han AJ, Bi XL, Xiong B, Yang WC (2010) Tumor inhibition by sodium selenite is associated with activation of c-Jun NH2-terminal kinase 1 and suppression of β-catenin signaling. Int J Cancer 127:32–42
Grimellec CL, Lesniewska E, Giocondi MC, Finot E, Vié V, Goudonnet JP (1998) Imaging of the surface of living cells by low-force contact-mode atomic force microscopy. Biophys J 75:695–703
Hu W, Kavanagh JJ (2003) Anticancer therapy targeting the apoptotic pathway. Lancet Oncol 4:721–729
Huang GY, Zhang Y, Zhang Q, Zhang B, Wen LP (2010) Vacuolization and apoptosis induced by nano-selenium in HeLa cell line. Sci China Chem 53:2272–2278
Huang CH, Jin H, Song B, Zhu X, Zhao HX, Cai JY, Lu YJ, Chen B, Lin YC (2012) The cytotoxicity and anticancer mechanisms of alterporriol L, a marine bianthraquinone, against MCF-7 human breast cancer cells. Appl Microbiol Biotechnol 93:777–785
Jin H, Huang X, Chen Y, Zhao HX, Ye HY, Huang FC, Xing XB, Cai JY (2010) Photoinactivation effects of hematoporphyrin monomethyl ether on Gram-positive and -negative bacteria detected by atomic force microscopy. Appl Microbiol Biotechnol 88:761–770
Kalber TL, Kamaly N, So PW, Pugh JA, Bunch J, McLeod CW, Jorgensen MR, Miller AD, Bell JD (2011) A low molecular weight folate receptor targeted contrast agent for magnetic resonance tumor imaging. Mol Imaging Biol 13:653–662
Ke JH, Lin JJ, Carey JR, Chen JS, Chen CY (2010) A specific tumor-targeting magnetofluorescent nanoprobe for dual-modality molecular imaging. Biomaterials 31:1707–1715
Koller LD, Exon JH (1986) The two faces of selenium-deficiency and toxicity are similar in animals and man. Can J Vet Res 50:297–306
Kong L, Yuan Q, Zhu HR, Li Y, Guo QY (2011) The suppression of prostate LNCaP cancer cells growth by selenium nanoparticles through Akt/Mdm2/AR controlled apoptosis. Biomaterials 27:6515–6522
Laskey RA, Fairman MP, Blow JJ (1989) S phase of the cell cycle. Science 246:609–614
Leamon CP, Reddy JA (2004) Folate-targeted chemotherapy. Adv Drug Deliv Rev 56:1127–1141
Lee RJ, Wang S, Low PS (1996) Measurement of endosome pH following folate receptor-mediated endocytosis. BBA Mol Cell Res 1312:237–242
Lekka M, Laidler P (2009) Applicability of AFM in cancer detection. Nat Nanotechnol 4:72–73
Li GP, Li D, Zhang LX, Zhai JF, Wang E (2009) One-step synthesis of folic acid protected gold nanoparticles and their receptor-mediated intracellular uptake. Chem Eur J 15:9868–9873
Li Q, Chen TF, Yang F, Liu J, Zheng WJ (2010) Facile and controllable one-step fabrication of selenium nanoparticles assisted by L-cysteine. Mater Lett 64:614–617
Li YH, Li XL, Wong YS, Chen TF, Zhang HB, Liu CR, Zheng WJ (2011a) The reversal of cisplatin-induced nephrotoxicity by selenium nanoparticles functionalized with 11-mercapto-1-undecanol by inhibition of ROS-mediated apoptosis. Biomaterials 32:9068–9076
Li W, Zhao LN, Wei TT, Zhao YL, Chen CY (2011b) The inhibition of death receptor mediated apoptosis through lysosome stabilization following internalization of carboxyfullerene nanoparticles. Biomaterials 32:4030–4041
Lu M, Bi CS, Gong XG, Chen HM, Sheng XH, Deng TL, Xu KD (2007) Anti-proliferative effects of recombinant iron superoxide dismutase on HepG2 cells via a redox-dependent PI3k/Akt pathway. Appl Microbiol Biotechnol 76:193–201
Milosavljevic T, Kostic-Milosavljevic M, Jovanovic I, Krstic M (2002) Gastrointestinal and liver tumors and public health in Europe. Eur Rev Med Pharmacother 1:11–15
Muller RH, Jacobs C (2002) Buparvaquone mucoadhesive nanosuspension: preparation optimisation and long-term stability. Int J Pharm 237:151–161
Pan J, Feng SS (2008) Targeted delivery of paclitaxel using folate-decorated poly-(lactide)-vitamin E TPGS nanoparticles. Biomaterials 29:2663–2672
Patil JB, Kim J, Jayaprakasha GK (2010) Berberine induces apoptosis in breast cancer cells (MCF-7) through mitochondrial-dependent pathway. Eur J Pharmacol 645:70–78
Qiu Y, Liu Y, Wang LM, Xu LG, Bai R, Ji YL, Wu XC, Zhao YL, Li YF, Chen CY (2010) Surface chemistry and aspect ratio mediated cellular uptake of Au nanorods. Biomaterials 31:7606–7619
Raj L, Ide T, Gurkar AU, Foley M, Schenone M, Li XY, Tolliday NJ, Golub TR, Carr SA, Shamji AF, Stern AM, Mandinova A, Schreiber SL, Lee SW (2011) Selective killing of cancer cells by a small molecule targeting the stress response to ROS. Nature 475:231–234
Rayman MP (2000) The importance of selenium to human health. Lancet 356:233–241
Reid ME, Duffield-Lillico AJ, Garland L, Turnbull BW, Clark LC, Marshall JR (2002) Selenium supplementation and lung cancer incidence: an update of the nutritional prevention of cancer trial. Cancer Epidemiol Biomarkers 11:1285–1291
Rock C, Moos PJ (2009) Selenoprotein P regulation by the glucocorticoid receptor. Biometals 6:995–1009
Stéphanie L, Bénédicte C, Vincente LV, Katharina G, Corinne DL, Karine BL, Laure L, Dominique M, Minh-Dung N, Sara G, Brice D, Gérard Z, Florence J (2011) Evaluation of current practice: management of chemotherapy-related toxicities. Anti Cancer Drugs 9:919–925
Sudimack J, Lee RJ (2000) Targeted drug delivery via the folate receptor. Adv Drug Deliv Rev 41:147–162
Tabassum A, Bristow RG, Venkateswaran V (2010) Ingestion of selenium and other antioxidants during prostate cancer radiotherapy: a good thing? Cancer Treat Rev 3:230–234
Tang HL, Le AHP, Lung HL (2006) The increase in mitochondrial association with actin precedes Bax translocation in apoptosis. Biochem J 396:1–5
Trachootham D, Zhou Y, Zhang H, Demizu Y, Chen Z, Pelicano H, Chiao PJ, Achanta G, Arlinghaus RB, Liu JS, Huang P (2006) Selective killing of oncogenically transformed cells through a ROS-mediated mechanism by β-phenylethyl isothiocyanate. Cancer Cell 10:241–252
Wang H, Zhang JS, Yu HQ (2007) Elemental selenium at nano size possesses lower toxicity without compromising the fundamental effect on selenoenzymes: comparison with selenomethionine in mice. Free Radic Biol Med 42:1524–1533
Wang HJ, Zhao PQ, Liang XF, Gong XQ, Song T, Niu RF, Chang J (2010) Folate-PEG coated cationic modified chitosan–cholesterol liposomes for tumor-targeted drug delivery. Biomaterials 31:4129–4138
Wang LM, Liu Y, Li W, Jiang XM, Ji YL, Wu XH, Xu LG, Qiu Y, Zhao K, Wei TT, Li YF, Zhao YL, Chen CY (2011a) Selective targeting of gold nanorods at the mitochondria of cancer cells: implications for cancer therapy. Nano Lett 11:772–780
Wang M, Ruan YX, Chen Q, Li SP, Wang QL, Cai JY (2011b) Curcumin induced HepG2 cell apoptosis-associated mitochondrial membrane potential and intracellular free Ca2+ concentration. Eur J Pharmacol 650:41–47
Waterhouse NJ, Goldstein JC, Ahsen OV, Schuler M, Ne DD (2001) Cytochrome c maintains mitochondrial transmem-brane potential and ATP generation after outer mitochondrial membrane permeabilization during the apoptotic process. J Cell Biol 153:319–328
Xia YY (2007) Synthesis of selenium nanoparticles in the presence of silk fibroin. Mater Lett 61:4321–4324
Yoo HS, Park TG (2004) Folate-receptor-targeted delivery of doxorubicin nano-aggregates stabilized by doxorubicin-PEG-folate conjugate. J Control Release 2:247–256
Zhang Y, Kohler N, Zhang MQ (2002) Surface modification of superparamagnetic magnetite nanoparticles and their intracellular uptake. Biomaterials 23:1553–1561
Zhang ZP, Lee SH, Feng SS (2007) Folate-decorated poly- (lactide-co-glycolide)-vitamin E TPGS nanoparticles for targeted drug delivery. Biomaterials 28:1889–1899
Zheng JS, Zheng SY, Zhang YB, Yu B, Zheng WJ, Yang F, Chen TF (2011) Sialic acid surface decoration enhances cellular uptake and apoptosis-inducing activity of selenium nanoparticles. Colloid Surf B 83:183–187
Acknowledgments
This work was supported by National Basic Research Program of China (973 Program, 2010CB833603); Overseas, Hong Kong & Macao Cooperative Research Funds of China (31129002); National Natural Science Foundation of China (30872404); Jinan University’s Scientific Research Cultivation and Innovation Fund (21612601); and Jinan University’s Scientific Research Creativeness Cultivation Project for Outstanding Undergraduates Recommended for Postgraduate Study (50503594).
Author information
Authors and Affiliations
Corresponding author
Additional information
Jiang Pi, Hua Jin, and RuiYing Liu contribute equally to this paper and share the first authorship.
Rights and permissions
About this article
Cite this article
Pi, J., Jin, H., Liu, R. et al. Pathway of cytotoxicity induced by folic acid modified selenium nanoparticles in MCF-7 cells. Appl Microbiol Biotechnol 97, 1051–1062 (2013). https://doi.org/10.1007/s00253-012-4359-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-012-4359-7