
Abstract
Fermentation-derived products are in greater demand to meet the increasing global market as well as to overcome environmental problems. In this work, Escherichia coli has been metabolically engineered with acrylate pathway genes from Clostridium propionicum for the conversion of d-lactic acid to propionic acid. The introduced synthetic pathway consisted of seven genes encoding the enzymes propionate CoA-transferase (Pct), lactoyl-CoA dehydratase (Lcd) and acryloyl-CoA reductase (Acr). The engineered strain synthesised propionic acid at a concentration of 3.7 ± 0.2 mM upon fermentation on glucose. This low production level could be attributed to the low activity of the recombinant enzymes in particular the rate-limiting enzyme, Acr. Interestingly, the recombinant pathway caused an increased lactate production in E. coli with a yield of 1.9 mol/mol of glucose consumed along with a decrease in other by-products. Down-regulation of the pfl (pyruvate formate lyase) genes and a possible inhibition of Pfl activity by the acrylate pathway intermediate, acryloyl-CoA, could have reduced carbon flow to the Pfl pathway with a concomitant increase in lactate production. This study reports a novel way of synthesising propionic acid by employing a non-native, user-friendly organism through metabolic engineering.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Chemicals produced via the fermentation process are gaining attention to circumvent the hitches of petroleum-based production. Products such as 3-hydroxypropionic acid, 1,3-propanediol, glutaconic acid, butanol and 1-butanol have been synthesised through fermentation route by introducing synthetic pathways in microbe with the help of improved understanding of biochemical networks and vast availability of gene manipulation tools (Atsumi et al. 2007; Gokarn et al. 2007; Inui et al. 2008; Raj et al. 2008; Djurdjevic et al. 2011). One such chemical, propionic acid preferred to be synthesised by fermentation route, is widely used in food and feed preservation. Propionic acid is also used in the synthesis of herbicides, cellulose plastics, pharmaceuticals, plasticisers, perfumes and artificial fruit flavours (Coral et al. 2008).
In microbes, propionic acid production occurs via two routes, the dicarboxylic acid pathway and the acrylic acid pathway. The dicarboxylate pathway is functional in several species of propionibacteria whereas the acrylic acid pathway is restricted to few microbes like Clostridium propionicum, Megasphaera elsdenii and Prevotella ruminicola (Strobel 1992; Hino and Kuroda 1993; Hetzel et al. 2003). The acrylate pathway proceeds via the formation of d-lactic acid, d-lactoyl-CoA, acryloyl-CoA and propionyl-CoA to yield propionic acid (Hetzel et al. 2003).
The current biotechnological processes for propionic acid synthesis using these native producers are constrained by factors like very slow growth rates of the microbes, inhibition of cell growth by propionic acid and expensive product recovery. Hence, there is a demand to explore other user-friendly microbes for viable propionic acid synthesis. Among the key lactic acid producers, E. coli shall be the ideal host of choice since this organism is extensively employed for industrial production of various organic acids and other chemicals by metabolic engineering, due to its manifold advantages (Millard et al. 1996; Chang et al. 1999; Cirino et al. 2006; Moon et al. 2008; Heuser et al. 2009).
However, the construction of an efficient synthetic pathway in E. coli is often limited by various factors. The key factors that could limit product synthesis include insufficient supply of precursors, cofactors and rate-limiting enzyme concentrations required for the pathway (Berríos-Rivera et al. 2003; Atsumi et al. 2007; Moon et al. 2008). It has been shown that product concentrations have been improved remarkably by enhancing the activity of crucial enzymes in the recombinant pathway (Atsumi et al. 2010; Yim et al. 2011). Further, the differences in relative in vivo activities of recombinant enzymes could lead to intermediate accumulation and its regulation could also play a crucial role in controlling the synthesis of the product (Atsumi et al. 2007).
Metabolic robustness and tight coupling of various biochemical pathways resist the introduction of new metabolic routes leading to low product synthesis. Upon genetic perturbation, the fluxes at fragile nodes are actively redistributed to maintain the cellular functions. Flux redistribution is essentially attained by flux control at hierarchical (via transcription, translational or post-translation modifications) or at metabolic (through interaction between metabolite and enzyme) levels (Cakir et al. 2004). Therefore, for successful metabolic engineering of pathways, identification of bottlenecks for optimum functioning of the pathways as well as understanding the metabolic perturbations owing to the foreign pathway have to be addressed.
In this work, we have metabolically engineered E. coli with genes from acrylate pathway of C. propionicum to synthesise propionic acid. Metabolite redistribution exhibited by the host due to the recombinant pathway is analysed by studying the variations in transcript levels of host genes by quantitative real-time polymerase chain reaction (qRT-PCR) studies. Further, the functionality of the pathway is confirmed by expression analyses including enzyme activities.
Materials and methods
Bacterial strains and plasmids
The bacterial strains and plasmids used in this study are listed in Table 1.
Chemicals and reagents
The enzymes used in the cloning experiments namely NcoI, SalI, BamHI, PstI, BglII, KpnI, PhusionTM Flash High-Fidelity PCR Master Mix, M-MuLV reverse transcriptase and T4 DNA ligase have been procured from New England Biolabs (Manassas, USA). DNeasy® Blood & Tissue kit, Plasmid miniprep spin kit, PCR purification kit, RNAprotect Bacteria Reagent, RNeasy Mini Kit, RNase-Free DNase Set, random hexamers, dNTP Mix (PCR Grade) and Proteinase K have been purchased from Qiagen (Germany). Chloramphenicol, ampicillin, kanamycin and lysozyme were bought from Biobasic Inc (USA). Primers were procured from VBC-Biotech (Austria). The other media components and chemicals were procured from HiMedia Laboratories (India).
Construction of recombinant plasmids
The genomic DNA of C. propionicum DSM 1682 was isolated using DNeasy® Blood & Tissue kit. Using this genomic DNA as template, the genes encoding Pct (GenBank accession number: AJ276553), Lcd (GenBank accession no: JN244651-3) and Acr (GenBank accession number: JN244654-6) were PCR amplified using PhusionTM Flash High-Fidelity PCR master mix. The oligonucleotides used for amplification of different gene fragments are listed in Table 2. The PCR conditions for amplification of the gene fragments were—an initial denaturation at 98 °C (10 s), followed by 25 cycles of the program: denaturation at 98 °C (3 s), annealing at varying temperatures (5 s), extension at 72 °C (varying times) and a final extension at 72 °C (1 min). The annealing temperatures were varied for different gene fragments, 55 °C (pct), 55 °C (lcd) and 50 °C (acr). The extension time used was 25 s (pct) and 60 s (lcd and acr). The three genes encoding Acr were inserted at the BamHI/PstI site in MCS1 of pCOLADuet-1 to generate the recombinant plasmid, pVK1. Subsequently, the gene encoding Pct was inserted at the BglII/KpnI site in MCS2 of pVK1 to generate the recombinant plasmid pVK2. The genes encoding Lcd were cloned at the NcoI/SalI site in MCS1 of the pETDuet-1 vector to form the recombinant plasmid pVK3. E. coli Rosetta-gami 2(DE3)pLysS was transformed individually with pVK2 and pVK3 to generate the recombinant strains, E. coli VK-2 and VK-3, respectively. Further, these recombinant plasmids were co-transformed into E. coli Rosetta-gami 2(DE3)pLysS to generate the recombinant strain VK-23.
Relative quantitation of mRNA abundance
RNA extraction
Batch bioreactor samples were treated with RNA Protect Bacteria reagent according to the manufacturer’s protocol and stored at −80 °C until extraction. Total RNA was isolated using RNeasy mini kit and on-column DNase digestion was performed with RNase-Free DNase set from Qiagen according to the supplier’s protocol. RNA quality was checked by non-denaturating gel electrophoresis. The purity and concentration of RNA were determined by measuring A260 and A260/A280 using nanodrop (Thermo Scientific, USA). The concentration of RNA was calculated using the formula, concentration (μg/mL) = A260 × 40 × dilution factor.
cDNA synthesis
The cDNA was synthesised using the M-MuLV reverse transcriptase in a total reaction volume of 60 μl containing 1 μg RNA, 0.5 mM dNTPs and 0.5 μg random hexamers. The reaction mixture was incubated at 37 °C for 60 min. Further, the reverse transcriptase was inactivated by heating the reaction mixture at 65 °C for 10 min. The cDNA was diluted three times and stored at −20 °C until use.
qRT-PCR analysis
Quantitative real-time polymerase chain reaction was performed in HT Fast Realtime PCR, model no 7900 (Applied Biosystems, USA) using Power SYBR Green PCR master mix (Applied Biosystems, USA). The list of oligonucleotides used for qRT-PCR studies are listed in Table 3. PCR was carried out using 96-well plate with a total reaction volume of 20 μl. The reaction mixture contained 1 μl each of forward primer and reverse primer (5 pmol), 1 μl of cDNA (50 ng/μl) and 10 μl of SYBR Green mix (2×) made up to 20 μl with sterile water. The reaction mixture was incubated at 95 °C for 10 min, followed by 40 cycles of denaturation (95 °C) for 15 s, annealing (50 °C) 2 min and extension of 1 min at 60 °C. The reactions were performed in triplicates for each gene.
C T values were calculated using the RQ manager software (Applied Biosystems, USA) for each gene amplification. The rrsA gene was used as reference gene which codes for the 16S ribosomal RNA. It was used to normalise the amount of cDNA used for each reaction. ΔC T was calculated as the difference between C T of the gene of interest and C T of the reference gene. ΔΔC T was calculated as difference between the ΔC T of a gene before induction and ΔC T of the same gene at various time points, after induction with IPTG. The fold change was calculated using the formula 2-ΔΔC t (Livak and Schmittgen 2001).
SDS-PAGE analysis
The SDS-PAGE was carried out on a 12 % polyacrylamide gel (Laemmli 1970). Protein estimation was performed by the Bradford method (Bradford 1976) using bovine serum albumin as standard. Coomassie Brilliant Blue G-250 (0.025 %, w/v) was used to stain the proteins on the gel. The recombinant proteins were quantitated by densitometric analysis of the gel using Image Lab 3.0 software (Biorad, CA).
Enzyme activity analysis
Cells were grown under anoxic conditions in 10 ml LB in sealed serum bottles. When OD600 reached 0.6, the cultures were induced with 1 mM IPTG. After 2 h of induction, the cells were harvested, suspended in 0.1 M Tris pH 7.0 and lysed by three passages through a French press. Briefly, the cell suspension was sucked into a pre-cooled French pressure cell (American Instruments, Maryland, USA) and a pressure of 100 MPa was applied. Broken cells were collected into serum bottles and refilled into the pressure cell via the needle for another cycle of disruption. The cell lysate was centrifuged for 1 h at 100,000×g at 4 °C in the ultracentrifuge and cell-free extract was used for enzyme assays. The CoA substrates for the assay were synthesised by acylation of the corresponding anhydrides with CoASH as described previously (Hetzel et al. 2003; Simon and Shemin 1953). The propionate CoA-transferase assay was carried out in analogy to that of glutaconate A transferase using lactoyl-CoA and acetate as substrates (Buckel et al. 1981). Lactoyl-CoA dehydratase and acryloyl-CoA reductase activities were measured according to Parthasarathy et al. (2010) and Hetzel et al. (2003).
Batch bioreactor studies
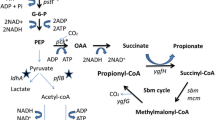
Batch bioreactor studies were carried out in a 2-L fermenter (KLF 2000—Bioengineering AG, Switzerland) containing LB medium supplemented with glucose (20 g/L). The seed culture was grown in LB broth with appropriate antibiotics until OD600 reached 1.5. For batch cultivation in LB medium a 10 % (v/v) inoculum was used. The pH was kept at 7.0 by addition of 3 M NaOH. Temperature and agitation were maintained at 37 °C and 600 rpm, respectively. The dissolved oxygen was continuously monitored by using a DO probe. Initially, the culture was grown under aerobic condition (with 1 vvm of aeration) to achieve the required cell density of 0.4 g/L followed by imposition of anaerobic condition by sparging sterile nitrogen gas. When growth under anaerobic conditions reached 1.05 g/L, the culture was induced with 1 mM IPTG. The culture was maintained for 6 h after induction. The samples were withdrawn periodically for analysis of biomass. For qRT-PCR experiments the culture was extracted with RNA Protect Bacteria reagent and stored at −80 °C until analysis. The supernatants collected at various time intervals were stored at −20 °C for analysing glucose and products of mixed acid fermentation (Fig. 1).

Mixed acid fermentation of E. coli with the introduced propionate synthetic pathway. Abbreviations: Pfl pyruvate formate lyase, LdhA lactate dehydrogenase, AckA acetate kinase, AdhE alcohol dehydrogenase, Pct propionate CoA-transferase, Lcd lactoyl-CoA dehydratase, Acr acryloyl-CoA reductase
HPLC analysis
Samples were filtered through 0.2 μm PVDF membranes before HPLC analysis. Further, they were analysed with Aminex HPX-87 H column (300 × 78 mm, Bio-Rad, USA) in HPLC (LC-10Avp) that was equipped with a refractive index detector. Glucose, succinate, lactate, formate, acetate and ethanol were analysed using 5 mM sulphuric acid as mobile phase with a flow rate of 0.6 ml/min at 65 °C oven temperature. Since the propionic acid peak was merging with a medium component under these conditions, it was separated by altering the temperature of the oven and flow rate of the mobile phase to 35 °C and 0.4 ml/min, respectively.
Results
Expression analysis of acrylate pathway genes in metabolically engineered E. coli
The E. coli Rosetta-gami 2(DE3)pLysS strain has been engineered with acrylate pathway genes from C. propionicum for the conversion of d-lactic acid to propionic acid as described in Fig. 2. The functionality of the acrylate pathway has not been studied in E. coli so far and therefore active expression of the genes was ascertained by SDS-PAGE, qRT-PCR and enzyme activity analyses. For carrying out SDS-PAGE analysis, shake-flask experiments were conducted with E. coli strains, VK-2 (with lcd genes), VK-3 (with acr and pct genes) to visualise the expression of individual subunits of the recombinant enzymes and VK-23 (with lcd, acr and pct genes) using the native strain as control. The culture was induced at 0.6 OD600 with 1 mM IPTG for 3 h. SDS-PAGE analysis of these strains, revealed the expression of acrylate pathway genes which were absent in the control (Fig. 3). Densitometric analysis of the SDS-PAGE of recombinant enzymes has shown that the expression of lcd and pct genes were significantly greater than that of the acr genes and all the three enzymes were synthesised in abundant quantity (Fig. 4). The CoA transferase, Pct was synthesised in the amounts of around 10 % of the total protein. The two subunits and the activator comprising dehydratase achieved protein levels 10–20 % while each subunit of heterotrimeric Acr produced was less than 5 % of the total protein.

Construction of the recombinant plasmids pVK-2 and pVK-3. The position and direction of transcription of recombinant genes are indicated by arrows; Kan R kanamycin resistance gene, Ap ampicilin resistance gene

SDS-PAGE analysis of E. coli strains expressing acrylate pathway genes. a E. coli strain VK-2 depicting the expression lcd genes (Lcd EI, LcdEIIα and LcdEII β). b E. coli VK-3 showing the expression of pct and acr genes (acr α, acr β and acr γ). c E. coli strainVK-23 showing the expression of pct, lcd and acr genes. M protein molecular mass marker, 1 control strain, 2 recombinant strain

Relative expression analysis of acrylate pathway genes in E. coli. Open bars represent relative increase in mRNA abundance 45 min after induction. Filled bars represent the amount of protein synthesised with respect to the total protein of E. coli
In concordance with the differential expression of acrylate pathway genes at protein levels, significant differences were also noted at the mRNA transcript levels (Fig. 4). Transcript-level analyses were carried out using quantitative real-time PCR during pre- and post-induction phase. The uninduced culture showed leaky expression of all the seven genes. pct was expressed at moderate levels with less than 10-fold increase in mRNA abundance after induction. lcd showed varying expression levels (10- to 35-fold increase) with the gene encoding EII β subunit being expressed at low levels in spite of relative higher synthesis of its protein. Amongst Acr’s subunits, β and γ subunit showed lesser expression (lesser than 5-fold increase) while Acr α subunit showed higher expression levels (greater than 25-fold) as opposed to low level synthesis of its protein possibly due to the lesser stability of its mRNA. Though all the recombinant genes were cloned under the same T7 promoter in medium copy number plasmids, they exhibited varied expression levels.
Further, the functionality of the propionate synthetic pathway was ascertained by enzyme activity analysis of the VK-23 strain, with the native strain as control. Pct from C. propionicum has been actively expressed in E. coli in earlier studies (Yang et al. 2009). However, this is the first report on the functional expression of the other two enzymes of the pathway namely Lcd and Acr in a heterologous host. The activities of all the acrylate pathway enzymes were found to be lower as compared to their activities in the native producer, C. propionicum (Table 4). Despite showing higher levels of enzyme synthesis, Pct and Lcd exhibited lower activity by 14- and 10-fold, respectively. Whereas, Acr was synthesised at 12 % of total protein as opposed to the higher level (around 33 %) of this enzyme in C. propionicum (Hetzel et al. 2003), with a 14-fold lower activity in recombinant E. coli as compared to the native producer.
Metabolic impact of expressed acrylate pathway genes in E. coli
We evaluated the impact of the introduced non-native pathway on growth, substrate consumption and metabolite formation in VK-23 strain, using native strain as control. Batch bioreactor studies were conducted using glucose as a carbon source. The recombinant strain exhibited slower growth with a μ max of 0.77 which is 18 % lower than the control (Fig. 5a). The slower growth rate was reflected in decreased biomass formation of VK-23 strain (1.3 g DCW/l) in comparison with the control (1.6 g DCW/l). The consumption of glucose (initial 20 g/L, 110 mM) also decreased in the recombinant strain with a considerable amount of glucose being left unconsumed (3 g/L, 17 mM). This is in contrast to the complete consumption of glucose by the control strain.

Time course of glucose consumption, biomass growth and by-product formation of recombinant (solid lines) and native (dotted line) strains of E. coli. a Biomass (diamond) and glucose (multiplication sign). b Lactate (triangle), formate (circle) and succinate (diamond). c Acetate (triangle) and ethanol (square).The culture was induced at sixth hour
Further, the analysis of secreted metabolites has divulged the influence of the recombinant acrylate pathway enzymes on E. coli’s metabolism (Fig. 6). The metabolite distribution at the pyruvate node showed significant change with diversion of pyruvate away from pyruvate formate lyase, towards lactate formation. The lactate yield from glucose reached 1.9 mol/mol (0.94 g/g) in the post-induction phase, which is 1.9-fold higher than that of the control strain. The yield obtained is comparable with metabolically engineered E. coli strains for lactate hyper-production (Chang et al. 1999; Zhu et al. 2007; Liu et al. 2010). The other by-products namely formate, ethanol, acetate and succinate showed a considerable decrease in production levels. In particular, formate and succinate synthesis ceased after induction, whereas in the control strain the synthesis continued until the 12th hour. At the acetyl-CoA node, the recombinant strain showed a decrease in the ethanol to acetate ratio (1:1.3) indicating that more of acetyl-CoA is channelled for ATP formation rather than to regenerate NAD+ via ethanol synthesis. In contrast, the control strain produced equimolar concentrations of ethanol and acetate mimicking typical mixed acid fermentation of E. coli.

Time course of propionic acid synthesis (diamond) and substrate consumption (triangle) during the anaerobic production phase of E. coli VK-23 strain after induction. Biomass was built up to 3 g/L in the aerobic phase followed by the anaerobic phase. The culture was induced 2 h after adaptation in the anaerobic phase
Propionic acid was synthesised by VK-23 strain at 4 h of post-induction phase. A concentration of 1.0 ± 0.2 mM was reached and thereafter it remained constant until the end of fermentation. However, lactate, the key intermediate of the acrylate pathway was constantly synthesised in ample amounts. Further, to check if increasing biomass in batch cultivation could improve product titres, biomass concentration was increased to 3 g/L in aerobic phase followed by anaerobic production phase containing 30 g/L glucose. There was no biomass growth in production phase though glucose was consumed in considerable amount (Fig. 6). Propionate was not detected before induction and a maximum concentration of 3.7 mM (274 mg/L) was achieved at 10 h of post-induction phase after which the synthesis ceased.
Effect of acrylate pathway enzymes on the expression of host genes at transcript level
To analyse the metabolic shifts in VK-23 strain owing to the expression of non-native pathway genes at transcript level, qRT-PCR studies were conducted. The genes involved in pyruvate node (ldhA, pflA and pflB), acetyl-CoA node (adhE and ackA) and global regulator (fnr) were analysed during pre- and post-induction phase (Fig. 1). In general, the abovementioned host genes considered for the analysis were down-regulated (excluding fnr) when recombinant genes were expressed (Table 5). The pflA and pflB genes involved in the conversion of pyruvate to formate and acetyl-CoA were down-regulated several folds, which corroborates with ceased formate synthesis observed in VK-23 strain during post-induction phase. Expression of ldhA was also moderately down-regulated, which is in contrast to the increased lactate flux from pyruvate node exhibited by the recombinant strain. Acetyl-CoA is channelised to ethanol and acetate formation by the enzymes AdhE and AckA, respectively. The adhE gene was down-regulated to a greater extent as compared to the ackA gene. The down-regulation of adhE gene is reflected in decreased ethanol to acetate ratio during post-induction phase. In contrast to repression of the host genes analysed, Fnr, a global transcriptional regulator which activates the expression of anaerobic genes and also represses aerobic genes has been found to be up-regulated by 2-fold during post-induction phase.
Discussion
E. coli is known to undergo mixed acid fermentation when grown anaerobically on glucose (Chang et al. 1999). The carbon flow from the substrate to each of these acids occurs in a particular ratio in order to generate ATP as well as to regenerate the NAD+ which is being consumed during glycolysis. Therefore to achieve energy and redox balance during genetic perturbations, E. coli responds by altering the metabolic pathways and cellular physiology. The alterations in metabolic pathways can be accomplished by the cell either at expression levels or by the control exerted on the enzyme activities (terKulie and Westerhoff 2001; Cakir et al. 2004). Interestingly, in the VK-23 strain, the introduction of acrylate pathway had profoundly altered the native anaerobic glucose metabolism, redirecting more than 90 % of glucose metabolised to lactic acid, with concomitant decrease in other by-products. This shift in metabolism has occurred due to the changes in transcript levels of the genes involved in pyruvate and acetyl-CoA node (Table 5) in response to expression of acrylate pathway genes.
VK-23 strain flaunted differential down-regulation of the host genes when the acrylate pathway genes were expressed. Though the reason behind the down-regulation of the host gene is not clear, it could be possibly attributed to regulation rendered by acrylate pathway intermediates on host metabolism. Amongst the down-regulated genes, pflA and pflB showed remarkable decrease in transcript levels. This might have led to decreased flux in Pfl pathway with concomitant diversion of flux towards lactate pathway. Disruption of pfl genes has been previously correlated with decreased formate levels and increased lactate production in E. coli (Liu et al. 2010). Earlier studies in E. coli have also shown that disruption of pfl pathway rather than ldhA over-expression is crucial for lactate overproduction (Yang et al. 1999). This is in tune with the tremendous increase in lactate flux despite ldhA down-regulation observed in our study (Table 5).
In addition to changes at transcript levels of pfl genes, we also envisage that activity of Pfl enzyme could also have been inhibited by the acrylate pathway intermediate, acryloyl-CoA. It is a strong electrophile which has the potential to inhibit thiols and many enzymes (Hermann et al. 2004). Acryloyl-CoA could possibly act as a scavenger of the glycyl radical of pyruvate formate lyase, thus disrupting its activity. However, further investigation would be needed to ascertain this hypothesis. At acetyl-CoA node, the adhE (encoding NADH-dependent alcohol dehydrogenase) showed dramatic decrease in mRNA abundance during post-induction phase. This could be attributed to increased regeneration of NAD+ via the lactate pathway. Previous studies have demonstrated that increase in cellular NAD+/NADH ratio down-regulates the expression of adhE gene in E. coli (Leonardo et al. 1993; Leonardo et al. 1996).
E. coli has been found to tolerate propionic acid up to a concentration of 250 mM at neutral pH in anaerobic conditions (data not shown). However, the VK-23 strain synthesised only low levels of propionic acid. This could be attributed to low activities of the heterologous enzymes, especially Acr (acryloyl-CoA reductase). This low activity of Acr is further compounded by its low turnover number under physiological conditions, reportedly due to its tight binding to the product propionyl-CoA. As a compensation for its poor turnover number, large amounts of the enzyme are usually synthesised in the cell, as observed in C. propionicum (Hetzel et al. 2003). Also the recent annotation of genome sequence of C. propionicum revealed the presence of three copies of acr genes (data not published). This again reinforces the greater requirement of Acr for efficient reduction of pyruvate to propionic acid as well as to prevent accumulation of toxic intermediate, acryloyl-CoA. However, the recombinant E. coli strain described here seems to suffer from relatively inadequate Acr levels (Fig. 4), limiting the efficiency of the introduced pathway. Further, discrepancy in activities of the constituent enzymes (relatively high Pct and low Lcd and Acr levels) could lead to imbalance in the pathway. This would result in accumulation of intermediates and depletion of the free CoA pool in the cell. It has been postulated in a similar work involving CoA intermediates that balancing of each reaction step would be vital for improved product formation (Atsumi et al. 2007). Also, the conversion of glucose to lactic acid in E. coli is a redox-balanced pathway. Two moles of NAD+ consumed during glycolysis are recycled via formation of 2 mol lactate. The additional consumption of NADH by the introduced pathway (Fig. 1) would create deficiency of reducing equivalents in the cell.
In summary, this work has indicated the possibility to engineer a non-native pathway for propionic acid production in a user-friendly host like E. coli. Additional co-factor engineering by introducing NADH-forming enzymes like phosphite dehydrogenase/formate dehydrogease (van der Donk and Zhao 2003) would convert the pathway into a redox-neutral process. Also expression of acrylate pathway genes from other microbes like Megasphaera elsdenii and Prevotella ruminicola (Strobel 1992; Gokarn et al. 2007) could be tried to enhance product levels. As the relative activities of enzymes is found to be critical, kinetic modelling of the pathway and metabolic control analysis would provide further leads for improving product titres.
References
Atsumi S, Cann AF, Connor MR, Shen CR, Simth KM, Brynildsen MP, Chou KJY, Hanai T, Liao JC (2007) Metabolic engineering of Escherichia coli for 1-butanol production. Metab Eng 10:305–311
Atsumi S, Wu TY, Eckl EM, Hawkins SD, Buetler T, Liao JC (2010) Engineering the isobutanol biosynthetic pathway in Escherichia coli by comparison of three aldehyde reductase/alcohol dehydrogenase genes. Appl Microbiol Biotechnol 85:651–657
Berríos-Rivera SJ, San K-Y, Bennett GN (2003) The effect of carbon sources and lactate dehydrogenase deletion on 1,2-propanediol production in Escherichia coli. J Ind Microbiol Biotechnol 30:34–40
Bradford MM (1976) Rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein–dye binding. Anal Biochem 72:248–254
Buckel W, Dorn U, Semmler R (1981) Gluctaconate CoA-transferase from Acidaminococcus fermentans. Eur J Biochem 118:315–321
Cakir T, Kirdar B, Ulgen KO (2004) Metabolic pathway analysis of yeast strengthens the bridge between transcriptomics and metabolic networks. Biotechnol Bioeng 86:251–260
Chang D-E, Jung H-C, Rhee J-S, Pan J-G (1999) Homofermentative production of d- or l-lactate in metabolically engineered Escherichia coli RR1. Appl Environ Microbiol 65:1384–1389
Cirino PC, Chin JW, Ingram LO (2006) Engineering Escherichia coli for xylitol production from glucose-xylose mixtures. Biotechol Bioeng 95:1167–1176
Coral J, Karp SG, Vandenberghe LPdS, Parada JL, Pandey A, Soccol CR (2008) Batch fermentation of propionic acid by Propionibacterium acidipropionici in different carbon sources. Appl Biochem Biotechnol 151:333–341
Djurdjevic I, Zelder O, Buckel W (2011) Production of glutaconic acid in a recombinant Escherichia coli strain. Appl Envion Microbiol 77:320–322
RR Gokarn, OV Selifonova, HJ Jessen, SJ Gort, T Selmer, W Buckel (2007) 3-Hydroxypropionic acid and other organic compounds. Patent no. 7,18,6541
Hermann G, Selmer T, Jessen HJ, Gokarn RR, Selifonova O, Gort SJ, Buckel W (2004) Two beta-alanyl-CoA:ammonialyases in Clostridium propionicum. FEBS J 272:813–821
Hetzel M, Brock M, Selmer T, Pierik AJ, Golding BT, Buckel W (2003) Acryloyl-CoA reductase from Clostridium propionicum. An enzyme complex of propionyl-CoA dehydrogenase and electron-transferring flavoprotein. Eur J Biochem 270:902–910
Heuser F, Marin K, Kaup B, Bringer S, Sahm H (2009) Improving d-mannitol productivity of Escherichia coli. Impact of NAD, CO2 and expression of a putative sugar permease from Leuconostocpseudomesenteroides. Metab Eng 11:178–183
Hino T, Kuroda S (1993) Presence of lactate dehydrogenase and lactate racemase in Megasphaera elsdenii grown on glucose or lactate. Appl Environ Microbiol 59:255–259
Inui M, Suda M, Kimura S, Yasuda K, Suzuki H, Toda H, Yamamot S, Okino S, Suzuki N, Yukawa H (2008) Expression of Clostridium acetobutylicumbutanol synthetic genes in Escherichia coli. Appl Microbiol Biotechnol 77:1305–1316
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685
Leonardo MR, Cunningham PR, Clark DP (1993) Anaerobic regulation of adhE gene, encoding the fermentative alcohol dehydrogenase of Escherichia coli. J Bacteriol 175:870–878
Leonardo MR, Dailly Y, Clark DP (1996) Role of NAD in regulating the adhE gene of Escherichia coli. J Bacteriol 178:6013–6018
Liu H, Kang J, Qingsheng Qi, Chen G (2010) Production of lactate in Escherichia coli by redox regulation genetically and physiologically. Appl Biochem Biotechnol 164:162–169
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔC t method. Methods 25:402–408
Martínez I, Bennett GN, San K-Y (2010) Metabolic impact of the level of aeration during cell growth on anaerobic succinate production by an engineered Escherichia coli strain. Metab Eng 12:499–509
Millard CS, Chao Y-P, Liao JC, Donnelly MI (1996) Enhanced production of succinic acid by overexpression of phosphoenolpyruvate carboxylase in Escherichia coli. Appl Environ Microbiol 62:1808–1810
Moon TS, Yoon S-H, Lanza AM, Roy-Mayhew JD, Prather KLJ (2008) Production of glucaric acid from synthetic pathway in recombinant Escherichia coli. Appl Environ Microbiol 75:589–595
Parthasarathy A (2009) Substrates and mechanism of 2-hydroxyglutaryl-CoA-dehydratase from Clostridium symbiosum. Dissertation, Philipps-Universität Marburg
Parthasarathy A, Buckel W, Smith DM (2010) On the thermodynamic equilibrium between (R)-2-hydroxyacyl-CoA and 2-enoyl-CoA. FEBS J 277:1738–1746
Raj SM, Rathnasingh C, Jo J-E, Park S (2008) Production of 3-hydroxypropionic acid from glycerol by a novel recombinant Escherichia coli BL21 strain. Process Biochem 43:1440–1446
Selmer T, Willanzheimer A, Hetzel M (2002) Propionate CoA-transferase from Clostridium propionicum. Cloning of gene and identification of glutamate 324 at the active site. Eur J Biochem 269:372–380
Simon E, Shemin D (1953) The preparation of S-succinyl-CoA coenzyme A. J Am ChemSoc 75:2520
Strobel HJ (1992) Vitamin B12-dependent propionate production by the ruminal bacterium Prevotella rumincola 23. Appl Environ Microb 58:2331–2333
terKulie BH, Westerhoff HV (2001) Transcriptome meets metabolome: hierarchical and metabolic regulation of the glycolytic pathway. FEBS Lett 500:169–171
van der Donk WA, Zhao H (2003) Recent developments in pyridine nucleotide regeneration. Curr Opin Biotechnol 14:421–426
Yang TH, Kim TW, Kang HO, Lee S-H, Lee EJ, Lim S-C, Oh SO, Song A-J, Park SJ, Lee SY (2009) Biosynthesis of polylactic acid and its copolymers using evolved propionate CoA transferase and PHA synthase. Biotechnol Bioeng 105:150–160
Yang Y-T, San K-Y, Bennett GN (1999) Redistribution of metabolic fluxes in Escherichia coli with fermentative lactate dehydrogenase overexpression and deletion. Metab Eng 1:141–152
Yim H, Haselbeck R, Niu W, Pujol-Baxley C, Burgard A, Boldt J, Khandurina J, Trawick JD, Osterhout RE, Stephen R, Estadilla J, Teisan S, Schreyer HB, Andrae S, Yang TH, Lee SY, Burk MJ, Van Dien S (2011) Metabolic engineering of Escherichia coli for direct production of 1,4-butanediol. Nat Chem Biol 7:445–452
Zhu Y, Eiteman MA, DeWitt K, Altman E (2007) Homolactate fermentation by metabolically engineered Escherichia coli strains. Appl Environ Microbiol 73:456–464
Acknowledgements
We gratefully acknowledge Dr. Mark A. Eiteman, University of Georgia for his valuable comments on this study. We also thank the Genomics Laboratory, Göttingen, for sequencing the acr and lcd genes prior to depositing in the gene bank, Mr. Parthiban Singaravelu and Mr. Nadeem Ahmad, Centre for Biotechnology, Anna University, Chennai for their assistance in batch bioreactor studies. Part of this work was supported by the Max Planck Society, Germany, and the Zentrum für synthetische Mikrobiologie, Marburg, Germany. The first author would like to thank Department of Biotechnology, India for providing fellowship to pursue this study.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Kandasamy, V., Vaidyanathan, H., Djurdjevic, I. et al. Engineering Escherichia coli with acrylate pathway genes for propionic acid synthesis and its impact on mixed-acid fermentation. Appl Microbiol Biotechnol 97, 1191–1200 (2013). https://doi.org/10.1007/s00253-012-4274-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-012-4274-y