
Abstract
This study represents two different large-scale proteomic experiments analyzing the antibiotic response and the mechanisms of production of β-lactamases in the nosocomial pathogen Stenotrophomonas maltophilia. Two-dimensional gel electrophoresis on the cytoplasmic protein fraction, together with iTRAQ® differential labeling and 2-D liquid chromatographic separation (2D-LC) MS/MS on the enriched membrane protein fraction, revealed 73 proteins with a change in abundance upon imipenem challenge. These proteins belong to several different functional pathways. We observe an increase in β-lactamase production as well as in proteins important for their function in the periplasm. The up-regulation of the L1 and L2 β-lactamases, along with their activator LysR transcriptional factor AmpR, is linked to an increase in proteins responsible for peptidoglycan remodeling and stress response. The interesting identification of an increase in abundance after treatment of the two-component GGDEF signaling protein and an integral membrane sensor signal transduction histidine kinase, indicates that induction of the β-lactamases is not restricted to the ampR-ampD-ampG pathway. This is the first proteomic study in S. maltophilia upon imipenem stimulation to further unravel the cellular adaptation resulting in β-lactamase production.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Antibiotic resistance is a public health issue worldwide. A growing group of Gram-negative non-fermenting bacteria has become unsusceptible to most currently used broad-spectrum antibiotics. They are typically responsible for severe nosocomial infections in critically ill patients and lead to reduced recovery rates. We therefore need a thorough understanding of the responses of these bacteria to antibiotics, which will enable us to develop new strategies to keep pace with this phenomenon of rapidly increasing antibiotic resistance (McGowan 2006; Livermore 2009; Fournier and Raoult 2011; Wecke and Mascher 2011).
The signature of protein expression, characterizing the adaptation of the bacteria to changes in growth conditions for survival, is increasingly being used to find molecular mechanisms of action of known and potentially new antibiotic compounds (Brötz-Oesterhelt et al. 2005; Wenzel and Bandow 2011). Two-dimensional gel electrophoresis (2-DE) based approaches are commonly found in applications for bacterial quantitative proteomics, also for the study of the antibiotic response (Jungblut 2001; Bandow et al. 2003; Lin et al. 2010; Vashist et al. 2010; Wenzel et al. 2011). Unfortunately, membrane proteins are frequently under-represented in 2-DE maps because of their hydrophobic and low abundant nature (Poetsch and Wolters 2008; Thein et al. 2010). The involvement of these membrane proteins in antibiotic resistance is also very interesting to study because they are located at the first bacterial barriers antibiotics encounter and they can be the drug target themselves. However, shotgun proteomics combined with differential isotopic labeling for quantitation offers an attractive alternative to 2-DE to study these membrane proteins. One of these techniques is the Isobaric Tag for Relative and Absolute Quantitation (iTRAQ®) method, where peptides from different samples are labeled by amine-specific reagents (Ross et al. 2004). As much as eight different samples can be labeled and processed together in order to reduce technical bias. The reliability of protein identification as well as the proteome coverage are also improved by tagging almost all peptides, due to the specific reaction of the label with amines after tryptic digestion. This technique was used by Yun et al. (2011), who combined it with other label-free proteomic approaches, thus identifying 484 proteins of common and antibiotic-specific protein responses to tetracycline and imipenem in a clinical Acinetobacter baumanii strain. Another group analyzed the membrane protein profile of Escherichia coli stimulated with an antimicrobial peptide, by combining iTRAQ® with a classical two-dimensional liquid chromatographic separation coupled with tandem mass spectrometry (2D LC-MS/MS) approach (Zhou and Chen 2011).
Here, we focus on the third most frequently found Gram-negative non-fermenting bacterium in severe nosocomial infections, i.e., Stenotrophomonas maltophilia (Sader and Jones 2005). The organism is associated with bacteraemia, pneumonia, skin, and soft-tissue infections in immunocompromised patients. The risk factors include prolonged hospital stay, mechanical ventilation, previous exposure to broad-spectrum antibiotics, malignancy, and even transplantation. The association of high mortality with these infections is largely due to the intrinsic and acquired resistance of S. maltophilia to many of the currently utilized antibiotics, including β-lactams (Senol 2004; Paez and Costa 2008; Looney et al. 2009; Ryan et al. 2009; Sanchez et al. 2009; Abbott et al. 2011). In the present study, the β-lactam compound imipenem (carbapenem) was used. This drug inhibits penicillin-binding proteins (PBPs), and therefore, the peptidoglycan synthesis leading to bacterial lysis. It is widely used in clinical settings, but increased resistance is observed against it. Drug resistance determinants were also found to shield the recently determined genome of the clinical S. maltophilia strain K279a (Crossman et al. 2008). This resistance can be attributed to an increased impermeability of the outer membrane, the production of efflux pumps, and the presence of β-lactam hydrolyzing enzymes. L1 and L2 β-lactamases are two known examples of the latter in S. maltophilia. The L2 enzyme is a class A serine β-lactamase that is susceptible to clavulanic acid, a clinically used β-lactamase inhibitor, whereas the L1 enzyme is not. The L1 enzyme belongs to the zinc-dependent class B metallo-β-lactamases. The expression of both β-lactamases is induced during β-lactam challenge, even to a different degree, and confers resistance to these β-lactams (Avison et al. 2002; Mercuri et al. 2002). The mechanisms resulting in chromosomal β-lactamase production are of great interest to counteract the increased resistance against those broadly used β-lactams but are currently not fully understood. The induced expression of the L1 and L2 β-lactamases is regulated, to a different degree, by a LysR transcriptional factor (ampR gene) found upstream of the L2 gene (bla L2) (Hu et al. 2008a; Okazaki and Avison 2008; Lin et al. 2009). The N-acetylmuramoyl-l-alanine amidase ampDI is reported to be involved in the regulation of ampR by degrading the ampR activatory ligand, and subsequently repressing the L1 and L2 β-lactamase production during normal growth (Yang et al. 2009). Huang et al. (2010) also demonstrated that the membrane-bound permease ampG, and its upstream gene ampN, are essential for L1 and L2 β-lactamase induction. However, this operon does not show a gene dosage effect on the expression of the β-lactamases. The inactivation of the PBP1a gene mrcA causes a higher basal production of L1 and L2 β-lactamases in S. maltophilia KJ but does not increase their induction. This derepression depends on a functional ampN-ampG permease and on ampR (Lin et al. 2011). The regulation of both β-lactamases thus appears to be closely coupled with peptidoglycan recycling, but it is not yet known whether the regulation by the LysR transcriptional factor (ampR gene) is directly affecting both β-lactamases and whether it is the only regulation system.
In order to get more insight into the physiological changes upon antibiotic stress, and in an attempt to better understand the mechanisms resulting in β-lactamase production and bacterial adaptation, we have chosen a proteomic set up, that provides a broad view on the state of cells. We used two different strategies to analyze the bacterial response of S. maltophilia to the β-lactam antibiotic imipenem: the differential expression profiles of the soluble proteins were investigated using 2-DE, while changes at the membrane protein level, the target area of imipenem, were examined by 2D-LC separation and Matrix Assited Laser Desorption Ionization (MALDI)—mass spectrometry (MS) after iTRAQ® differential labeling.
Materials and methods
Bacterial cell culture
The imipenem-resistant S. maltophilia strain 44/98 was recovered from the Clinical Microbiology Unit of the Varese University hospital in Italy (Mercuri et al. 2002) and is available at the BCCM/LMG bacteria collection with accession number LMG 26824. It was grown aerobically overnight as separate cultures in Luria Bertani (LB) medium in a rotatory shaker at 37 °C, up to the stationary phase, which corresponds to an Optical Density (OD) at 600 nm of approximately 2. The cells were then diluted ten times in fresh LB medium and allowed to grow further in the same conditions. When they reached the mid-exponential growth phase (OD600nm = 0.65–0.75), half of the cultures were stimulated with 25 μg/mL imipenem (Minimal Inhibitory Concentration, MIC > 32 μg/mL) (Howe et al. 1997; Mercuri et al. 2002). After 3 h of incubation, cells were harvested by centrifugation for 15 min at 4,000×g and washed with 50 mM Tris–HCl, pH 8 (Mercuri et al. 2002). The cell pellets were then stored at −80 °C until further use.
β-Lactamase assay
Two milliliters of six biological replicate cultures sampled at different time points after induction with 25 μg/mL imipenem were collected by centrifugation for 10 min at 13,000×g. The cell pellets were dissolved in 2 mL of 10 mM HEPES–NaOH, pH 7.5, containing a (EDTA-free) protease inhibitor mixture (Roche Applied Science, Vilvoorde, Belgium), and were then sonicated for 30 s (Branson Digital Sonifier® S-250D, Danbury, CT, US; horn frequency of ∼20 kHz, power of 60 W). The protein solution was subsequently centrifuged for 20 min at 1,500×g and the supernatant was retained. Protein concentrations for each of these bacterial extracts were determined in triplicate using the BCA™ protein assay (Thermo Scientific, San Jose, CA, US) on a 680XR micro plate reader (Bio-Rad, Hercules, CA, US). Appropriate dilutions of protein extracts were employed in the enzymatic reaction. β-lactamase activity of the cells was measured at different time points after induction with imipenem (0 min, 30 min, 1, 2, 3, 4, and 5 h) by monitoring the initial rate of hydrolysis of 100 μM CENTA™ β-lactamase substrate (Δε 405 = +6,400 M−1 cm−1 , Calbiochem, Darmstadt, Germany) at 25 °C using an Uvikon XL spectrophotometer (Secomam, Ales, France) (Bebrone et al. 2001). L1 and L2 β-lactamase activities were differentiated by adding 50 mM EDTA to the protein solution and incubation for 30 min at 4 °C (Gould et al. 2006; Hu et al. 2008b). One unit of β-lactamase activity was defined as the hydrolysis of 1 nmol of substrate per minute, at 25 °C.
2-D gel electrophoresis of the soluble protein fraction
Six out of twelve independently grown bacterial cultures were incubated with imipenem as described above. The cultures were centrifuged (4,000×g) and the pellets were resuspended in a solution consisting of 9 M urea, 4 % CHAPS (3-3[(3-cholamidopropyl)dimethylamonio]-1-propanesulfonate), 1 % dithiothreitol (DTT), 20 mM Tris, 0.5 mg/mL bovine pancreas DNase I, 0.25 mg/mL bovine pancreas RNase A, 50 mM MgCl2 and the protease inhibitor mixture. A volume of 1.5 mL of this solution was added per gram of biomass. The cells were then vortexed and sonicated to homogeneity, on ice, for 30 s (Branson Digital Sonifier® S-250D; horn frequency of ∼20 kHz, power of 60 W). After 20 min of centrifugation at 16,100×g, the soluble protein fraction was retained and the concentration determined using the Coomassie Plus Bradford™ Assay kit after appropriate dilution (Thermo Scientific). 2-DE was carried out as described in Supplement Section 1.
The Proteomweaver software v.4.0 (Bio-Rad) was used to determine the ratio of difference in protein expression between cells induced with imipenem and those without. Additional statistical analysis was carried out using SPSS v.15. Normal distribution of the resulting quantitative ratios and coefficients of variation (CV) were evaluated with the Kolmogorov–Smirnov test, Shapiro–Wilk statistics, and Q–Q plots. The reproducibility of the gels was assessed with a Pearson's correlation test. The following analysis set was employed in Proteomweaver: the protein spots had to be present in 80 % of the replicate gels and the intensities had to be higher or lower in all of the treated gels compared to those of the control experiment (Mann–Whitney–Wilcoxon test, p = 0.00). The significant difference in intensity distribution of spots between the two groups was determined by a Kolmogorov–Smirnov statistic (p ≤ 0.05). Only the significantly different spots were retained for further investigation, taking into account a cut-off of twofold increase or decrease in spot intensity. Spots absent in either the control group or the β-lactam treated group were also included for further investigation.
Protein identification of differential spots by mass spectrometry
The spots of interest were identified as described by Van Mulders et al. (2011). The peptide mixture was analyzed by MALDI-TOF/TOF (time-of-flight) mass spectrometry and/or nano-LC-ESI-MS (electrospray ionization) (Supplement Section 1). For protein identification, an in-house Mascot server (version 2.1.0) was used with a database containing the NCBI protein sequences of S. maltophilia strains K279a (4,386 entries) and R551-3 (4,039 entries); the shuffled decoy sequences created for all those sequences, as well as a list of commonly found contaminants. This database contained a total of 17,012 entries. The shuffled decoy database was created with the Decoy Database Builder software (Peakardt) (Reidegeld et al. 2008). For the MALDI MS and MS/MS spectra, noise filtering with a coefficient of 0.9 and background subtraction (baseline fit peak width = 32, baseline fit flexibility = 0.5, baseline fit degree = 0.1) were performed in Data Explorer (v.4.9, ABSciex, Darmstadt, Germany), prior to MASCOT analysis. For the MASCOT database search, the number of allowed missed cleavages was set to 2. Carbamidomethylation of cysteine and oxidation of methionine were selected as variable modifications. The selected MS accuracies were 100 ppm for MALDI MS data and 1.5 Dalton (Da) for quadrupole-ion trap (QTRAP) MS data; for MS/MS data it was 0.5 Da. The expectation value was set to be maximally 0.05 for the data generated by the QTRAP mass spectrometer. The functional annotation of the differential proteins was retrieved from the KEGG database and complemented with information about function and conserved domains from the Uniprot and NCBI protein databases. The predicted cellular localization of the proteins was derived from PSORTb v 3.0.2 (Yu et al. 2010).
Isolation of the membrane protein fraction
The membrane proteins of eight S. maltophilia cultures (four controls and four imipenem-treated) were isolated according to an adaption of the protocols used by Pessione et al. (2003) and Molloy (2008). Cells were collected by centrifugation and resuspended in 5 mL 50 mM Tris–HCl, pH 8, to which 0.5 mg/mL bovine pancreas DNase I, 0.25 mg/mL bovine pancreas RNase A, 50 mM MgCl2, and the protease inhibitor mixture were added. After sonication on ice for 1 min (Branson Digital Sonifier® S-250D; ∼20 kHz, 60 W), unbroken cells were centrifuged at 2,500×g for 8 min at 4 °C. The supernatant, S1, was collected, and the cell pellet subjected to another round of resuspension, sonication, and centrifugation, resulting in the collection of supernatant S2. Both supernatants were mixed and 20 mL of ice-cold 0.1 M Na2CO3 at pH 11 was added. The mixture was then stirred on ice for 1 h and subsequently centrifuged for 1 h at 100,000×g using the Avanti J-301 centrifuge (Beckman Coulter, Fullerton, CA, US). The resulting pellet, containing the membrane proteins, was solubilized in 40 mM Tris–HCl, pH 8, containing 7 M urea, 2 M thiourea, 1 % amidosulfobetaine 14 (ASB-14), 0.5 % Triton-X-100, 0.1 % sodium dodecyl sulfate (SDS), 0.5 mg/mL bovine pancreas DNase I, 0.25 mg/mL bovine pancreas RNase A, 50 mM MgCl2, and protease inhibitor mixture.
Labeling with iTRAQ® Reagents
The membrane proteins were labeled with the iTRAQ® Reagents 8-plex kit (ABSciex), according to the manufacturer's instructions, with some modifications. The proteins were precipitated by acetone and resuspended in 60 % methanol/30 mM ammonium bicarbonate and 0.1 % SDS, prior to trypsin digestion, to remove any contaminants. The protein concentration was assessed after resuspension, using the Coomassie Plus Bradford™ Assay kit. An amount of 25 μg of protein was taken for digestion and labeling. The imipenem-treated S. maltophilia peptide samples were labeled with iTRAQ® Reagents 114, 116, 117, and 121, and the control samples with the Reagents 113, 115, 118, and 119. After 2 h of labeling at room temperature, the eight samples were mixed, dried in a Speedvac (SC110, Thermo Savant, Holbrook, NY, US), and stored at −20 °C until analysis.
2-D liquid chromatographic separation
A reversed phase (RP) high pressure liquid chromatographic (HPLC) separation of the peptides at pH 10, followed by a separation at pH 3, was chosen as the 2D-LC strategy (Gilar et al. 2005). Some 100 μg of combined labeled peptide samples were first injected, utilizing a 100 μL loop, on a Luna 5u C18 column (150 × 2.0 mm, 5 μm, 100 Å; Phenomenex, CA, US) placed in an Ettan LC chromatograph (GE Healthcare, Diegem, Belgium). The LC pump was operated at a flow rate of 0.2 mL/min. The mobile phases were 20 mM ammonium formate, pH 10, in 2 % acetonitrile (ACN) as solvent A, and with the same salt in 80 % ACN as solvent B. The separation gradient consisted of a linear increase from 0 % to 50 % solvent B over 35 min, followed by 50 % to 100 % solvent B over 15 min. This percentage was maintained for 10 min prior to a return to the starting conditions. The eluted peptides were fractionated in a 96-well plate at a 1 min interval (resulting in 62 fractions) and conditioned for the next separation by drying in a Speedvac followed by resuspension in 5 % ACN with 0.1 % trifluoroacetic acid (TFA). The column effluent was registered at 214, 220, and 280 nm by a UV-900 monitor (GE Healthcare). The number of fractions was restricted to 36 by pooling some fractions after manual inspection of the chromatogram profile based on peak intensity. Of these fractions, 10 μL was loaded by a Famos microautosampler (Dionex-LC Packings, Hercules, CA, US) onto a RP trapping column (Pepmap C18, 800 μm × 5 mm) using the Switchos device, equipped with two micro 2-position/10 port valves (Dionex-LC Packings). The loading pump operated at 10 μL/min, using 0.05 % TFA in 5 % ACN as the mobile phase. After 8 min, the valve was switched and the sample was eluted onto the analytical separation column (Pepmap C18, 75 μm × 150 mm), in a back flush mode, using the nano-LC pump operating at 250 nL/min. Here, the mobile phases were 0.1 % TFA in 5 % ACN (solvent A) and 0.1 % TFA in 80 % ACN (solvent B). Peptides were resolved by gradient elution with a linear increase from 0 % to 50 % solvent B over 25 min, followed by 50 % to 100 % solvent B over 10 min. This was maintained for another 10 min before returning to the initial conditions. The column effluent was monitored at 214 nm using a 3 nL UV flow cell (Dionex-LC Packings). The effluent was directly spotted by the Probot device (Dionex-LC Packings) on an OptiTOF™ LC/MALDI target plate, at 30 s intervals, together with matrix flowing at a rate of 1.029 μL/min. The matrix consisted of 4 mg/mL α-cyano-4-hydroxycinnamic acid, 70 % ACN, 1 % TFA, 10 mM dibasic ammonium citrate and 0.01 pmol/μL Glu-fibrinopeptide B.
Offline LC-MALDI mass spectrometric analysis
The MALDI plate was analyzed with the 4800 Plus MALDI TOF/TOF Analyzer (ABSciex) as described in Supplement Section 1. While the mass spectrometric identification of a peptide was made in the MS mode using the combined peak intensity of all iTRAQ® labeled samples, the relative quantitation of that peptide was accomplished in the MS/MS mode using the peak intensity of the eight different iTRAQ® reporter ions. ProteinPilot™ (v. 3.0.), involving the Paragon and Pro-Group algorithms from ABSciex, was the software used for identification and quantitation of the proteins (Shilov et al. 2007). The same database was searched as in the 2-DE experiment, with the exception of the shuffled decoy part, since this was created on the fly in ProteinPilot™(17,162 entries). The tolerance utilized for matching was based on information of the mass accuracy of the instrument: MS accuracy at 0.5 m/z, MS/MS accuracy at 1 m/z. The selected parameters were: iTRAQ-label modification at the peptide level, fixed MMTS modification of cysteine, variable biological modifications, and amino acid substitutions. Background correction was employed. In addition, a false discovery rate (FDR) analysis was performed automatically by the software. Following this analysis, the proteins with a local FDR < 5 % were exclusively retained. Of these proteins, only proteins with an “unused score” greater than 1.0 (identification of 90 % confidence) were further investigated. The ratios of protein abundance in imipenem-treated bacteria compared to untreated cells were automatically corrected for experimental bias by calculating the median average protein ratio of every labeled sample and correcting the median to unity. This correction factor was then applied to all quantitation results. Excel was employed to analyze the peptide reporter area distribution and the reproducibility between the different labels. Finally, only proteins with a statistically significant difference in abundance in the induced S. maltophilia cultures compared to the non-induced cultures (ProteinPilot™, p ≤ 0.05), in at least two of the four biological replicates, were kept as results. The functional annotation and cellular localization of the differential proteins were derived similarly as for the 2-DE results.
Validation with quantitative real-time PCR (qPCR)
Interesting protein differences were validated at the mRNA level by qPCR. Total RNA from three control and three induced S. maltophilia cultures was extracted using the RNA protect Bacteria Reagent and RNeasy Plus Mini kit (QIAGEN, Hilden, Germany) as described by the manufacturer, with some modifications (Supplement Section 1). To remove residual DNA, a DNase I treatment was then performed for 2 h at 37 °C in a final volume of 200 μL containing 100 U of RNase-free DNase I, 40 U of RNase inhibitor, and DNase I incubation buffer. Subsequently, DNase I was inactivated and the total RNA was further purified by standard phenolization and precipitation techniques (Sambrook et al. 1989). After precipitation, the RNA pellet was solubilized in 50 μL of RNase-free water and stored at −80 °C until further use (Werbrouck et al. 2006). A NanoDrop ND 1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE, US) was employed to determine RNA concentrations and purity. Afterwards, cDNA was synthesized from 0.75 μg RNA using the Transcriptor First Strand cDNA synthesis kit with random hexamer primers, according to the manufacturer's instructions (Roche Diagnostics). To assess the presence of remaining DNA, each RNA sample was subjected to a cDNA synthesis reaction without addition of “Transcriptor reverse transcriptase enzyme” (NoRT).
Primers for the genes of interest from the S. maltophilia strain 44/98 were designed on the basis of the genome of S. maltophilia K279a and R551-3, as well as of some sequence information from strain 44/98 itself, with the Primer3Plus software (Untergasser et al. 2007) (Supplement Section 1; Supplement Section 2, Table S1). qPCR was carried out on a CFX96™ instrument (Bio-Rad) with each reaction containing IQ SYBR Green Supermix (Bio-Rad), forward and reverse primers of an optimized concentration, and 5 μL of 1:10 diluted cDNA, making up a final volume of 15 μL. The cycling conditions were as follows: initial denaturation at 95 °C for 10 min, followed by 40 cycles of 15 s at 95 °C, and 60 s at 60 °C; detection was of the SYBR Green signal. After the final cycle, melting curve analysis was performed to examine the specificity of each reaction. The green fluorescence of the PCR products was continuously monitored throughout a temperature gradient from 60 °C to 95 °C, with 0.3 °C increments of 15 s. All measurements were done in triplicate, and each assay included a triplicate no-reverse transcriptase (NoRT) and no-template control (NTC) for every primer pair. The reaction efficiency of the primers was derived from a standard curve generated by serial dilution of a cDNA pool of all S. maltophilia 44/98 samples (Supplement Section 2, Table S2).
The crossing point (Cq) values were determined by the single threshold method, using an auto calculated baseline threshold and baseline subtracted curve fit (CFX Manager software version 1.1.308.1111, Bio-Rad). The Cq values were exported to qbasePLUS v.1.5 (Biogazelle, Ghent, Belgium) and corrected for primer-specific amplification (Supplement Section 2, Table S2), applying the standard curve amplification efficiencies (Hellemans et al. 2007). The expression ratios from the three biological replicates were normalized using three stable reference genes (Supplement Section 2, Tables S3 and S4), selected as most stable ones at the protein level for the tested conditions (Vandesompele et al. 2002). After assessing the quality of the qPCR experiment with qbasePLUS, the resulting normalized ratios were exported to Excel. Furthermore, a two-tailed Student's t test was performed on the logarithmic transformed ratios after determination of equality of variance between the groups by an F test (Supplement Section 2, Table S5).
Results
Induction of L1- and L2-β-lactamases upon stimulation
Production of the L1 and L2 β-lactamases was investigated by measuring the initial hydrolysis of CENTA™ β-lactamase substrate in crude extracts of six biological replicates. There is a low-level basal total β-lactamase activity and a rapid increase upon exposure to a sub-lethal concentration of imipenem (25 μg/ml). The strongest activity was seen 3 h after stimulation with imipenem (Fig. 1), while the cells still grew exponentially, similarly to the untreated cells. Consequently, the cells were collected at this time point for the proteomic and qPCR experiments. Upon addition of 50 mM of the zinc-chelator EDTA, the activity of the L1 metallo-β-lactamase was inhibited. In all time-related samples, the β-lactamase activity was reduced by more than 90 % after incubation with EDTA, showing that the hydrolysis of imipenem was mainly due to the L1 β-lactamase (Mercuri et al. 2002).

Total and L2 β-lactamase activity in units per microgram of protein (left vertical axis) of S. maltophilia 44/98, in function of the incubation time (hours) with 25 μg/mL (83.5 pM) imipenem (MIC > 32 μg/mL). The error bars represent the standard error of the mean (SEM) of six biological replicates. One unit of β-lactamase activity is defined as the hydrolysis of 1 nmol of substrate per minute at 25 °C. The corresponding growth curve, where the optical density (OD) was measured at 600 nm, is also shown (right vertical axis)
Changes in abundance of soluble proteins induced by imipenem
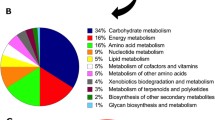
Soluble proteins from six control and six imipenem-treated S. maltophilia 44/98 cultures were subjected to 2-DE. After scanning of the gel images, one gel of the control group was eliminated because of a skewed pattern and very low spot detection. The remaining cropped images were investigated with Proteomweaver, which detected between 500 and 600 spots per gel (Supplement Section 3, Table S1). The reproducibility of the gel images (Supplement Section 3, Fig. S1, Table S2) and the distribution of the coefficients of variation (Supplement Section 3, Fig. S2–S3, Tables S3–S4) were analyzed with SPSS v. 15. The mean coefficient of variation of the spot intensities were 20.4 % and 24.5 % in the imipenem-treated and control group, respectively. Proteins that showed a regulation factor ≥ 2 (26 spots) or ≤ 0.5 (7 spots) were excised for in-gel trypsin digestion and MS identification (Supplement Section 3, Tables S5–S6). Spots that were completely absent in the other group were also excised. One spot was not found in the induced group, whereas 38 spots were missing in the control group (Supplement Section 3, Tables S7–S8). Of the 72 excised spots, 42 proteins were identified with 95 % confidence (p ≤ 0.05) (Supplement Section 3, Tables S9–S10). The excised spots represent 1.64 % of all predicted proteins from S. maltophilia strain K279a, while 0.96 % was also identified with enough confidence (1.78 % and 1.04 %, respectively, for strain R551-3). The excised spots that did not result in a confident identification by mass spectrometry, mostly belonged to the very faint spots after fluorescent staining and were not abundant enough to be detected. As expected, only a small proportion of the proteome showed changes in protein abundance upon adaptation to the imipenem challenge (Bandow et al. 2003; Lin et al. 2010; Vashist et al. 2010). Some of the predicted proteins will not be visible on the 2-DE gels because they are low abundant, have isoelectric points that are not resolved by the first dimensions, or they are too hydrophobic (Brötz-Oesterhelt et al. 2005). Therefore, the hydrophobic membrane proteins were enriched from the samples and separated by a gel-free 2D-LC MS method after stable isotope labeling for quantitative proteomics. The proteins identified by 2-DE as differentially expressed (Fig. 2) after imipenem treatment can be found in Tables 1 and 2, classified according to their most important function.

Average 2-DE gel images of Stenotrophomonas maltophilia 44/98 treated with (a) and without (b) 25 μg/mL imipenem. The differential spots, identified by mass spectrometry, are indicated by a circle and arrows, in addition to the spot number
One of those proteins, exclusively observed in the imipenem-treated cultures, was the metallo-β-lactamase L1, which confirmed the increased activity seen in the β-lactamase assay. The L2 enzyme was not found in this experiment. This protein has a pI of 9.6, and although we used IPG-strips with a broad pI (3–10) range, it is commonly observed that proteins with such a high pI are not well resolved in a 2-DE experiment. Furthermore, the induced β-lactamase activity is mainly due to the L1-metallo-β-lactamase and only a small increase of the L2 enzyme activity is observed. Anyway, the L2-β-lactamase exhibits a low activity against imipenem and a higher activity against extended-spectrum cephalosporins (Walsh et al. 1997; Mercuri et al. 2002). The UDP-N-acetylmuramoylalanyl-d-glutamyl-2,6-diaminopimelate/d-alanyl-d-alanyl ligase protein (MurF protein) was more abundant after treatment with the antibiotic. This points to an increase in peptidoglycan biosynthesis, an expected phenomenon as a compensation for the action of imipenem on the inhibition of the cell wall synthesis (Typas et al. 2012). Furthermore, stimulation affected mainly protein translation, carbohydrate metabolism and amino acid metabolism. Except for a putative aminopeptidase and three ribosomal proteins, the proteins involved in these three pathways showed an increased abundance. Other pathways were increased after imipenem stimulation, such as the metabolism of nucleotides and lipids, the energy metabolism and the machineries for replication and repair as well as of transcription. There are some conflicting results on the changes in protein abundance of the GroEL protein involved in general protein folding events during stress response, a property they share with the increased expression of DnaK chaperone protein (Frydman 2001). Different spot intensity ratios were observed for the GroEL protein, probably reflecting modified or processed forms of GroEL, for instance phosphorylation (Sherman and Goldberg 1992). We also detected an increased amount of two components of the protein quality pathway involving Clp protease, namely the two ATP-dependent Clp subunits ClpX and ClpA (Camberg et al. 2011). Similarly, the putative UvrABC system protein B (UvrB protein) was increased upon antibiotic challenge, reflecting an oxidative stress response linked to adaptation to antibiotic stress (Crowley and Hanawalt 1998; Cirz et al. 2005). Contrary to the other proteins involved in stress response, the abundance of the universal stress protein A (UspA domain protein) was found to be decreased (Kvint et al. 2003). It is not clear at this stage whether this concerns the non-phosphorylated form. Besides those general pathways, there was an interesting increased abundance of a two-component system response regulator GGDEF signaling protein upon antibiotic challenge (Hoch 2000). Furthermore, the experiment revealed an elevated abundance of a flagellin protein, which suggests biofilm formation as an additional antibiotic resistance mechanism (de Oliveira-Garcia et al. 2002). Finally, we observed a reduced expression of bacterioferritin, which plays a role in the cellular iron homeostasis.
Changes in abundance of membrane proteins induced by imipenem
Membrane proteins from control and treated S. maltophilia 44/98 cultures, with similar OD600nm values, were analyzed by differential labeling followed by RP-RP LC and MALDI-TOF/TOF analysis. The samples were randomly labeled with iTRAQ® Reagents. All duplicate RP-LC MALDI-TOF/TOF MS runs, resulting in 13,102 non-empty spectra, were analyzed at once by ProteinPilot™. When an identity confidence cut-off of 90 % was chosen, 3,102 distinct peptides originating from 383 proteins were identified (44.8 % of total spectra; Supplement Section 4, Table S1, S4, and S5, Fig. S1). Of these, two were contaminant bovine proteins and eight were false positive identifications. Applying a “Local False Discovery Rate (FDR)” threshold <5 %, calculated by the ProteinPilot™ FDR analysis (Supplement Section 4, Table S7), 349 proteins were retained. This corresponds to 7.96 % of the predicted proteome of S. maltophilia strain K279a (8.65 % for strain R551-3). This workflow resulted in 27 % of the 349 proteins to be localized in the cytoplasmic membrane and 12 % in the outer membrane. In comparison, the cellular prediction of all S. maltophilia K279a proteins in our database with PSORTb resulted in 22.62 % of cytoplasmic membrane proteins and 3.58 % of outer membrane proteins (24.29 % and 3.99 %, respectively, for strain R551-3). This discrepancy is due to contamination of the membrane fraction by highly abundant cytoplasmic proteins, such as the GroEL protein and hydrophobic ribosomal proteins. This was unavoidable and similar to what is observed in other studies (Thein et al. 2010). Finally, 24 proteins in this experiment were found to have a consistent statistically significant increased abundance after treatment with imipenem, while 7 proteins displayed a decreased abundance (Tables 3 and 4; Supplement Section 4, Table S6) or 0.71 % of the predicted proteome of S. maltophilia strain K279a (0.77 % for strain R551-3). In accordance to the results of the 2-DE experiment, a small proportion of proteins show changes in abundance upon antibiotic challenge and are involved in the energy and nucleotide metabolism, in transcription as well as in replication and repair. A change in peptidoglycan biosynthesis and metabolism was also observed by the increase in UDP-N-acetylglucosamine-N-acetylmuramyl-(pentapeptide) pyrophosphoryl-undecaprenol N-acetylglucosamine transferase (MurG protein) (Typas et al. 2012). Moreover, the increased level of an integral membrane sensor signal transduction histidine kinase indicates, once more, the involvement of another two-component system in the adaptation of S. maltophilia to imipenem (Hoch 2000). Besides these confirmations, the iTRAQ® experiment on the enriched membrane protein fraction also identified changes in the membrane transport and secretion pathways, in which the putative outer membrane usher protein precursor (MrkC protein), the putative RND/Acr family transmembrane transporter (SmeO protein) (Crossman et al. 2008), the putative protein-export membrane protein (SecD protein) (Pradel et al. 2009) and the parB-like partition protein (ParB protein) were more abundant after antibiotic stimulation. In contrast, a TonB-dependent receptor and the putative RND family acriflavine resistance protein A precursor (SmeG protein) were less abundant (Koebnik 2005; Crossman et al. 2008). The putative histone-like protein and two ribosomal proteins were found to be less present in the induced cultures.
Two peptidases similar to PepO peptidase had an increased abundance observed in the iTRAQ® experiment, while a leucyl aminopeptidase was found to be absent in the imipenem-stressed bacteria in the 2-DE experiment. In addition, the proteins encoded by the GroEL and GroES operon (De Carolis et al. 2010) were decreased in abundance according to the iTRAQ® experiment, while some modified forms of the GroEL protein were found to be increased according to the 2-DE experiment, probably truncated or phosphorylated GroEL (Sherman and Goldberg 1992). In the iTRAQ® experiment there is ,however, no differentiation between the modified forms of a protein possible, so the quantitation is done based on the average quantity of all unique peptides from all possible forms of a protein.
Validation of some proteins by qPCR
Of all proteins showing a differential abundance after treatment with imipenem, 12 were retained for validation at the mRNA level by qPCR (Supplement Section 2, Table S1) because of their functional annotation and possible implication in the antibiotic response, as discussed later. Three reference genes for normalization were chosen from both proteomic experiments as being the most stable under both conditions used, namely the putative survival protein surA (surA, Smlt0820), ribosomal protein L15 (rplO, Smlt0925) and putative delta-aminolevulinic acid dehydratase (hemB, Smlt4497). The primer sequences as well as the amplification efficiencies, reference gene stabilities, resulting normalization factors and ratios derived from the qbasePLUS analysis can be found in Supplement Section 2, Tables S1–S5. An increased abundance of L1 metallo-β-lactamase (Smlt2667), L2 β-lactamase (Smlt3722), and UDP-N-acetylmuramoylalanyl-d-glutamyl-2,6-diaminopimelate/d-alanyl-d-alanyl ligase (murF) was confirmed at the RNA level (Fig. 3). A significant increase in L2 β-lactamase on the mRNA level but not on the protein level can be explained by its low activity against imipenem. It is minimally responsible for the increased β-lactamase activity, as shown in the assay, but it is still induced upon imipenem challenge. The primers designed for murF were spanning a region from murF (Smlt0752) as well as from murE (Smlt0751). The up-regulation observed for the murEF gene after induction with imipenem can be extrapolated to the whole operon (Smlt0750–Smlt0761). This operon is homologous to the well-studied Mra operon, containing cell envelope biosynthesis and division genes, in E. coli and Pseudomonas aeruginosa. Therefore, not only the increased abundance of the MurF protein, but also that of the MurG protein was validated on the RNA level. A TonB-dependent receptor (Smal_3321, Smlt3905) was observed to be decreased (Fig. 3). The flagellin fliC was shown to have a down-regulation at the mRNA level (Fig. 3), contrary to the findings at the protein level in the 2-DE experiment. These discrepancies might indicate complex post-transcriptional regulation mechanisms. A good correlation between transcriptome and proteome quantification is often solely found for high abundant “housekeeping” proteins. Differences between proteome and transcript ratios have been found in other experiments as well (Gygi et al. 1999; Griffin et al. 2002). In addition, we analyzed the abundance of some transcripts from genes already known to be implicated in β-lactamase induction in other S. maltophilia strains. In accordance, we observed a correlated up-regulation of the putative HTH and LysR transcriptional factor (ampR, Smlt3723) and the L2 β-lactamase (bla L2) operon in the S. maltophilia 44/98 cultures that were grown for 3 h after induction with imipenem (Fig. 3; Supplement Section 2, Fig. S1, Table S6) (Okazaki and Avison 2008; Lin et al. 2009). Even though the putative N-acetylmuramoyl-l-alanine amidase (ampDI) was shown to be involved in the derepression of L1 and L2 β-lactamases (Yang et al. 2009), we observed that neither ampDI (Smlt1562) nor ampDII (Smlt0154) mRNA levels were regulated by imipenem induction. There was also no significant effect on the expression of the operon constituted of the known putative β-lactamase induction signal transducer ampG (Smlt0413) and the putative endonuclease/exonuclease/phosphatase family protein ampN (Smlt0412) (Huang et al. 2010).

Bar chart of the average calibrated normalized relative quantity (CNRQ) in logarithmic scale (Log (10) ) of the mRNA levels of the selected genes, normalized to the geometric mean of three reference genes, in the three control cultures compared to the three cultures induced with 25 μg/mL imipenem. The error bars represent the average Log (10) standard error. Significant changes are depicted by an asterisk (Student's t test, p < 0.05). The CNRQ values and corresponding standard error were derived from qbasePLUS
Discussion
This study represents two different large-scale quantitative proteomic experiments investigating the antibiotic response and the mechanisms of production of β-lactamases in S. maltophilia 44/98 upon stimulation with imipenem. The growth phases, in presence or absence of the antibiotic, were not significantly different for both conditions, so differences in the proteomes can be linked to the imipenem challenge alone. The analysis of the soluble protein fraction by 2-DE, followed by MS identification resulted in 42 proteins having a change in abundance. Additionally, the investigation of the membrane proteins with iTRAQ® labeling, followed by RP (pH10)/RP (pH3) separation and subsequent MALDI-TOF/TOF analysis identified 31 proteins with differential protein abundance. The bacterial adaptation to the imipenem challenge can be seen in the change of protein abundances in different functional pathways.
β-lactamase production
The increased β-lactamase activity upon stimulation with imipenem is essentially due to the L1 metallo-enzyme, which is in agreement with the existence of different induction patterns for the L1 and L2 β-lactamases, dependent on the strain and antibiotic used (Avison et al. 2002; Mercuri et al. 2002). Moreover, the L1 metallo-β-lactamase was exclusively detected in the imipenem-treated cultures in the 2-DE experiment (Table 1).
The increase of β-lactamase production is accompanied by that of proteins important for its folding and secretion, such as the MreB, DnaK, and SecD proteins (Tables 1 and 3). The metallo-β-lactamase is exported to the periplasm by the Sec-dependent translocation with the help of the bacterial cytoskeleton “putative rod shape-determining protein” MreB (Pradel et al. 2009). Concerning the DnaK protein, it is known that the L1 β-lactamase precursor is protected during transport from cytoplasmic proteases, being assisted by the DnaK chaperone system, of which an increased truncated form is observed after stimulation (Morán-Barrio et al. 2009). Additionally, the production of the putative protein-export membrane protein SecD, which promotes the release of the mature protein into the periplasm in the Sec-dependent protein export machinery (Pradel et al. 2009), also appears to be increased. We also observe an increase in the Hflc protein (Table 3), shown to inhibit SecY protein degradation by the FtsH protein, resulting in more Sec-dependent translocation (Kihara et al. 1996). In conclusion, S. maltophilia reacts to the imipenem challenge by mainly increasing its L1 metallo-β-lactamase production and that of the proteins needed to protect and export this β-lactamase to the periplasm, where it can hydrolyze the imipenem.
ampR-ampDI-ampG dependent β-lactamase induction
The proteome results were complemented with results from other studies on the induction of β-lactamase activity. These studies addressed the issue by disrupting or knocking out genes and observing the influence of this loss on the production of β-lactamase in other S. maltophilia strains and with different types of antibiotics as well as at different time points after stimulation. Our study shows an up-regulation of ampR after imipenem treatment in S. maltophilia 44/98 (Fig. 3). This results coincide with the role of ampR as an activator of L1 and L2 β-lactamase production in an induced state (Okazaki and Avison 2008; Lin et al. 2009). As was shown for cephalosporin-induced S. maltophilia KJ, the mRNA level of ampDI was not influenced by antibiotic challenge, even though it is associated with β-lactamase derepression (Yang et al. 2009). In another study, the ampN and ampG operon was found to be essential for β-lactamase in S. maltophilia KJ, but an increased amount of ampG did not alter the basal nor the induced β-lactamase activity (Huang et al. 2010). We do not observe an increase in mRNA from ampG upon imipenem challenge. These proteins, essential for β-lactamase induction, could be activated upon antibiotic challenge without necessarily having to be up-regulated. The possibility exists that β-lactamase induction is more complex and triggered by additional unknown mechanisms, besides the elevated levels of peptidoglycan intermediates that specifically activate ampR derepression by ampDI.
Peptidoglycan remodeling
Interestingly, the MurF and MurG proteins are more abundant after treatment with imipenem (Tables 1 and 3). These proteins belong to the same operon (Smlt0750–Smlt0761), which is homologous to the well-studied Mra operon of E. coli and P. aeruginosa responsible for proteins involved in cell envelope biosynthesis and cell division. This links the regulation of both β-lactamases with peptidoglycan recycling. The involvement of the peptidoglycan enzymes and intermediates in β-lactam resistance was also demonstrated by Moya et al. (2009), who linked the inactivation of PBP4 to the overproduction of the chromosomal β-lactamase AmpC in P. aeruginosa. In contrast, Lin et al. (2011) showed that the inactivation of PBP1a, mrcA, caused a basal L1 and L2 β-lactamase hyperproduction after antibiotic challenge but no increased induction. However, both research groups clearly link peptidoglycan cell wall enzymes and intermediates to ampR-dependent overexpression of β-lactamases, similar to what we observe in our study. We also demonstrate an increase of the FtsZ protein partner putative cell division protein ZipA homolog (Table 3), which recruits the FtsZ protein to the cell membrane for making it a partner in the cell division apparatus (Hale and de Boer 1997). A similar response for the ZipA protein after imipenem treatment was observed in A. baumanii (Yun et al. 2011). Apparently, the β-lactam resistance response and stress response at the cell envelope are interacting.
Two-component systems
The role and underlying mechanisms of two-component systems (TCS) in β-lactam response, and more particularly in β-lactamase induction, is still uncertain. On the one hand, Moya et al. (2009) clearly demonstrated a role for the CreBC two-component system in β-lactam resistance and induction of the AmpC β-lactamase in P. aeruginosa, of which the exact mechanism and function is not fully understood yet. On the other hand, Lin et al. (2011) showed that this TCS is not involved in mrcA protein nor ampDI protein derived β-lactamase hyperproduction in S. maltophilia KJ, but they could not rule out a role for this CreBC TCS in β-lactam resistance nor the implication of other TCS systems. Furthermore, Tayler et al. (2010) found evidence that the inhibition of PBP4 by antibiotic challenge caused an direct or indirect elevated level of the activator ligand for the BlrAB-TCS and a concomitant overproduction of β-lactamase. In conclusion, there is a particularly interesting link between two-component systems and β-lactam response, either through β-lactamase production or through other mechanisms not elucidated yet. This link is also present in our study, where we show that a sensor histidine kinase (Table 3) and a GGDEF response regulator (Table 1), that are part of two different TCS, display an increased abundance upon imipenem challenge. This GGDEF response regulator protein is activated by the corresponding sensor histidine kinase (Hoch 2000). The stimulus and downstream function of the two TCS proteins identified in our study are unknown. Further research on the implication of phosphorylation and the system of phosphorelay, and thus on the involvement of the two-component system on β-lactam resistance, are needed to elucidate these pathways.
Multi-drug efflux pumps and other membrane transporters
Some secretion and membrane transport proteins were also found to be increased in the iTRAQ® experiment, the putative RND/Acr family transmembrane transporter (SmeO protein) being the most interesting one (Table 3). The SmeO protein, most likely, forms a complex with the SmeP protein with whom it constitutes an expressed operon. S. maltophilia hosts a large number of similar multidrug efflux pumps of which the precise function is not well established (Crossman et al. 2008). The smeC protein, for example, has been indicated as being hyperexpressed in resistant strains (Li et al. 2002), but it is arguable that strain differences exist in the expression of these efflux pumps. Surprisingly, another efflux pump, the SmeG protein, shows a decreased abundance (Table 4), which indicates that the expression of efflux pumps is dependent on the nature of the target compound. A TonB-dependent receptor was confirmed to be downregulated at the transcription (Fig. 3) and protein level (Table 4). It encodes a porin and its downregulation may reflect a reduced intake of imipenem (Koebnik 2005). A similar protein is also downregulated in imipenem-treated A. baumanii (Yun et al. 2011). Overall, our results indicate drug and strain-specific changes in efflux pumps and transporters, of which the exact function remains unknown.
Stress response and protein quality pathway
An increased amount of the protein quality pathway ATP-dependent Clp subunits, the ClpX and ClpA proteins, was detected in our 2-DE experiment (Table 1). They use ATP to unfold proteins and deliver them to the ClpP proteases for targeted protein degradation. Interestingly, ClpP-deficient mutants of Streptococcus mutans are more sensitive to imipenem and other cell-wall oriented antibiotics (Chattoraj et al. 2010). Moreover, E. coli ClpX and ClpP proteins were recently observed to be involved in controlled FtsZ degradation, playing a major role in cell division (Camberg et al. 2011). However, it has been shown that the ClpA proteins can act as a protein translocase in the absence of the ClpP protein and may then be involved in membrane fusion and transport (Rajendar and Lucius 2010). In addition, the UvrB protein, induced by the SOS response, is increased upon antibiotic challenge, which points to bacterial adaptation through mutation (Crowley and Hanawalt 1998; Cirz et al. 2005). The β-lactam resistance response and the stress response and protein quality pathway are thus potentially linked.
In conclusion, our study provides a first large-scale quantitative proteomic analysis of the antibiotic response of S. maltophilia in an attempt to unravel the mechanisms of β-lactamase induction. With the use of large-scale protein abundance profiling techniques, multiple proteins of one pathway, as well as of different pathways, can be studied together, which provides a broad view on the different factors and possible interactions playing a role in the imipenem response. A good understanding of this antibiotic response will stimulate the development of new therapeutic agents needed to counteract the increasing amount of untreatable nosocomial infections caused by this opportunistic pathogen.
References
Abbott IJ, Slavin MA, Turnidge JD, Thursky KA, Worth LJ (2011) Stenotrophomonas maltophilia: emerging disease patterns and challenges for treatment. Expert Rev Anti Infect Ther 9:471–488
Avison MB, Higgins CS, Ford PJ, von Heldreich CJ, Walsh TR, Bennett PM (2002) Differential regulation of L1 and L2 β-lactamase expression in Stenotrophomonas maltophilia. J Antimicrob Chemother 49:387–389
Bandow JE, Brötz H, Leichert LIO, Labischinski H, Hecker M (2003) Proteomic approach to understanding antibiotic action. Antimicrob Agents Chemother 47:948–955
Bebrone C, Moali C, Mahy F, Rival S, Docquier JD, Rossolini GM, Fastrez J, Pratt RF, Frère JM, Galleni M (2001) CENTA as a chromogenic substrate for studying β-lactamases. Antimicrob Agents Chemother 45:1868–1871
Brötz-Oesterhelt H, Bandow JE, Labischinski H (2005) Bacterial proteomics and its role in antibacterial drug discovery. Mass Spectrom Rev 24:549–565
Camberg JL, Hoskins JR, Wickner S (2011) The interplay of ClpXP with the cell division machinery in Escherichia coli. J Bacteriol 193:1911–1918
Chattoraj P, Banerjee A, Biswas S, Biswas I (2010) ClpP of Streptococcus mutans differentially regulates expression of genomic islands, mutacin production, and antibiotic tolerance. J Bacteriol 192:1312–1323
Cirz RT, Chin JK, Andes DR, de Crécy-Lagard V, Craig WA, Romesberg FE (2005) Inhibition of mutation and combating the evolution of antibiotic resistance. PLoS Biol 3:e176
Crossman LC, Gould VC, Dow JM, Vernikos GS, Okazaki A, Sebaihia M, Saunders D, Arrowsmith C, Carver T, Peters N, Adlem E, Kerhornou A, Lord A, Murphy L, Seeger K, Squares R, Rutter S, Quail MA, Rajandream MA, Harris D, Churcher C, Bentley SD, Parkhill J, Thomson NR, Avison MB (2008) The complete genome, comparative and functional analysis of Stenotrophomonas maltophilia reveals an organism heavily shielded by drug resistance determinants. Genome Biol 9:R74
Crowley DJ, Hanawalt PC (1998) Induction of the SOS response increases the efficiency of global nucleotide excision repair of cyclobutane pyrimidine dimers, but not 6–4 photoproducts, in UV-irradiated Escherichia coli. J Bacteriol 180:3345–3352
De Carolis E, Posteraro B, Florio AR, Colonna B, Prosseda G, Bugli F, Lorenzetti SR, Fiscarelli E, Inzitari R, Iavarone F, Castagnola M, Fadda G, Sanguinetti M (2010) Analysis of heat-induced changes in protein expression of Stenotrophomonas maltophilia K279a reveals a role for GroEL in the host-temperature adaptation. Int J Med Microbiol 301:273–281
de Oliveira-Garcia D, Dall’Agnol M, Rosales M, Azzuz ACGS, Martinez MB, Girón JA (2002) Characterization of flagella produced by clinical strains of Stenotrophomonas maltophilia. Emerg Infect Dis 8:918–923
Fournier PE, Raoult D (2011) Prospects for the future using genomics and proteomics in clinical microbiology. Annu Rev Microbiol 65:169–188
Frydman J (2001) Folding of the newly translated proteins in vivo: the role of molecular chaperones. Annu Rev Biochem 70:603–647
Gilar M, Olivova P, Daly AE, Gebler JC (2005) Two-dimensional separation of peptides using RP-RP-HPLC system with different pH in first and second separation dimensions. J Sep Sci 28:1694–1703
Gould VC, Okazaki A, Avison MB (2006) β-Lactam resistance and β-lactamase expression in clinical Stenotrophomonas maltophilia isolates having defined phylogenetic relationships. J Antimicrob Chemother 57:199–203
Griffin TJ, Gygi SP, Ideker T, Rist B, Eng J, Hood L, Aebersold R (2002) Complementary profiling of gene expression at the transcriptome and proteome levels in Saccharomyces cerevisiae. Mol Cell Proteomics 1:323–333
Gygi SP, Rochon Y, Franza BR, Aebersold R (1999) Correlation between protein and mRNA abundance in yeast. Mol Cell Biol 19:1720–1730
Hale CA, de Boer PA (1997) Direct binding of FtsZ to ZipA, an essential component of the septal ring structure that mediates cell division in E. coli. Cell 88:175–185
Hellemans J, Mortier G, De Paepe A, Speleman F, Vandesompele J (2007) qBase relative quantification framework and software for management and automated analysis of real-time quantitative PCR data. Genome Biol 8:R19
Hoch JA (2000) Two-component and phosphorelay signal transduction. Curr Opin Microbiol 3:165–170
Howe RA, Wilson MP, Walsh TR, Millar MR (1997) Susceptibility testing of Stenotrophomonas maltophilia to carbapenems. J Antimicrob Chemother 40:13–17
Hu RM, Chiang KH, Lin CW, Yang TC (2008a) Modified nitrocefin-EDTA method to differentially quantify the induced L1 and L2 β-lactamases in Stenotrophomonas maltophilia. Lett Appl Microbiol 47:457–461
Hu RM, Huang KJ, Wu LT, Hsiao YJ, Yang TC (2008b) Induction of L1 and L2 β-lactamases of Stenotrophomonas maltophilia. Antimicrob Agents Chemother 52:1198–1200
Huang YW, Lin CW, Hu RM, Lin YT, Chung TC, Yang TC (2010) AmpN-AmpG operon is essential for expression of L1 and L2 β-lactamases in Stenotrophomonas maltophilia. Antimicrob Agents Chemother 54:2583–2589
Jungblut PR (2001) Proteome analysis of bacterial pathogens. Microbes Infect 3:831–840
Kihara A, Akiyama Y, Ito K (1996) A protease complex in the Escherichia coli plasma membrane: HflKC (HflA) forms a complex with FtsH (HflB), regulating its proteolytic activity against SecY. EMBO J 15:6122–6131
Koebnik R (2005) TonB-dependent trans-envelope signalling: the exception or the rule? Trends Microbiol 13:343–347
Kvint K, Nachin L, Diez A, Nyström T (2003) The bacterial universal stress protein: function and regulation. Curr Opin Microbiol 6:140–145
Li XZ, Zhang L, Poole K (2002) SmeC, an outer membrane multidrug efflux protein of Stenotrophomonas maltophilia. Antimicrob Agents Chemother 46:333–343
Lin CW, Huang YW, Hu RM, Chiang KH, Yang TC (2009) The role of AmpR in regulation of L1 and L2 β-lactamases in Stenotrophomonas maltophilia. Res Microbiol 160:152–158
Lin XM, Yang JN, Peng XX, Li H (2010) A novel negative regulation mechanism of bacterial outer membrane proteins in response to antibiotic resistance. J Proteome Res 9:5952–5959
Lin CW, Lin HC, Huang YW, Chung TC, Yang TC (2011) Inactivation of mrcA gene derepresses the basal-level expression of L1 and L2 β-lactamases in Stenotrophomonas maltophilia. J Antimicrob Chemother 66:2033–2037
Livermore DM (2009) Has the era of untreatable infections arrived? J Antimicrob Chemother 64:29–36
Looney WJ, Narita M, Mühlemann K (2009) Stenotrophomonas maltophilia: an emerging opportunist human pathogen. Lancet Infect Dis 9:312–323
McGowan JE (2006) Resistance in nonfermenting gram-negative bacteria: multidrug resistance to the maximum. Am J Infect Control 34:S29–S37, S64–73
Mercuri PS, Ishii Y, Ma L, Rossolini GM, Luzzaro F, Amicosante G, Franceschini N, Frère JM, Galleni M (2002) Clonal diversity and metallo-β-lactamase production in clinical isolates of Stenotrophomonas maltophilia. Microb Drug Resist 8:193–200
Molloy MP (2008) Isolation of bacterial cell membranes proteins using carbonate extraction. Methods Mol Biol 424:397–401
Morán-Barrio J, Limansky AS, Viale AM (2009) Secretion of GOB metallo-β-lactamase in Escherichia coli depends strictly on the cooperation between the cytoplasmic DnaK chaperone system and the Sec machinery: completion of folding and Zn(II) ion acquisition occur in the bacterial periplasm. Antimicrob Agents Chemother 53:2908–2917
Moya B, Dötsch A, Juan C, Blázquez J, Zamorano L, Haussler S, Oliver A (2009) β-lactam resistance response triggered by inactivation of a nonessential penicillin-binding protein. PLoS Pathog 5:e1000353. doi:10.1371/journal.ppat.1000353
Okazaki A, Avison MB (2008) Induction of L1 and L2 β-lactamase production in Stenotrophomonas maltophilia is dependent on an AmpR-type regulator. Antimicrob Agents Chemother 52:1525–1528
Paez JIG, Costa SF (2008) Risk factors associated with mortality of infections caused by Stenotrophomonas maltophilia: a systematic review. J Hosp Infect 70:101–108
Pessione E, Giuffrida MG, Prunotto L, Barello C, Mazzoli R, Fortunato D, Conti A, Giunta C (2003) Membrane proteome of Acinetobacter radioresistens S13 during aromatic exposure. Proteomics 3:1070–1076
Poetsch A, Wolters D (2008) Bacterial membrane proteomics. Proteomics 8:4100–4122
Pradel N, Delmas J, Wu LF, Santini CL, Bonnet R (2009) Sec- and Tat-dependent translocation of β-lactamases across the Escherichia coli inner membrane. Antimicrob Agents Chemother 53:242–248
Rajendar B, Lucius AL (2010) Molecular mechanism of polypeptide translocation catalyzed by the Escherichia coli ClpA protein translocase. J Mol Biol 399:665–679
Reidegeld KA, Eisenacher M, Kohl M, Chamrad D, Körting G, Blüggel M, Meyer HE, Stephan C (2008) An easy-to-use Decoy Database Builder software tool, implementing different decoy strategies for false discovery rate calculation in automated MS/MS protein identifications. Proteomics 8:1129–1137
Ross PL, Huang YN, Marchese JN, Williamson B, Parker K, Hattan S, Khainovski N, Pillai S, Dey S, Daniels S, Purkayastha S, Juhasz P, Martin S, Bartlet-Jones M, He F, Jacobson A, Pappin DJ (2004) Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol Cell Proteomics 3:1154–1169
Ryan RP, Monchy S, Cardinale M, Taghavi S, Crossman L, Avison MB, Berg G, van der Lelie D, Dow JM (2009) The versatility and adaptation of bacteria from the genus Stenotrophomonas. Nat Rev Microbiol 7:514–525
Sader HS, Jones RN (2005) Antimicrobial susceptibility of uncommonly isolated non-enteric Gram-negative bacilli. Int J Antimicrob Agents 25:95–109
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning: a laboratory manual, vol 1, second edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York, pp 7.6–7.9
Sanchez MB, Hernandez A, Martinez JL (2009) Stenotrophomonas maltophilia drug resistance. Future Microbiol 4:655–660
Senol E (2004) Stenotrophomonas maltophilia: the significance and role as a nosocomial pathogen. J Hosp Infect 57:1–7
Sherman M, Goldberg AL (1992) Heat shock in Escherichia coli alters the protein-binding properties of the chaperonin groEL by inducing its phosphorylation. Nature 357:167–169
Shilov IV, Seymour SL, Patel AA, Loboda A, Tang WH, Keating SP, Hunter CL, Nuwaysir LM, Schaeffer DA (2007) The paragon algorithm, a next generation search engine that uses sequence temperature values and feature probabilities to identify peptides from tandem mass spectra. Mol Cell Proteomics 6:1638–1655
Tayler AE, Ayala JA, Niumsup P, Westphal K, Baker JA, Zhang L, Walsh TR, Wiedemann B, Bennett PM, Avison MB (2010) Induction of β-lactamase production in Aeromonas hydrophila is responsive to β-lactam-mediated changes in peptidoglycan composition. Microbiology 156:2327–2335
Thein M, Sauer G, Paramasivam N, Grin I, Linke D (2010) Efficient subfractionation of Gram-negative bacteria for proteomics studies. J Proteome Res 9:6135–6147
Typas A, Banzhaf M, Gross CA, Vollmer W (2012) From the regulation of peptidoglycan synthesis to bacterial growth and morphology. Nat Rev Microbiol 10:123–136
Untergasser A, Nijveen H, Rao X, Bisseling T, Geurts R, Leunissen JAM (2007) Primer3Plus, an enhanced web interface to Primer3. Nucleic Acids Res 35:W71–W74
Van Mulders SE, Stassen C, Daenen L, Devreese B, Siewers V, van Eijsden RG, Nielsen J, Delvaux FR, Willaert R (2011) The influence of microgravity on invasive growth in Saccharomyces cerevisiae. Astrobiology 11:45–55
Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F (2002) Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol 3:research 0034.1–0034.11
Vashist J, Tiwari V, Kapil A, Rajeswari MR (2010) Quantitative profiling and identification of outer membrane proteins of β-lactam resistant strain of Acinetobacter baumannii. J Proteome Res 9:1121–1128
Walsh TR, McGowan AP, Bennett PM (1997) Sequence analysis and enzyme kinetics of the L2 serine β-lactamase from Stenotrophomonas maltophilia. Antimicrob Agents Chemother 41:1460–1464
Wecke T, Mascher T (2011) Antibiotic research in the age of omics: from expression profiles to interspecies communication. J Antimicrob Chemother 66:2689–2704
Wenzel M, Bandow JE (2011) Proteomic signatures in antibiotic research. Proteomics 11:3256–3268
Wenzel M, Patra M, Albrecht D, Chen DY, Nicolaou KC, Metzler-Nolte N, Bandow JE (2011) Proteomic signature of fatty acid biosynthesis inhibition available for in vivo mechanism of action studies. Antimicrob Agents Chemother 55:2590–2596
Werbrouck H, Grijspeerdt K, Botteldoorn N, Van Pamel E, Rijpens N, Van Damme J, Uyttendaele M, Herman L, Van Coillie E (2006) Differential inlA and inlB expression and interaction with human intestinal and liver cells by Listeria monocytogenes strains of different origins. Appl Environ Microbiol 72:3862–3871
Yang TC, Huang YW, Hu RM, Huang SC, Lin YT (2009) AmpD(I) is involved in expression of the chromosomal L1 and L2 β-lactamases of Stenotrophomonas maltophilia. Antimicrob Agents Chemother 53:2902–2907
Yu NY, Wagner JR, Laird MR, Melli G, Rey S, Lo R, Dao P, Sahinalp SC, Ester M, Foster LJ, Brinkman FSL (2010) PSORTb 3.0: Improved protein subcellular localization prediction with refined localization subcategories and predictive capabilities for all prokaryotes. Bioinformatics 26:1608–1615
Yun SH, Choi CW, Kwon SO, Park GW, Cho K, Kwon KH, Kim JY, Yoo JS, Lee JC, Choi JS, Kim S, Kim SI (2011) Quantitative proteomic analysis of cell wall and plasma membrane fractions from multidrug-resistant Acinetobacter baumannii. J Proteome Res 10:459–469
Zhou Y, Chen WN (2011) iTRAQ-coupled 2-D LC-MS/MS analysis of membrane protein profile in Escherichia coli incubated with apidaecin IB. PLoS One 6:e20442. doi:10.1371/journal.pone.0020442
Acknowledgments
The authors are indebted to the Belgian Federal Government-Interuniversity Attraction Pole Action P6/19. B. Devreese is indebted to the “Agentschap voor Innovatie door Wetenschap en Technologie (IWT)”—Vlaanderen, through the SBO-program BIOSURF and to the ‘Bijzonder Onderzoeksfonds’ from the Ghent University for a concerted action grant.
Conflict of interest
The authors have declared no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Van Oudenhove, L., De Vriendt, K., Van Beeumen, J. et al. Differential proteomic analysis of the response of Stenotrophomonas maltophilia to imipenem. Appl Microbiol Biotechnol 95, 717–733 (2012). https://doi.org/10.1007/s00253-012-4167-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-012-4167-0