
Abstract
Furfural from lignocellulosic hydrolysates is the prevalent inhibitor to microorganisms during cellulosic ethanol production, but the molecular mechanisms of tolerance to this inhibitor in Zymomonas mobilis are still unclear. In this study, genome-wide transcriptional responses to furfural were investigated in Z. mobilis using microarray analysis. We found that 433 genes were differentially expressed in response to furfural. Furfural up- or down-regulated genes related to cell wall/membrane biogenesis, metabolism, and transcription. However, furfural has a subtle negative effect on Entner–Doudoroff pathway mRNAs. Our results revealed that furfural had effects on multiple aspects of cellular metabolism at the transcriptional level and that membrane might play important roles in response to furfural. This research has provided insights into the molecular response to furfural in Z. mobilis, and it will be helpful to construct more furfural-resistant strains for cellulosic ethanol production.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Although lignocellulosic biomass provides an abundant and renewable source for the fermentative production of liquid fuels and valuable chemicals, biomass pretreatments are necessary to make carbohydrates available for enzymatic hydrolysis and further fermentation. As a result, by-products such as furfural and 5-hydroxymethylfurfural (HMF), organic acids, vanillin and lignin monomers will be produced during the pretreatment process of acid-catalyzed hydrolysis of lignocelluloses, which will have a negative effect on the consequent enzymatic hydrolysis and ethanol fermentation (Almeida et al. 2007; Liu 2006). One approach to overcome the inhibition caused by pretreatment processes is to remove the inhibitor from the hydrolyzate by physical, chemical, or biochemical method (Zhang et al. 2010), which will increase costs, add complexity to the process, and generate extra waste products of manufacturing cellulosic ethanol (Liu 2006). Engineered ethanologenic strains’ tolerance to hydrolysate by-products is another attractive method for improving lignocellulosic bioethanol production based on the mechanisms of stress response. Recently, extensive reviews were concentrated on inhibitors formed by the pretreatment of lignocellulosic materials and their inhibition of ethanol production in yeast and bacteria (Pienkos and Zhang 2009).
Furfural is one of the major inhibitors in lignocellulosic hydrolysates. Although the mechanism of furfural stress has been studied intensively on DNA damage (Barciszewski et al. 1997; Khan et al. 1995), metabolite analysis, enzyme activity analysis, metabolic flux analysis, kinetic analysis, genomics, and comparative proteomic in Saccharomyces cerevisiae or Escherichia coli (Lin et al. 2009a, b; Palmqvist and Hahn-Hagerdal 2000), the molecular mechanism involved in the tolerance and adaptation of ethanologenic strains to inhibitors represented in lignocellulosic hydrolysate is still unclear.
S. cerevisiae, E. coli, and other microorganisms have been shown to contain enzymes that catalyze the reduction of furfural to the less toxic product (e.g., furfuryl alcohol, etc.) (Almeida et al. 2008; Gutiérrez et al. 2006; Koopman et al. 2010; Liu et al. 2008; Zaldivar et al. 2000). Previous studies (Koopman et al. 2010) at the gene level showed that furfural tolerance in S. cerevisiae requires a functional pentose phosphate pathway (PPP) and induces the accumulation of reactive oxygen species (Allen et al. 2010; Gorsich et al. 2006). Comparative proteomics also indicated that central carbon metabolism, levels of alcohol dehydrogenases, the redox balance, and other general stress response (such as unfolded protein response, oxidative stress, osmotic and salt stress, DNA damage, and nutrient starvation) may be related to the tolerance of ethanologenic yeast for and its adaptation to furfural (Lin et al. 2009a). These insights into the response of yeast to the presence of furfural will benefit the design and development of inhibitor-tolerant ethanologenic yeast by metabolic engineering or synthetic biology. For example, overexpression of zwf1 of PPP pathway and Lsm protein allowed S. cerevisiae to grow at furfural concentrations that are normally toxic (Gorsich et al. 2006; Yang et al. 2010).
Cellulosic hydrolysate toxicity and tolerance mechanisms in E. coli have also been reviewed by Ryan T. Gill (Mills et al. 2009). Previous studies in E. coli suggested that furfural may detrimentally affect multiple glycolytic enzymes essential to central metabolism (Zaldivar et al. 1999). Furan tolerance in E. coli primarily depends on lower expression of yqhD and dkgA. Furan tolerance was also increased by adding plasmids encoding a NADPH/NADH transhydrogenase (pntAB). Together, Ingram LO proposed a hypothesis that the NADPH-dependent reduction of furan by yqhD and dkgA inhibits growth by competing with biosynthesis for this limiting cofactor (Miller et al. 2010). This hypothesis was verified by deleting yqhD and dkgA gene (Miller et al. 2009). Another study revealed that overexpression of NADH-dependent oxidoreductase FucO could also improve furfural tolerance in E. coli (Wang et al. 2011). These extensive studies on furfural stress response to yeast and E. coli will play an important role in strain improvement by metabolic engineering or synthetic biology.
Zymomonas mobilis is a candidate microorganism for converting cellulosic biomass into ethanol or other valuable chemicals for its special Entner–Doudoroff (ED) pathway and desirable industrial characteristics. By using ED pathway to metabolize glucose, 1 mol of ATP is produced per molecule of glucose (Swings and Deley 1977). Different engineered Z. mobilis strains have also been successfully constructed by introducing desirable genes including C5 (arabinose and xylose) metabolic pathway (including five arabinose-metabolized related genes—araA, araB, araD, talB, and tktA, four xylose-metabolized related genes—XI, XK, tkt, and tal, respectively) (Deanda et al. 1996; Zhang et al. 1995) and cellulolytic enzymes (Linger et al. 2010) to convert cellulosic biomass into ethanol. Seo et al. reported the first genome sequence for Z. mobilis ZM4 (Seo et al. 2005). The complete genome of Z. mobilis ZM4 contains a 2,056,416-bp circular chromosome and five circular plasmids. To date, the US Department of Energy’s Joint Genome Institute (JGI) has also published the complete genome sequence of Z. mobilis NCIMB11163, 29192, and 10988 (Kouvelis et al. 2009, 2011; Pappas et al. 2011). The other strains of this organism have been hitherto sequenced for comparative genome analysis (http://www.ncbi.nlm.nih.gov/sites/entrez?db=bioproject&cmd=Retrieve&dopt=Overview&list_uids=12354). With the completed genome from different Z. mobilis strains on hand, comparative genomics or global expression analysis should reveal ways to improve the performance of Z. mobilis, and more approaches to strain improvement will certainly be identified in the future (Jeffries 2005). In addition, recent achievements to improve transformation efficiency by modifying DNA restriction–modification systems (Kerr et al. 2010), as well as the genome-scale modeling and in silico analysis were completed (Widiastuti et al. 2011). These extensive studies will also aid future metabolic engineering and synthetic biology in strain improvement.
However, few studies have examined the response of Z. mobilis to various stresses. For example, furfural was examined for toxicity against CP4 (pZB5) for the first time in 1997 (Ranatunga et al. 1997). The influence of furfural on the recombinant Z. mobilis strain CP4 (pZB5) was also investigated in 2005, which showed that furfural led to decrease by 30 and 60 % (w/v) in biomass production and 19 and 76 % (v/v) in ethanol production in Z. mobilis CP4 (pZB5) when 0.475 and 1.9 g/l furfural was added in the fermentation media, respectively (Gutierrez-Padilla and Karim 2005). Zhang’s group at National Renewable Energy Laboratory, USA, have also presented an effective high-throughput method to evaluate the impact of inhibitory compounds from lignocellulosic hydrolysate on the growth of Z. mobilis (Franden et al. 2009). However, only one research has examined response to furfural stress at the gene level. Steven D. Brown et al. showed that Z. mobilis Hfq (ZMO0347) plays important roles in resisting multiple lignocellulosic pretreatment inhibitors including acetate, vanillin, furfural, and HMF (Yang et al. 2010). It offers the possibility to manipulate hfq for Z. mobilis strain improvement. However, the physiological basis and genetic mechanisms involved in furfural tolerance for Z. mobilis are poorly understood.
In this study, microarray technology was used to investigate the expression profiling of the ethanologenic Z. mobilis in response to furfural stress. The results showed that 433 genes were expressed as up- or down-regulated. These data will help us to understand the molecular mechanisms and provide a global insight into strain improvement by metabolic engineering or synthetic biology.
Materials and methods
Bacterial strains and fermentation conditions
Z. mobilis ZM4 (ATCC31821) was cultured in rich media (Goodman et al. 1982) at 30 °C without shaking. Cultures were maintained on glucose agar (20.0 g/l glucose, 10.0 g/l yeast extract, and 15.0 g/l agar). The organism was subcultured to fresh inoculum media for 24 h at 30 °C before being inoculated into the fermentation medium. The inoculum medium (g/l) consisted of 10.0 g yeast extract, 1.0 g MgCl2, 1.0 g (NH4)2SO4, 1.0 g KH2PO4, and 20.0 g glucose. The concentration of furfural in lignocellulosic hydrolysates was measured in the range from 0.5 to 11 g/l (Almeida et al. 2007). Our primary experiment showed that Z. mobilis ZM4 was not able to grow on the medium added with 1.5–2.0 g/l of furfural (initially added). Therefore, the final concentration of furfural was set up at 1.0 g/l for the study of the response of Z. mobilis to the presence of furfural under an extreme set of conditions. The optical density was measured with a spectrophotometer at 600 nm with an initial OD600 of 0.05 when the inoculum was added to each flask (with or without 1.0 g/l furfural).
Cell growth, glucose, and ethanol analysis
Cell growth was determined by monitoring the optical density at 600 nm by using Multi Scanner Spectrometer (Thermo Inc.) at 4-h intervals. Fermentation supernatant was prepared by passing through a 0.2-μm membrane (Millipore) and used to determine the concentrations of glucose and ethanol. Ion chromatography (Metrohm Bio-Scan 871, Switzerland) was applied to measure the concentration of glucose with sodium hydroxide (0.1 M) as mobile phase at a flow rate of 1 ml/min. Sugars were quantified by comparing their peak areas with standard sugar of known concentrations. Ethanol was assayed using GC122 gas chromatography with a glass column (0.26 × 200 cm) filled with Porapak Type QS (80–100 mesh, Waters, Milford, MA, USA) at 150 °C and a FID detector at 80 °C. N2 was used as the carrier gas (30 ml/min), and butylacetate was added as inner reference.
RNA isolation and preparation of fluorescein-labeled cDNA
Total RNA was isolated essentially as described previously (Chomczynski 1993). Briefly, two samples from Z. mobilis 24-h culture were harvested by centrifugation and TRIzol reagent (Invitrogen) was used to extract total cellular RNA. Each total RNA preparation was further purified using NucleoSpin® RNA clean-up (MACHEREY-NAGEL, Germany) according to the manufacturer’s instructions. The RNA quality was assessed by formaldehyde agarose gel electrophoresis and quantitated at OD260 and OD280 by a spectrophotometer, respectively. The purified RNA from each sample was used as the template to generate cDNAs while labeled with either Cy3-dUTP or Cy5-dUTP (CapitalBio). In a duplicate set of cDNA synthesis reactions, the fluorescent dyes were reversed for each sample so that the effects of a specific dye were minimized.
Microarray hybridization, scanning, image quantification, and data analyses
Z. mobilis microarrays were constructed by CapitalBio Corporation (Beijing, China) using coding sequences predicted by The Institute for Genomic Research (TIGR,http://www.tigr.org/). Microarray hybridization, washing, scanning, and data analysis were carried out according to NimbleGen’s Expression user’s guide. Gene expression analysis was performed using six independent microarray experiments (two dye reversal reactions × three biological replicates) with each microarray containing one to two probes each per predicted coding sequence.
Hierarchical clustering with the average linkage method was performed with Cluster 3.0 software and the cluster result was visualized through the Treeview software. In a comparison analysis, two-class unpaired method in the Significant Analysis of Microarray software (SAM, version 3.02) was performed to identify significantly differentially expressed genes between furfural-treated and control (furfural-untreated) groups. Genes were determined to be significantly differentially expressed with a selection threshold of false discovery rate, FDR <5 % and fold change ≥1.2 (significant induction) or ≤0.8 (significant repression). Raw data were log2-transformed and imported.
Quantitative-PCR analysis
Real-time quantitative-PCR (RT-qPCR) was carried out to assess the microarray expression results for the selected genes as described earlier. Total RNA was isolated by using TRIzol reagent (Invitrogen). The RNA samples were reverse-transcribed by using Protoscript First Strand cDNA Synthesis Kit (MBI, Fermentas Inc.) as described in the manufacturer’s protocol. Sixteen genes representing different functional categories and a range of gene expression values, based on microarray hybridizations, were analyzed using qPCR (iQ5 Real-Time PCR System, Bio-Rad) from cDNAs derived from different treatment samples. Optimized primers were designed using Beacon Designer 7 software (length from 18 to 20 nucleotides and predicted annealing temperatures ranging from 55 to 57 °C) to amplify about 90–120 bp of the target genes, and the oligonucleotide sequences of the 16 genes selected for qPCR analysis are listed in Table 1. First-strand cDNA was synthesized using a cDNA synthesis kit (MBI, Fermentas Inc.). PCR conditions were 10 min at 94 °C, followed by 40 cycles of heating at 94 °C for 20 s and 60 °C for 30 s, and final extension at 72 °C for 5 min. PCR amplification was detected by SYBR fluorescence dye (Takara). The rrsA gene (ZMOr009), encoding the 16 S ribosomal RNA gene, served as an endogenous control to normalize for differences in total RNA quantity.
Results
Profiling of cell growth, glucose utilization, and ethanol yield under furfural stress
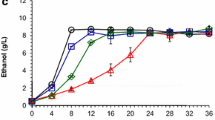
The presence of furfural in the medium led to negative impacts on cell growth, glucose consumption, and ethanol production of Z. mobilis ZM4 (Fig. 1). In the furfural-untreated culture, maximal cell density (OD600) reached to 4.5 approximately 16 h post-inoculation, while the time needed for Z. mobilis to reach its highest cell density of 3.62 (OD600) was delayed until 24 h after initial inoculation under furfural stress conditions.

Z. mobilis fermentations under normal and furfural stress conditions. The data come from mean values of triplicate experiments
Z. mobilis also consumed glucose more slowly under furfural stress conditions, with more than half of the initial aerobic glucose concentration (10.8 g/l) remaining after 12 h of incubation. In opposite, 99.4 % of glucose has been utilized at this time point under normal conditions. When Z. mobilis growth reached its peak after 24 h under stress conditions, 14 % of glucose remained in the culture. As a result, the yield of ethanol was also decreased by 20.5 % under furfural stress conditions (Fig. 1).
Global gene expression patterns in response to furfural
Since the growth and other phenotypes of Z. mobilis were significantly affected by addition of furfural in the medium, we may ask how it happened in the transcriptome. In this study, microarray technology was adopted to answer this question. Based on the genome data of Z. mobilis (Seo et al. 2005), 1,800 gene fragments were amplified by PCR and spotted onto the glass slide. With the sophisticated microarray, the global transcriptional response of Z. mobilis ZM4 to furfural stress was examined at 24 h post-inoculation under normal (media with no furfural) and stress conditions (media with 1.0 g/l furfural). Of the 1,800 genes examined by microarray analysis, 433 genes (24 % of the total number of open reading frames represented on the array) were identified as being significantly up- or down-regulated (P ≤ 0.05) during furfural stress condition (Figs. 2 and 3; Tables S1 and S2). Two hundred sixteen genes were up-regulated 24 h post-inoculation under furfural stress condition and 217 genes were down-regulated (Fig. 3; Tables S1 and S2). Figure 4 provides a summary of the percentage of differentially expressed genes grouped by functional categories according to TIGR’s annotation of the Z. mobilis genome (Seo et al. 2005). We have deposited the entire microarray data at Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE37848) database so interested parties can conduct their analyses. Approximately two thirds of the genes down-regulated in the presence of furfural were related to metabolism. In the presence of furfural, about 49.53 % of the genes related to regulation, cell processes, transport, and unknown function showed greater expression as compared to normal conditions. Nearly 25 % of the genes including plasmid-encoding genes showing greater expression under stress condition remain uncharacterized. However, the genes in ED pathway such as glk, zwf, pgl, pgk, and eno, as well as ethanol fermentation-related genes like pdc and adhB, were shown to be little differentially expressed under stress conditions, indicating that furfural yielded, at most, a subtle negative effect on ED pathway under low concentration of furfural.

Volcano plot result from JMP genomics analysis showing significantly differentially expressed genes under furfural stress condition. Red dots indicate down-regulated genes and green dots indicate up-regulated genes. Black-colored dots were not considered as significantly differentially expressed. The X-axis shows the difference values between furfural stress and normal conditions based on a log2 scale. The Y-axis shows statistical significance values for expression values, based on a−log10 p-value. The gray line shows the statistical significance cutoff used in this study

Hierarchical cluster analysis of significantly differentially expressed ZM4 genes for normal and furfural stress condition at 24 h. Gene expression values were clustered based on their log2-based expression values. Negative numbers (colored red) indicates less relative gene expression under normal conditions and positive numbers (colored green) indicate greater relative gene expression under furfural stress

Number of differentially expressed genes according to the Z. mobilis genome database. Columns: C energy production and conversion, D cell cycle control, mitosis, and meiosis; E amino acid transport and metabolism, F nucleotide transport and metabolism, G carbohydrate transport and metabolism, H coenzyme transport and metabolism, I lipid transport and metabolism, J translation, K transcription, L replication, recombination, and repair, M cell wall/membrane biogenesis, N cell motility, O post-translational modification, protein turnover, chaperones, P inorganic ion transport and metabolism, Q secondary metabolites biosynthesis, transport, and catabolism, R general function prediction only, S function unknown, T signal transduction mechanisms, U intracellular trafficking and secretion, V defense mechanisms, NC not in COGs, PD plasmid encoding genes. Columns above 0 represent the genes whose expression increased in response to 1.0 g/l furfural; columns below 0 represent the genes whose expression decreased in response to furfural stress
To confirm the microarray results, 16 genes involved in metabolism and information transfer, plasmid-encoding genes, and hypothetical proteins were chosen for the qPCR analysis. The data showed that qPCR was more sensitive with greater differences in comparison to the microarray results (Table 1; Fig. 5), which was in keeping with previous reports (Yang et al. 2009). On the other hand, genes involved in ED and pyruvate pathways were also chosen for the qPCR analysis, which showed the same results as in the microarray (data not shown).

Comparison of stationary growth phase gene expression measurements by microarray and qPCR. The gene expression ratios for wild-type Z. mobilis ZM4 under furfural and normal conditions after 24 h of fermentation were log-transformed in base 2. The microarray ratio values were plotted against the qPCR values. A comparison of the two methods indicated a high level of concordance (R = 0.84)
Induction and repression of cell envelope components under furfural stress
As expected, genes involved in cell motility and cell wall/membrane biogenesis were showed to be down-regulated under furfural stress, such as flagellar-related types (flhA, fliE, fliG, flgH, flgL, and ZMO0619), lipoproteins (ZMO0285, ZMO0780, ZMO1525, and oprM), capsule polysaccharide biosynthesis protein kpsC (ZMO0307), efflux protein (ZMO0779), porin protein oprB (ZMO0064), cell division protein ftsQ (ZMO0835), penicillin-binding protein (pbp and ZMO0197), and ion channel protein mscS (ZMO1331). However, the transcripts encoding flagellin domain-containing protein fliC (ZMO0629), rod shape-determining protein MreC (ZMO0356), ion channel protein MscS (ZMO0996), membrane protein (ZMO0216, ZMO1174), organic solvent tolerance protein ostA (ZMO1311), and lipoprotein lgt (ZMO0291) were shown to be more abundant under stress condition by microarray analysis (see Table S1). This suggests that Z. mobilis might minimize furfural-induced damage by altering or modifying the composition and structure of the cell membrane. Especially, gene encoding putative organic solvent tolerance protein ostA may play an important role under stress conditions. Further work may be performed by overexpression of this gene to get more tolerant Z. mobilis strains for producing valuable chemicals.
Repression of protein synthesis under furfural stress
In this study, about 39 genes related to protein synthesis were down-regulated in response to furfural. They belong to ribosomal proteins (such as rpsD, rpsF, rplI, rbsR, frr, and rbfA), tRNA synthetases (proS, alaS, leuS, glyS, pheT, and valS), and amino acid metabolism-related genes (glnA, trpA, trpB, argG, gltB, ilvE, glnB, serA, and serC). Only one amino acid biosynthetic gene (leuC) was induced by 1.5-fold under furfural stress (Tables S1 and S2). Further work about metabolomics data of detected amino acids may be verified by these array results. The repression of these genes may indicate an arrest of overall protein synthesis presumably contributing to the diminished growth, while cells apparently redirected energy toward the increased expression of genes more directly involved in the protective response to an external environment.
Repression of terpenoid biosynthesis under furfural stress
Hopanoids are a class of pentacyclic triterpenoid lipids that occur in a wide range of Gram-negative and Gram-positive bacteria. Squalene–hopene cyclase (shc) is the only enzyme involved in the biosynthesis of hopanoid lipids that has been characterized on the genetic level in Z. mobilis. There are five open reading frames (designated as hpnA-E) in close arrangement with the shc gene (ZMO0872) (Perzl et al. 1998). In this study, the genes such as hpnD (ZMO0870) and hpnE (ZMO0871) involved in hopanoid biosynthesis pathway were shown to be down-regulated (0.7-fold) in the presence of 1.0 g/l furfural after 24 h of incubation. Other terpenoid biosynthesis-related genes, such as dxs (ZMO1234 and ZMO1598) and dxr (ZMO1150), also exhibited a down-regulated expression pattern. The importance of hopanoids for ethanol tolerance of Z. mobilis was firstly demonstrated by an increased growth inhibition of cells with reduced hopanoid content by ethanol (Horbach et al. 1991). Recently, a further study indicated that hopanoids play an important role in maintaining membrane integrity and pH homeostasis in Rhodopseudomonas palustris TIE-1 (Welander et al. 2009). Taken together, these data suggest that furfural has a negative effect on terpenoid biosynthesis and then may damage the cell membrane of Z. mobilis.
Transcripts of gene related to DNA replication, recombination, and repair
Previous studies have demonstrated that furfural may cause DNA damage (Barciszewski et al. 1997; Khan et al. 1995). As expected, seven genes related to DNA replication, recombination, and repair (putative DNA helicase ZMO0351, double-strand break repair helicase addA, double-strand break repair protein addB, rnhB, uracil-DNA glycosylase ung, ZMO1185 and ZMO1562) were revealed as being down-regulated in the presence of 1.0 g/l furfural after 24 h of incubation. However, transcripts encoding the other genes related to DNA replication, recombination, and repair were found to be more abundant under furfural stress (see Table S1), such as intZ (ZMO1930), dnaA (ZMO1356), DNA repair protein radC (ZMO1426), uvrA (ZMO1588), uvrB (ZMO0362), recJ (ZMO1231), recF (ZMO1584), DNA mismatch repair enzyme mutL (ZMO0354), and topA (ZMO1193). This suggests that Z. mobilis might minimize furfural-induced DNA damage by inducing some genes related to DNA replication, recombination, and repair.
Transcripts of universal stress response gene under furfural stress
In addition, universal stress genes, such as groES–groEL (ZMO1928 and ZMO1929), were not affected significantly under furfural stress (data not shown). However, other universal stress genes, such as chaperone protein dnaJ (ZMO0661 and ZMO1545) and ATP-dependent protease lon (ZMO0376), showed higher expression under furfural stress condition.
Transcripts of respiratory chain genes
Five respiratory chain genes were not differentially expressed under furfural stress condition, including ndh (ZMO1113, encoding NADH dehydrogenase), ZMO0022 (putative Fe-S oxidoreductase), ZMO1571 (cytochrome bd-type quinol oxidase subunits 1), ZMO1572 (cytochrome bd-type quinol oxidase subunits 2), and ZMO1844 (oxidoreductase gene). On the other hand, adhA (alcohol dehydrogenase I, ZMO1236), adhB (alcohol dehydrogenase II, ZMO1596), and adhC (ZMO1722) were also not differentially expressed (data not shown). These suggest that the NADH flux from respiration to ethanol synthesis did not redirect under furfural stress (Kalnenieks, 2006). Interestingly, transcripts of the putative respiratory gene rnfA and rnfB (ZMO1814 and ZMO1813), encoding a putative NADH/ubiquinone oxidoreductase subunit, were illustrated to express more greatly (1.9-fold) under furfural stress condition. Another putative respiratory gene rnfC also showed a higher expression level (1.3-fold) under the stress. Previous studies on transcriptomic profiling of Z. mobilis during aerobic and anaerobic fermentation have also verified that rnfA was expressed more greatly (1.9-fold) under aerobic conditions (Yang et al. 2009). There are six genes encoding the putative NADH/ubiquinone oxidoreductase complex (RnfABCDGE) in the ZM4 genome, most homologous to the rnf operon of photosynthetic bacterium Rhodobacter capsulatus, which is involved in electron transport to nitrogenase (Schmehl et al. 1993). However, physiological information about the rnf gene products of Z. mobilis is unavailable (Sootsuwan et al. 2008).
Transcriptional regulation under furfural stress
Eight transcriptional regulators including LysR family (ZMO1336, ZMO0050, ZMO0774 and ZMO0471), LytR family (ZMO1738), GntR family (ZMO1944), TetR family (ZMO0281), LacI family (ZMO1283), and rpoD (ZMO1623) were up-regulated under furfural stress (see Table S1). On the other hand, MerR family (pbrR, ZMO0203), Fis family sigma54 specific transcriptional regulator (ZMO0631), and fliA (ZMO0626) showed more than 0.6-fold change under stress condition. Actually, the genome sequence shows that Z. mobilis has rpoH (ZMO0749, sigma-32 factor), rpoE (ZMO1404, sigma-24 factor), rpoN (ZMO0274, sigma-54 factor), rpoD, rpoA (ZMO0541), and fliA (ZMO0626, sigma F). However, only rpoD and fliA were differentially expressed in the microarray assay.
Other transcriptional regulators such as ZMO1107 (Lrp-like, sharing 40 % identity to E. coli global regulator Lrp) and ZMO0347 (sharing 60 % identity to E. coli global regulator Hfq) (Yang et al. 2009) were not differentially expressed in the studies (nearly 0.8-fold; data not shown). However, previous studies revealed that E. coli global regulator Lrp affected the expression of at least 10 % of all E. coli genes (Hung et al. 2002; Tani et al. 2002). Hfq is an RNA-binding protein that is common to diverse bacterial lineages and has key roles in the control of gene expression. Hfq also affected the translation and turnover rates of specific transcripts, which contributes to complex post-transcriptional networks (Vogel and Luisi 2011; Yang et al. 2009). Hfq could also be associated with E. coli motor protein Rho to mediate transcription antitermination via a novel transcription regulatory mechanism (Rabhi et al. 2011). Recently, studies indicated that Hfq may play an important role in sRNA network control (Hussein and Lim 2011; Storz et al. 2011). However, the mechanisms of translation, transcriptional, post-transcriptional, or sRNA network control in Z. mobilis mediated by Hfq, still remain to be elucidated. Yang et al. (2010) showed that Z. mobilis hfq (ZMO0347) contributes to tolerance against multiple lignocellulosic pretreatment inhibitors, which provide a fundamental example for further studies or industrial strain development in the future.
Two phage shock protein A and C (pspA and pspC) were shown to be more highly differentially expressed. The phage shock protein (Psp) response was originally discovered in P. Model’s laboratory at the Rockefeller University while studying filamentous phage f1 infection of E. coli (Brissette et al. 1990). Now, Psp protein is found out across Gram-positive bacteria, Gram-negative bacteria, and archaea to plants and might perceive cell membrane stress and signal to the transcription apparatus by using an ATP hydrolytic transcription activator to produce effector proteins to overcome the stress (Joly et al. 2010). There are four phage shock proteins in the ZM4 genome (ZMO1061, ZMO1063, ZMO1064, and ZMO1065), which may encode pspF, pspA, pspB, and pspC and consist of a psp regulon (Seo et al. 2005). However, ZMO1062 is a hypothetical protein, which may be included in the psp regulon of Z. mobilis. The function of psp regulon should be elucidated in response to stress in the future.
Induction of plasmid-encoding genes under furfural stress
Interestingly, 27 genes from ZM4 plasmids were shown to be more abundant (1.3–2.0 fold) under furfural stress condition. However, most of these genes encode hypothetical proteins, such as pzmob1_p05, pzmob1_p15, pzmob1_p16, pzmob1_p18, pzmob1_p19, pzmob1_p29, pzmob1_p33, and pzmob1_p35 (see Table S1).
Discussion
Z. mobilis has a number of positive attributes as an ethanologen for converting cellulosic biomass into ethanol or other valuable chemicals. In the present studies, we confirmed that the presence of furfural will negatively affect cell growth, glucose consumption, and ethanol production in Z. mobilis ZM4 fermentation. Furthermore, microarray analysis also revealed that furfural yields multiple effects on various aspects of cellular metabolism at the transcriptional level. This research has provided insights into the molecular response to furfural in Z. mobilis, and it will be helpful to construct more furfural-resistant strains for cellulosic ethanol production. Further work should be focused on functions of these more highly differentially expressed genes in response to stress condition.
Unexpectedly, ED pathway mRNAs such as glk, zwf, pgl, pgk, and eno as well as ethanol fermentation gene transcripts like pdc and adhB showed less differential expression under furfural stress condition (1.0 g/l) but decreased the yield of ethanol to some extent, which indicated that furfural has a subtle negative effect on ED pathway mRNAs under low concentration. These results were also confirmed by qPCR analysis, which showed no change in the two methods of measurement.
We also observed that two phage shock proteins A and C (pspA and pspC) were more highly differentially expressed. We deduce that there is a psp-like regulon in response to stress in Z. mobilis. In E. coli, the phage shock protein stress response system is responsible for repairing damage to the inner membrane of the cell and maintenance of the proton-motive force (pmf) across the inner membrane. The Psp stress response is diverse. For example, phage infection, secretin production, blockage of protein export or fatty acid/phospholipid biosynthesis, organic solvents, heat, osmotic, pH, etc., have been characterized in great detail (Huvet et al. 2011). However, function of psp regulon is still unclear, which should be elucidated in the future on a molecular level.
Seo et al. (2005) describe the Z. mobilis ZM4 (ATCC31821) genome as consisting of a single chromosome and plasmids (Seo et al. 2005), which we utilized for probe design and microarray fabrication. Therefore, the array data in the present study may fully represent the differences between furfural stress and normal conditions since these plasmid DNA sequences were available. We used a multiplex array format with an average probe length of 36 nucleotides and were able to detect significantly differentially expressed genes. Further work should also be focused on plasmid-encoding gene.
Genes involved in cell motility and cell wall/membrane biogenesis showed differential expression under furfural stress, such as ZMO0629, ZMO0356 ZMO0996, ZMO0216, ZMO1174, ZMO1311, ZMO0291, etc. On the other hand, the terpenoid biosynthesis-related genes, such as ZMO0870, ZMO0871, ZMO0872, ZMO1234, ZMO1598, and ZMO1150, have shown a down-regulated expression pattern, which indicated that furfural has a negative effect on terpenoid biosynthesis and then to make a damage on the cell membrane of Z. mobilis. Taken together, we conclude that cell membrane might play important roles in response to furfural stress.
References
Allen SA, Clark W, McCaffery JM, Cai Z, Lanctot A, Slininger PJ, Liu ZL, Gorsich SW (2010) Furfural induces reactive oxygen species accumulation and cellular damage in Saccharomyces cerevisiae. Biotechnol Biofuels 3(1):2–12
Almeida JRM, Modig T, Petersson A, Hahn-Hagerdal B, Liden G, Gorwa-Grauslund MF (2007) Increased tolerance and conversion of inhibitors in lignocellulosic hydrolysates by Saccharomyces cerevisiae. J Chem Tech Biotech 82(4):340–349
Almeida JRM, Roder A, Modig T, Laddan B, Liden G, Gorwa-Grauslund MF (2008) NADH- vs. NADPH-coupled reduction of 5-hydroxymethyl furfural (HMF) and its implications on product distribution in Saccharomyces cerevisiae. Appl Microbiol Biotechnol 78:839–945
Barciszewski J, Siboska GE, Pedersen BO, Clark BFC, Ratten SIS (1997) A mechanism for the in vivo formation of N-6-furfuryladenine, kinetin, as a secondary oxidative damage product of DNA. FEBS Lett 414:457–460
Brissette JL, Russel M, Weiner L, Model P (1990) Phage shock protein, a stress protein of Escherichia coli. P Natl Acad Sci USA 87:862–866
Chomczynski P (1993) A reagent for the single-step simultaneous isolation of RNA, DNA and proteins from cell and tissue samples. BioTechniques 15:532–537
Deanda K, Zhang M, Eddy C, Picataggio S (1996) Development of an arabinose-fermenting Zymomonas mobilis strain by metabolic pathway engineering. Appl Environ Microbiol 62(12):4465–4470
Franden MA, Pienkos PT, Zhang M (2009) Development of a high-throughput method to evaluate the impact of inhibitory compounds from lignocellulosic hydrolysates on the growth of Zymomonas mobilis. J Biotechnol 144(4):259–267
Goodman AE, Rogers PL, Skotnicki ML (1982) Minimal medium for isolation of auxotrophic Zymomonas mutants. Appl Environ Microbiol 44(2):496–498
Gorsich SW, Dien BS, Nichols NN, Slininger PJ, Liu ZL, Skory CD (2006) Tolerance to furfural-induced stress is associated with pentose phosphate pathway genes ZWF1, GND1, RPE1, and TKL1 in Saccharomyces cerevisiae. Appl Microbiol Biotechnol 71:339–349
Gutiérrez T, Ingram LO, Preston JF (2006) Purification and characterization of a furfural reductase (FFR) from Escherichia coli strain LY01-an enzyme important in the detoxification of furfural during ethanol production. J Biotechnol 121:154–164
Gutierrez-Padilla MGD, Karim MN (2005) Influence of furfural on the recombinant Zymomonas mobilis strain CP4 (pZB5) for ethanol production. J Am Sci 1(1):24–27
Horbach S, Neuss B, Sahm H (1991) Effect of azasqualene on hopanoid biosynthesis and ethanol tolerance of Zymomonas mobilis. FEMS Microbiol Lett 79(2–3):347–350
Hung SP, Baldi P, Hatfield GW (2002) Global gene expression profiling in Escherichia coli K12. The effects of leucine-responsive regulatory protein. J Biol Chem 277:40309–40323
Hussein R, Lim HN (2011) Disruption of small RNA signaling caused by competition for Hfq. Proc Natl Acad Sci USA 108(3):1110–1115
Huvet M, Toni T, Sheng X, Thorne T, Jovanovic G, Engl C, Buck M, Pinney JW, Stumpf MPH (2011) The evolution of the phage shock protein response system: interplay between protein function, genomic organization, and system function. Mol Biol Evol 28(3):1141–1155
Jeffries TW (2005) Ethanol fermentation on the move. Nat Biotechnol 2005(23):40–41
Joly N, Engl C, Jovanovic G, Huvet M, Toni T, Sheng X, Stumpf MPH, Buck M (2010) Managing membrane stress: the phage shock protein (Psp) response, from molecular mechanisms to physiology. FEMS Microbiol Rev 34(5):797–827
Kalnenieks U (2006) Physiology of Zymomonas mobilis: some unanswered questions. Adv Microb Physiol 51:73–117
Kerr AL, Jeon YJ, Svenson CJ, Rogers PL, Neilan BA (2010) DNA restriction–modification systems in the ethanologen, Zymomonas mobilis ZM4. Appl Microbiol Biotech 89(3):761–769
Khan QA, Shamsi FA, Hadi SM (1995) Mutagenicity of furfural in plasmid DNA. Cancer Lett 89:95–99
Koopman F, Wierckx N, de Winde JH, Ruijssenaars HJ (2010) Identification and characterization of the furfural and 5-(hydroxymethyl)furfural degradation pathways of Cupriavidus basilensis HMF14. Proc Natl Acad Sci USA 107(11):4919–4924
Kouvelis VN, Saunders E, Brettin TS, Bruce D, Detter C, Han C, Typas MA, Pappas KM (2009) Complete genome sequence of the ethanol producer Zymomonas mobilis NCIMB 11163. J Bacteriol 191(22):7140–7141
Kouvelis VN, Davenport KW, Brettin TS, Bruce D, Detter C, Han C, Nolan M, Tapia R, Damoulaki A, Kyrpides NC, Typas MA, Pappas KM (2011) Genome sequence of the ethanol-producing Zymomonas mobilis subsp. pomaceae lectotype ATCC 29192. J Bacteriol 193(18):5049–5050
Lin F-M, Qiao B, Yuan Y-J (2009a) Comparative proteomic analysis of tolerance and adaptation of ethanologenic Saccharomyces cerevisiae to furfural, a lignocellulosic inhibitory compound. Appl Environ Microbiol 75(11):3765–3776
Lin F-M, Tan Y, Yuan Y-J (2009b) Temporal quantitative proteomics of Saccharomyces cerevisiae in response to a nonlethal concentration of furfural. Proteomics 9:5471–5483
Linger JG, Adney WS, Darzins A (2010) Heterologous expression and extracellular secretion of cellulolytic enzymes by Zymomonas mobilis. Appl Environ Microbiol 76(19):6360–6369
Liu ZL (2006) Genomic adaptation of ethanologenic yeast to biomass conversion inhibitors. Appl Microbiol Biotechnol 73:27–36
Liu ZL, Moon J, Andersh BJ, Slininger PJ, Weber S (2008) Multiple gene-mediated NAD(P)H-dependent aldehyde reduction is a mechanism of in situ detoxification of furfural and 5-hydroxyfurfural by Saccharomyces cerevisiae. Appl Microbiol Biotechnol 81:743–753
Miller EN, Jarboe LR, Yomano LP, York SW, Shanmugam KT, Ingram LO (2009) Silencing of NADPH-dependent oxidoreductase genes (yqhD and dkgA) in furfural-resistant ethanologenic Escherichia coli. Appl Enviro Microbiol 75(13):4315–4323
Miller EN, Turner PC, Jarboe LR, Ingram LO (2010) Genetic changes that increase 5-hydroxymethyl furfural resistance in ethanol-producing Escherichia coli LY180. Biotchnol Lett 32(5):661–667
Mills TY, Sandoval NR, Gill RT (2009) Cellulosic hydrolysate toxicity and tolerance mechanisms in Escherichia coli. Biotchnol Biofuels 2:26–37
Palmqvist E, Hahn-Hagerdal B (2000) Fermentation of lignocellulosic hydrolysates. II: inhibitors and mechanisms of inhibition. Biores Technol 74:25–33
Pappas KM, Kouvelis VN, Saunders E, Brettin TS, Bruce D, Detter C, Balakireva M, Han C, Savvakis G, Kyrpides NC, Typas MA (2011) Genome sequence of the ethanol-producing Zymomonas mobilis subsp. mobilis lectotype ATCC 10988. J Bacteriol 193(18):5051–5052
Perzl M, Reipen IG, Schmitz S, Poralla K, Sahm H, Sprenger GA, Elmar L (1998) Cloning of conserved genes from Zymomonas mobilis and Bradyrhizobium japonicum that function in the biosynthesis of hopanoid lipids. Biochimica et Biophysica Acta (BBA) - Lipids and Lipid Metabolism 1393(1):108–118
Pienkos P, Zhang M (2009) Role of pretreatment and conditioning processes on toxicity of lignocellulosic biomass hydrolysates. Cellulose 16:743–762
Rabhi M, Espeli O, Schwartz A, Cayrol B, Rahmouni AR, Arluison V, Boudvillain M (2011) The Sm-like RNA chaperone Hfq mediates transcription antitermination at Rho-dependent terminators. Embo J 30(14):2805–2816
Ranatunga T, Jervis J, Helm R, McMillan J, Hatzis C (1997) Identification of inhibitory components toxic toward Zymomonas mobilis CP4(pZB5) xylose fermentation. Applied Biochem Biotech 67(3):185–198
Schmehl M, Jahn A, Vilsendorf AMz, Hennecke S, Masepohl B, Schuppler M, Marxer M, Oelze J, Klipp W (1993) Identification of a new class of nitrogen fixation genes in Rhodobacter capsulatus: a putative membrane complex involved in electron transport to nitrogenase. Mol Gen Genet 241:602–615
Seo JS, Chong H, Park HS, Yoon KO, Jung C, Kim JJ, Hong JH, Kim H, Kim JH, Kil JI, Park CJ, Oh HM, Lee JS, Jin SJ, Um HW, Lee HJ, Oh SJ, Kim JY, Kang HL, Lee SY, Lee KJ, Kang HS (2005) The genome sequence of the ethanologenic bacterium Zymomonas mobilis ZM4. Nat Biotechnol 23(1):63–68
Sootsuwan K, Lertwattanasakul N, Thanonkeo P, Matsushita K, Yamada M (2008) Analysis of the respiratory chain in ethanologenic Zymomonas mobilis with a cyanide-resistant bd-type ubiquinol oxidase as the only terminal oxidase and its possible physiological roles. J Mol Microbiol Biotechnol 14(4):163–175
Storz G, Vogel J, Wassarman Karen M (2011) Regulation by small RNAs in bacteria: expanding frontiers. Mol Cell 43(6):880–891
Swings J, Deley J (1977) Biology of Zymomonas. Bacterial Rev 41(1):1–46
Tani TH, Khodursky A, Blumenthal RM, Brown PO, Matthews RG (2002) Adaptation to famine: a family of stationary-phase genes revealed by microarray analysis. Proc Natl Acad Sci USA 99:13471–13476
Vogel J, Luisi BF (2011) Hfq and its constellation of RNA. Nat Rev Micro 9(8):578–589
Wang X, Miller EN, Yomano LP, Zhang X, Shanmugam KT, Ingram LO (2011) Increased furfural tolerance due to overexpression of NADH-dependent oxidoreductase FucO in Escherichia coli strains engineered for the production of ethanol and lactate. Appl Environ Microbiol 77(15):5132–5140
Welander PV, Hunter RC, Zhang L, Sessions AL, Summons RE, Newman DK (2009) Hopanoids play a role in membrane integrity and pH homeostasis in Rhodopseudomonas palustris TIE-1. J Bacteriol 191(19):6145–6156
Widiastuti H, Kim JY, Selvarasu S, Karimi IA, Kim H, Seo JS, Lee DY (2011) Genome-scale modeling and in silico analysis of ethanologenic bacteria Zymomonas mobilis. Biotechnol Bioeng 108(3):655–65
Yang S, Tschaplinski TJ, Engle NL, Carroll SL, Martin SL, Davison BH, Palumbo AV, Rodriguez M Jr, Brown SD (2009) Transcriptomic and metabolomic profiling of Zymomonas mobilis during aerobic and anaerobic fermentations. BMC Genomics 10:34
Yang S, Pelletier DA, Lu TY, Brown SD (2010) The Zymomonas mobilis regulator hfq contributes to tolerance against multiple lignocellulosic pretreatment inhibitors. BMC Microbiol 10:135
Zaldivar J, Martinez A, Ingram LO (1999) Effect of selected aldehydes on the growth and fermentation of ethanologenic Escherichia coli. Biotechnol Bioeng 65:24–33
Zaldivar J, Martinez A, Ingram LO (2000) Effect of alcohol compounds found in hemicellulose hydrolysate on the growth and fermentation of ethanologenic Escherichia coli. Biotechnol Bioeng 68:524–530
Zhang M, Eddy C, Deanda K, Finkelstein M, Picataggio S (1995) Metabolic engineering of a pentose metabolism pathway in ethanologenic Zymomonas mobilis. Science 267(5195):240–243
Zhang J, Zhu Z, Wang X, Wang N, Wang W, Bao J (2010) Biodetoxification of toxins generated from lignocellulose pretreatment using a newly isolated fungus, Amorphotheca resinae ZN1, and the consequent ethanol fermentation. Biotechnol Biofuels 3(1):26–41
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (Grant No. 31000028), Sichuan Key Technology R&D Program (Grant No. 2009NZ00045), and Sci-tech Fund Project of Chinese Academy of Agricultural Sciences (2009 and 2011).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 56 kb)
Rights and permissions
About this article
Cite this article
He, Mx., Wu, B., Shui, Zx. et al. Transcriptome profiling of Zymomonas mobilis under furfural stress. Appl Microbiol Biotechnol 95, 189–199 (2012). https://doi.org/10.1007/s00253-012-4155-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-012-4155-4