
Abstract
Ursodeoxycholic acid is an important pharmaceutical so far chemically synthesized from cholic acid. Various biocatalytic alternatives have already been discussed with hydroxysteroid dehydrogenases (HSDH) playing a crucial role. Several whole-cell biocatalysts based on a 7α-HSDH-knockout strain of Escherichia coli overexpressing a recently identified 7β-HSDH from Collinsella aerofaciens and a NAD(P)-bispecific formate dehydrogenase mutant from Mycobacterium vaccae for internal cofactor regeneration were designed and characterized. A strong pH dependence of the whole-cell bioreduction of dehydrocholic acid to 3,12-diketo-ursodeoxycholic acid was observed with the selected recombinant E. coli strain. In the optimal, slightly acidic pH range dehydrocholic acid is partly undissolved and forms a suspension in the aqueous solution. The batch process was optimized making use of a second-order polynomial to estimate conversion as function of initial pH, initial dehydrocholic acid concentration, and initial formate concentration. Complete conversion of 72 mM dehydrocholic acid was thus made possible at pH 6.4 in a whole-cell batch process within a process time of 1 h without cofactor addition. Finally, a NADH-dependent 3α-HSDH from Comamonas testosteroni was expressed additionally in the E. coli production strain overexpressing the 7β-HSDH and the NAD(P)-bispecific formate dehydrogenase mutant. It was shown that this novel whole-cell biocatalyst was able to convert 50 mM dehydrocholic acid directly to 12-keto-ursodeoxycholic acid with the formation of only small amounts of intermediate products. This approach may be an efficient process alternative which avoids the costly chemical epimerization at C-7 in the production of ursodeoxycholic acid.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Bile acids and their salts are a subgroup of steroids. Primary bile acids are synthesized from cholesterol in the liver, whereas secondary bile acids are metabolites of primary and secondary bile acids as a result of microbial conversion in the intestine (Hofmann et al. 1992).
The secondary bile acid ursodeoxycholic acid (UDCA) represents an industrial relevant bile acid that is used in the therapy of primary sclerosing cholangitis and the medicamentous, non-surgical dissolution of gallstones (Beuers et al. 1992; Gleeson et al. 1990; Igimi and Carey 1981). UDCA has two hydroxyl groups and is an epimer of chenodeoxycholic acid (CDCA). The hydroxyl group at C-3 is present in α-configuration and the hydroxyl group at C-7 in β-configuration.
In the chemical synthesis of UDCA, unconjugated cholic acid serves as raw material for the five-step preparation of CDCA using protective group chemistry (Hofmann 1963). The selective α/β epimerization of the hydroxyl group of C-7 is finalized by two chemical steps (Bortolini et al. 1997; Samuelsson et al. 1960; Sawada et al. 1982).
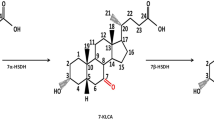
In the intestine, hydroxyl groups of bile acids are oxidized and epimerized by hydroxysteroid dehydrogenases (HSDH) of microbial inhabitants (Ridlon et al. 2006). The application of HSDH as a biocatalytic alternative route in the production of UDCA is shown to dramatically reduce the formation of by-products and incompletely epimerized substrates as they appear in the chemical procedure (Hofmann 1963; Sutherland et al. 1982). Many studies deal with chemo-enzymatic synthesis routes, which consist of the epimerization of the C-7 hydroxyl group of cholic acid as well as the oxidation of the 12α-hydroxyl group of cholic acid followed by the elimination of the carbonyl group through Wolff–Kishner reaction (Braun et al. 2011; Macdonald et al. 1982; Monti et al. 2009; Pedrini et al. 2006; Sutherland and Macdonald 1982). An alternative chemo-enzymatic strategy starts with the chemical oxidation of cholic acid to dehydrocholic acid (DHCA) which is followed by an asymmetric reduction of the 3-carbonyl to 3α-hydroxy group and of the 7-carbonyl to 7β-hydroxy group by the use of 3α-HSDH and 7β-HSDH (Carrea et al. 1992). A subsequent reduction of the 12-carbonyl group by Wolff–Kishner reduction leads to UDCA as depicted in Fig. 1.

HSDH differ in their molecular weight, pH optima, regio- and stereospecificity, and cofactor specificity, respectively (Ridlon et al. 2006). 3α-HSDH converts regio- and enantioselectively 3-carbonyl in 3α-hydroxyl groups of steroids and vice versa. They mostly utilize NAD(H) as cofactor and are found inter alia in Clostridium perfringens (Macdonald et al. 1976), Peptostreptococcus productus (Edenharder et al. 1989a, b), and Comamonas testosteroni (Skålhegg 1974; Skålhegg 1975). The 3α-HSDH from C. testosteroni was already successfully cloned, expressed in Escherichia coli, and its crystal structure was solved (Grimm et al. 2000; Maser et al. 2000; Möbus and Maser 1998).
7β-HSDH catalyze the reduction of the 7β-hydroxyl group as well as the back reaction. They normally utilize NADPH, but NADH-dependent 7β-HSDH are also known. NADPH-dependent representatives have been discovered in P. productus (Edenharder et al. 1989a, b; Hirano and Masuda 1982) and Clostridium species 25.11.c (Edenharder et al. 1989a, b); a NADH-dependent 7β-HSDH has been found in Xanthomonas maltophilia (Medici et al. 2002). Liu et al. (2011) succeeded in cloning and recombinantly expressing a NADPH-dependent 7β-HSDH from Collinsella aerofaciens in E. coli.
Biocatalytic processes involving HSDH require the presence of cofactors, e.g., the pyridine nucleotide cofactors NADH and NADPH. In order to achieve a cost-efficient process, these cofactors must be regenerated instead of using stoichiometric amounts of them. For enantioselective reductions, the enzyme-coupled approach by using formate dehydrogenases (FDH) is still a convenient method to regenerate pyridine nucleotide cofactors (van der Donk and Zhao 2003). The advantage over other systems is the inexpensive substrate formate and the volatile by-product CO2, which results in a favorable thermodynamic equilibrium. Drawbacks include the low specific activity of the FDHs, its sensitivity to organic solvents, and their typical NAD dependence (van der Donk and Zhao 2003). However, in recent works, NADP-accepting mutants of FDH could be developed and applied (Hölsch and Weuster-Botz 2010; Serov et al. 2002; Tishkov et al. 1999).
While isolated enzymes are mostly applied for hydrolysis and isomerization reactions, whole-cell biocatalysts are often used, if cofactors are required to be regenerated (Schmid et al. 2001). In the production of whole cells, the down-streaming process requires less effort (Carballeira et al. 2009). In whole-cell applications, enzymes are protected from the external environment and the cofactors are supplied by the cells (Carballeira et al. 2009; Schmid et al. 2001). Whole-cell biocatalysts also play an important role in the development of multi-enzymatic processes (Carballeira et al. 2009; Monti et al. 2009).
In the present work, a novel whole-cell biocatalyst was created, which catalyzes the stereoselective reduction of DHCA to 3,12-diketo-ursodeoxycholic acid (3,12-diketo-UDCA) with an integrated cofactor regeneration system. For this purpose, 7β-HSDH from C. aerofaciens and a NAD(P)-bispecific FDH mutant from Mycobacterium vaccae for cofactor regeneration were expressed in E. coli. Response surface methodology (RSM) was applied in order to achieve optimal process conditions in regard to maximal conversion and high product concentration in a simple batch biotransformation process. After RSM optimization, a 3α-HSDH from C. testosteroni was additionally integrated to the existing whole-cell system for the one-pot reduction of DHCA to 12-keto-ursodeoxycholic acid (12-keto-UDCA).
Material and methods
Chemicals, enzymes, bacterial strains, and expression vectors
Sodium dehydrocholate and sodium formate were obtained from Sigma-Aldrich (Munich, Germany); NADPH and all other chemicals were obtained from Carl Roth (Karlsruhe, Germany). All enzymes for molecular biological works were obtained from New England Biolab (Frankfurt, Germany); oligonucleotides were obtained from Metabion (Martinsried, Germany).
E. coli DH5α (F− endA1 glnV44 thi-1 recA1 relA1 gyrA96 deoR nupG Φ80dlacZΔM15 Δ(lacZYA-argF) U169, hsdR17(r −K m +K ), λ−; Invitrogen, Karlsruhe, Germany) was used for cloning works. A 7α-HSDH-knockout mutant (hdhA::KanR) of E. coli BL21(DE3) (F– ompT gal dcm lon hsdSB(r −B m −B ) λ(DE3) [lacI lacUV5-T7 gene 1 ind1 sam7 nin5]; Novagen, Darmstadt, Germany) was applied for whole-cell biotransformations.
Plasmids pET21a(+), pET22b(+), and the pET-compatible pCOLADuet (Novagen, Darmstadt, Germany) were used for expression. The vector pCOLADuet was modified by exchanging antibiotic resistance marker from kanamycin resistance to chloramphenicol resistance (pCOLA(mod)).
Molecular biological techniques
General molecular biological works and experiments were performed by standard methods (Sambrook and Russell 2001).
DNA manipulation
The vector pET21a(+)-fdh D221G was a gift from G. Gebhardt (Technische Universität München, Garching, Germany); pET22b(+)-7β-hsdh was provided by L. Liu (Universität Stuttgart, Stuttgart, Germany). Both plasmids were used as DNA template for a polymerase chain reaction (PCR) as well as expression vectors.
For PCR amplification of fdh D221G , the forward primer 5′-CCCaagcttAAGGAGATATATATGGCAAAGGTCCTGTGCGTTCTT-3′, which introduces a ribosomal binding site (rbs; underlined) upstream to the gene, and the reverse primer 5′-ATAAGAATgcggccgcTCAGACCGCCTTCTTGAACTTGG-3′ were used. The PCR fragment was inserted into pET22b(+)-7β-hsdh at the HindIII/NotI restriction sites downstream of the 7β-HSDH gene.
The 7β-HSDH gene was amplified using a forward primer 5′-GCgaattcGTGAAAGGAGATATACATGAACCTGAGGGAGAAGTACGG-3′, which introduced a rbs, and a reverse primer 5′-CCCaagcttCTAGTCGCGGTAGAACGA-3′. The fragment was digested with EcoRI/HindIII and cloned into pET21a(+)-fdh D221G downstream of the FDH gene. For cloning the 7β-HSDH gene into the pCOLA(mod), the procedure was followed as described above with the forward primer 5′-CATGccatggATGAACCTGAGGGAGAAGTACG-3′ comprising an NcoI restriction site that was used instead. Above-mentioned plasmid constructs are depicted in Fig. 2.

Overview of the plasmid constructs. As basis vectors pET22b (a, c), pET21a (b), and a modified pCOLADuet (d) were used (see “Material and methods”). The genes of the 7β-HSDH and formate dehydrogenase (FDH) are under the control of a T7 promoter. In addition, ribosomal binding sites (rbs) were inserted
For the construction of pCOLA(mod)-3α-hsdh, the vector pET22(+)-3α-hsdh was provided by Holger Beuttler (Universität Stuttgart, Stuttgart), which was digested with NdeI and BlpI. The resulting fragment containing the 3α-HSDH gene was cloned into the MCS2 of pCOLA(mod).
Preparation of the E. coli strain and its cultivation for expression and biotransformation
As host strain for the biocatalysts, a knockout strain of E. coli BL21(DE3) was generated in which the 7α-HSDH gene (hdhA) was disrupted by inserting a kanamycin resistance. The strain E. coli BL21(DE3) hdhA::KanR was constructed using the kit system TargeTron™ Gene Knockout System (Sigma-Aldrich, Munich, Germany).
For the cultivation of all investigated strains, 200 mL TB media (with antibiotics) was inoculated with a LB media overnight culture and cultivated at 37 °C in 1 L shaking flasks. At an optical density (OD600) of 0.6–0.8, the expression of recombinant proteins was induced with 1 mM isopropyl-β-d-thio-galactoside and cultivated at 25 °C for 21 h. A correlation of optical density and cell dry weight (CDW) of 0.462 gCDW L−1 OD −1600 was determined.
Activity assays
For enzyme activity measurements, cells pellets were resuspended in potassium phosphate buffer (50 mM, pH 8.0), and cell extracts were prepared. The enzyme activity was determined by measuring the change in UV absorbance at 340 nm in a 1-mL cuvette containing 50 mM potassium phosphate buffer (pH 8.0). Ten millimolars of DHCA and 0.1 mM NADPH for 7β-HSDH activity assays and 100 mM sodium formate and 0.25 mM NADP+ for FDH activity assays were used. One unit is defined as the amount of enzyme which converts 1 μmol NADP+/NADPH min−1 (λ = 340 nm, ε = 6.22 mM−1 cm−1; 24 °C).
Response surface methodology
Response surface methodology (RSM) is a statistical method for the optimization of problems in which several variables influence a response of interest. In the present work, three variables were studied ((1) substrate concentration, (2) cosubstrate concentration, and (3) pH) with regard to the response DHCA conversion.
A central composite design (CCD) was applied for optimal experimental design. The variables, their values, and the experimental are shown in Table 1. Afterward, a second-order polynomial model was fitted that describes the response η as a function of the three variables x j :
where η is the predicted response (relative DHCA conversion), x j the variables under study (x 1 represents DHCA concentration, x 2 formate concentration, and x 3 pH), β j the linear coefficient, β jj the quadratic coefficients, and β ij the interactions coefficients, respectively.
MATLAB 2010a (The Mathworks, Natick, MA, USA) was used for regression analysis of the polynomial model (least square method). Subsequently, the fitted model was refined and non-significant factors were removed.
Steroid reduction with whole-cell biocatalysts and free enzymes
Reductions of DHCA with the whole-cell biocatalyst were performed in parallel operated magnetically stirred (with a cross-shaped stir bar) 4-mL reaction tubes with a lid and a total reaction volume of 2 mL. Upscaled experiments were performed in magnetically stirred (with a cross-shaped stir bar) 20 mL reaction tubes with a lid and a total reaction volume of 10 mL.
At lower pH and high substrate concentrations, DHCA is in suspension; therefore, a defined amount of DHCA was mixed with 50 mM potassium phosphate buffer (for pH 6.0–8.0) and 50 mM Tris buffer (for pH 9.0), respectively, as well as sodium formate solution with the designated pH. The reaction was initiated by addition of cell suspension (9.2 g L−1 dry cell mass) and was incubated at 25 °C and stirring at 350 rpm.
For the use of free enzymes, the same amounts of cells were disrupted using glass beads in a vibratory mill. Resuspended cells were mixed with 50 % (v/v) glass beads and 1 mM phenylmethylsulfonyl fluoride and disrupted in the mill for 5 min at 30 Hz. Then the tubes were centrifuged at 4 °C and 17.880×g for 30 min to remove glass beads and cell debris.
Two-step biotransformation of dehydrocholic acid
Sodium dehydrocholate was converted in threaded bottles (V = 50 mL) with a total reaction volume of 10 mL and mixed with 600 mM sodium formate and potassium phosphate buffer (50 mM, pH 6.5). The reactions were initiated by addition of 18.5 g L−1 cells (dry mass), which were resuspended in potassium phosphate buffer (50 mM, pH 6.5). Reaction tubes were closed and incubated at 25 °C and 350 rpm.
Bile acid analysis
The conversion of experiments was monitored via high-performance liquid chromatography (HPLC) measurements. Methanol was spiked with HPLC samples and diluted with a methanol–water mixture (final ratio 70:30) until substrate and product were solved. After the separation of precipitated proteins, the sample was analyzed by UV detection at 200 nm using a mobile phase of water (pH 2.6 with phosphoric acid) and acetonitrile on a Purospher® STAR RP-18 column (Hibar® RT 125-4 Pre-Packed Column, Purospher® STAR RP-18 endcapped, Merck, Darmstadt, Germany). The quantification of conversion was carried out by comparing peak areas of the compounds.
Results
Strain development
Several plasmid systems were constructed for the reduction of DHCA to 3,12-diketo-UDCA, each of them containing 7β-HSDH and FDHD221G genes in different arrangements (see “Material and methods”). Modified E. coli BL21(DE3) hdhA::KanR, in which the natural occurring gene for 7α-HSDH was disrupted, were (co-)transformed with the plasmids in Fig. 2. These whole-cell biocatalysts were investigated regarding relevant enzyme activities and their capacity to catalyze the reduction of DHCA to 3,12-diketo-UDCA.
The 7β-HSDH and FDH activities strongly vary between the created strains, depicted in Fig. 3. Strains containing the plasmid pET21a(+)-fdh D221G_ 7β-hsdh show an approximately 10-fold higher FDH and a 2.5–5 lower 7β-HSDH activity than the strains containing pET22b(+)-7β-hsdh-fdh D221G due to the different arrangements of both genes. The increase of the gene copy number of 7β-hsdh by cotransforming the plasmid pCOLA(mod)-7β-hsdh results in 27 % higher activity of 7β-HSDH.

Enzyme activities and DHCA conversions of investigated E. coli strains with different plasmids. a Mean enzyme activities and standard errors of 7β-HSDH (black) and FDHD221G (gray) from triplicate samples; b mean DHCA conversion from duplicate samples of 25 mM DHCA and 100 mM sodium formate with 9.2 gCDW L−1 cells after 1 h (black) and 4.25 h (gray)
All constructed strains obtained higher DHCA conversions than the reference strain without FDH activity (pET22b(+)-7β-hsdh) in the standard experimental setup. Strains containing pET21a(+)-7β-hsdh-fdh D221G clearly achieved the highest DHCA conversions within 1 h and 4.25 h, which indicates that the cofactor regeneration is rate limiting. The additional 7β-HSDH gene on the pCOLA(mod)-vector exhibits an exiguous advantage, characterized by a conversion of 71.4 over 67.8 % within 1 h and 97.7 over 97.2 % within 4.25 h, respectively. Since the additional plasmid means the use of a further antibiotic and the resulting improvement was marginal, the strain E. coli BL21(DE3) hdhA::KanR pET21a(+)-fdh D221G -7β-hsdh was selected for further investigations.
Influence of pH
In order to examine the pH influence on the biotransformation activity of the biocatalyst, batch processes were performed a pH 6.0, pH 7.0, pH 8.0, and pH 9.0. The lower pH of 6.0 and pH 7.0 causes that part of the DHCA was not dissolved, which results in an aqueous substrate suspension.
At pH 6.0 and pH 7.0, no DHCA was detected after 24 h as depicted in Fig. 4, which indicates a complete conversion. At pH 9.0, a conversion of 79 % was observed. The reactions rates at pH 6.0 and pH 7.0 are increased compared to higher pH, shown by the conversion of 97 % (pH 6.0 and pH 7.0) compared to 71 % (pH 8.0) and 11 % (pH 9.0) after 1 h. The influence of the different buffer system at pH 9.0 (Tris buffer) was not clarified, but it can be summarized that conversion and reaction rate are higher at slightly acidic pH. A possible explanation could be pH dependencies of the involved enzymes as well as a substrate inhibition of the involved enzyme as only a lower amount of DHCA is dissolved. Furthermore, it was shown that the stirred tank reactor was sufficient for mixing as DHCA in suspension was fully converted. Substrate was substituted from the solid phase rapidly enough.

Influence of pH on whole-cell reduction of 25 mM DHCA. Mean DHCA conversion from duplicate samples after 1 h (black) and 24 h (gray) with 9.2 gCDW L−1 E. coli BL21(DE3) hdhA::KanR pET21a(+)-fdh D221G -7β-hsdh are shown
Model-based optimization of dehydrocholic acid reduction to 3,12-diketo-UDCA
In preliminary experiments, boundaries for statistical experimental design were determined. The influence of different DHCA, formate, and cell concentrations as well as of the pH on the DHCA conversion was investigated in single factor experiments (data not shown). As this procedure cannot take interactions between variables into account, a further optimization step with a RSM was performed. The initial substrate and cosubstrate concentration as well as the initial pH were selected as the most important influencing factors for the reduction of DHCA. The cell concentration was kept constant at 9.2 gCDW L−1.
Experiments were performed in parallel operated batch processes according to the optimal statistical design chosen (CCD). The obtained data (Table 1) was used for the identification of two second-order polynomial models. The models predict the substrate conversion after 3 and 48 h, respectively. This allows the appraisal of the initial reaction rate after 3 h as well as the final conversion after 48 h. Non-significant parameters were removed, p values for the remaining coefficients, and the model itself is listed in Table 4 in the “Appendix.” As a result, we obtain the following model equations:
-
1.
Relative conversion after 3 h (Fig. 5 a1):
$$ \eta = 0.273 - 0.232{x_1} + 0.094{x_2} - 0.115{x_3} + 0.104{x_1}{x_3} + 0.102x_1^2 $$(2) -
2.
Relative conversion after 48 h (Fig. 5 b1):
$$ \eta = 0.451 - 0.193{x_1} + 0.071{x_2} - 0.091{x_3} + 0.027{x_1}{x_3} + 0.072x_1^2 $$(3)

Simulation of the conversion and product concentration in the whole-cell batch reduction of DHCA to 3,12-diketo-UDCA. The results after 3 h (a) and after 48 h (b) are shown. In the top row (1), the conversion for pH levels of 6.0, 6.4, 7.0, 7.6, and 8.0 is shown and the experimental data. In the middle (2), the final product concentration is estimated. In the bottom (3), the product concentration is shown as a function of initial DHCA (substrate) and initial formate (cosubstrate) concentration and the color-coded conversion at an initial pH of 6.4, including the experimental data
Based on Eqs. (2) and (3) and the initial substrate concentration, it is possible to estimate the final product concentrations after a process time of 3 and 48 h, respectively. These are displayed in Fig. 5 a2, b2. Obviously low initial substrate concentrations, high cosubstrate concentrations, and lower initial pH result in higher conversions.
However, the model estimation for an initial pH 6 is not as well (Table 3), which is not surprising as we chose a circumscribed design with only one experimental data point at pH 6. Another reason for the poor accuracy at pH 6 might be the low buffer capacity of the phosphate buffer at this pH range. Consequently, the model quality at this border of the model space is not as good. Therefore, the application of the empirical model was limited to an initial pH of higher than 6.4.
Figure 5 a3, b3 displays both product concentration and conversion (color-coded) in one figure for this optimal pH. Maximal product concentrations can be obtained up to a substrate concentration of ~90 mM. A further increase results in a lower conversion, even at high cosubstrate concentrations or longer process time (48 h).
Both identified second-order polynomials were explored with the objective of finding optimal operating states for maximal product concentrations at a given minimum conversion threshold. Optimal operating states for achieving 95 and 99 % conversion in the simple batch process are shown in Table 2. Initial concentrations of 62.5 and 72 mM DHCA are estimated to be nearly completely converted (99 % conversion) within 3 and 48 h, respectively. A 97 % conversion of 70 mM DHCA is estimated to be achieved within 3 h and 97.9 % of 80 mM DHCA within 48 h, respectively. All identified operating points exhibit an optimal pH of 6.4.
Model validation
Both the 3- and 48-h empirical batch models show good coefficients of determination (R 2 = 0.983, 0.938) and adjusted coefficients of determination (\( \overline{R} ^{2} \) = 0.974, 0.903). Further statistics (p values) and the identified parameters of the refined polynomials are summarized in the “Appendix.”
In order to ensure a rigorous validation, both empirical models were verified experientially with randomly generated operating states in the design space. Furthermore, two experiments were repeated as control of reproducibility (R1 and R2). The new experimental data and the predicted model conversion are summarized in Table 3.
Both R1 and R2 are reproduced very well. For the experimental validation, some minor deviations could be observed, but overall the model prediction is accurate. Figure 6 shows the performance of both models. After a process time of 3 h, a very good correlation between predicted and experimental values (R 2 = 0.943) was observed. Only data point 7 at pH 6.0 diverged. However, this point is at the edge of the model space and should therefore not further be considered, as discussed before. After a process time of 48 h, the correlation is not as good (R 2 = 0.873). All conversions were estimated slightly higher compared to the experimental data, indicating a loss of the biocatalytic performance of the biocatalyst.

Validation of the empirical models for the reduction of DHCA in batch processes. The estimated conversions (y-axis) are plotted against the average values of measured conversions (x-axis) in triplicate. The data used for parameter identification (white points) and model validation (black points) after 3 h (a) and 48 h (b) are indicated
Batch conversion on a 10-mL scale
Optimal process conditions to achieve a minimum conversion of 95 % are estimated at an initial substrate concentration of 72 mM DHCA, 500 mM formate, and pH 6.4 (see Table 2). An experimentally validated operating state near the optimum (experiment no. 3 in Table 1) was chosen for batch conversion on a 10-mL scale, as it provides comparable conversions of 96.2 ± 0.3 % within 3 h and 99.2 ± 0.1 % within 48 h. Furthermore, the use of whole cells and crude extract are compared. A defined cell suspension was split: One part was applied as whole-cell biocatalyst; the other part was disrupted using glass beads. Afterward, the identical volume of either cell suspension or crude extract was added to the reaction mixture. Two reactions with free enzymes were investigated with and without the addition of 0.1 mM NADPH (Fig. 7).

Reduction of DHCA to 3,12-diketo-UDCA in batch processes at 10-mL scale. The mean conversions of 70 mM DHCA with 400 mM formate at pH 6.4 and 25 °C in a duplicate approach were compared; 9.2 gCDW L−1 whole cells (black points) as well as an equivalent of crude cell extract without (black squares) and with the addition of 0.1 mM NADPH (white squares) were applied
When whole cells were applied, no DHCA was detected after a process time of 1 h, indicating 100 % conversion. At 2-mL scale, slightly lower conversions have been observed (96.2 ± 0.3 and 99.2 ± 0.1 %) within 3 and 48 h, respectively. These deviations may be explained by the quality changes of the whole-cell biocatalyst. Batch processes with crude extracts proceeded with clearly lower reaction rate as shown in Fig. 7 (squares). Without cofactor addition, a conversion of 53 % was measured after 24 h. With the addition of 0.1 mM NADPH, the conversion of 70 mM DHCA was significantly increased to 87 % after 24 h.
Two-step whole-cell reduction of dehydrocholic acid with recombinant 7β-HSDH and 3α-HSDH
For direct preparation of 12-keto-UDCA with a single whole-cell biocatalyst, an additional 3α-HSDH from C. testosteroni was transformed into E. coli BL21(DE3) hdhA::KanR pET21a(+)-fdh D221G -7β-hsdh. At this point, the use of the NAD/NADP-bispecific formate dehydrogenase mutant D221G from M. vaccae N10 proves advantageous, as it regenerates NADPH, which is required by 7β-HSDH, as well as NADH, which is required by 3α-HSDH. For the two-step whole-cell batch reduction of 50 mM DHCA, the biocatalyst concentration was increased to 18.5 gCDW L−1. Bile acid compositions were quantified via HPLC.
As shown in Fig. 8, 96 % of DHCA conversion was observed after 1 h. The vast majority of dehydrocholic acid was reduced to 12-keto-UDCA, which accounts for 92 % (46 mM) of the involved reactants. Furthermore, the two possible intermediates, 3,12-diketo-UDCA (1.9 %) and 7,12-diketo-UDCA (2.6 %), were also detected. The ratio of the reactants changed only slightly within the following 3 h, which indicates a complete reaction after 1 h. The reasons could be the consumption of formate as well as an inactivation of the biocatalyst. Nevertheless, the results clearly demonstrate that the biocatalyst E. coli BL21(DE3) hdhA::KanR pET21a(+)-fdh D221G -7β-hsdh pCOLA(mod)-3α-hsdh is capable of reducing both keto groups of dehydrocholic acid, C-3 and C-7, resulting in the formation of 12-keto-UDCA.

Bile acid composition in the two-step batch reduction of 50 mM dehydrocholic acid with 600 mM formate and 18.5 gCDW L−1 biocatalyst at pH 6.5. The concentrations of dehydrocholic acid (black bars), 7,12-diketo-ursodeoxycholic acid (gray bars), 3,12-diketo-ursodeoxycholic acid (white bars), and 12-keto-ursodeoxycholic acid (patterned bars) are shown at different process times
Discussion
Chemo-enzymatic preparation of UDCA may be an alternative to pure chemical preparation, which represents the state of the art. One major step in the chemo-enzymatic route is the reduction of DHCA to 3,12-diketo-UDCA. We developed a novel whole-cell biocatalyst for this reaction by integrating a recently identified 7β-HSDH and a mutant FDH into E. coli BL21(DE3) hdhA::KanR, which had been created especially for this purpose. Several biocatalysts were created and investigated, each having different arrangements of FDH and 7β-HSDH on different vectors. However, both genes remained under the control of the same induction system. This approach is preferable to the use of different induction systems for modifying gene expression levels, since inducers have an influence on cell metabolism. Therefore, it is desirable to select comparable induction conditions for obtaining comparable results.
The biotransformation using E. coli BL21(DE3) hdhA::KanR pET21a(+)-fdh D221G -7β-hsdh showed a clear pH dependency. At pH 6–7, higher conversions were achieved, which is comparable with findings in the work of Liu et al. (2011). At this pH range, higher concentrations of the substrate will be in suspension due to the protonated carboxyl group. It is expected that the product 3,12-diketo-UDCA is more soluble than the substrate. During the process, the substrate is further dissolved. The advantage of the suspension can be explained by both the pH dependence of the enzymes, as well as a circumvention of a possible substrate inhibition of the 7β-HSDH.
The reduction of DHCA to 3,12-UDCA was successfully optimized in a model-based approach. The 3-h batch model can give advices concerning the initial reaction rate; the 48-h batch model allows for the estimation of final conversions. The identified second-order polynomial for the batch processes shows an interaction of initial substrate concentration and initial pH as well as a quadratic influence of the initial substrate concentration on the final product concentration. The linear model coefficient of the cosubstrate concentration indicates a higher conversion using high initial formate concentrations. The empirical batch model can be applied for identifying optimal operating states for maximum product concentrations at arbitrary thresholds of the conversion.
Making use of the data of the 2-mL scale at the 10-mL scale, the bioconversion was successful in a stirred tank. After 1 h, 70-mM DHCA were completely converted in a simple batch process. The comparison of the whole-cell batch bioreduction results with an equivalent of disrupted recombinant E. coli cells showed lower reaction rates, even after addition of 0.1 mM NADPH. The addition of feasible amounts of cofactor increased the reaction rate and the achieved conversion, which, however, remained considerably below the values of the whole-cell approach. The decreased reaction rate of free enzymes could be caused by inactivation of the involved enzymes. The production of biocatalysts for the use as isolated enzymes always causes time-consuming and cost-intensive downstream processing steps (Duetz et al. 2001). In addition, it incurs costs for required cofactors (Koeller and Wong 2001). In contrast, mass transport limitations across the cell membrane could occur in whole-cell systems, lowering space-time yields. Considering the reaction processes as depicted in Fig. 7, it can be assumed that the mass transport across the membrane may not be limiting. Furthermore, the results demonstrate the advantage of whole cells applied in a batch process for the reduction of dehydrocholic acid.
A 3α-HSDH was further integrated into the 7β-HSDH and FDH overexpressing biocatalyst, which makes it capable of catalyzing the two-step reduction of DHCA to 12-keto-UDCA. Here, the advantage of the bispecific formate dehydrogenase mutant can be exploited, as it was not necessary to introduce a second enzyme for cofactor regeneration. In this way, 50-mM DHCA was directly converted to 12-keto-ursodeoxycholic acid within 1 h process time, with only small formation of intermediates.
In a following synthesis step, 12-keto-UDCA can be directly converted to UDCA by Wolff–Kishner reduction. Starting from cholic acid, Monti et al. (2009) obtained a product concentration of 12.5 mM within 5 h in one biocatalytic step using a multi-enzymatic synthesis. Higher initial concentrations were not fully converted. Moreover, five different enzymes are applied in this case, usually requiring five production processes. Therefore, the batch process described in this study with integrated cofactor regeneration could offer an economically advantageous alternative. DHCA can be obtained by oxidizing cholic acid with CrO3. However, this reaction is easy to realize as no stereo- or regioselectivity must be considered (Carrea et al. 1992). DHCA has already been reduced with isolated enzymes without any detectable intermediates by using a glucose dehydrogenase for cofactor regeneration. In this previous approach, 12.5 mM was applied (Carrea et al. 1992). From this, it becomes clear that the simple batch process using whole cells with integrated cofactor regeneration described in this study provides a substantial improvement over previous methods.
References
Beuers U, Spengler U, Kruis W, Aydemir U, Wiebecke B, Heldwein W, Weinzierl M, Pape GR, Sauerbruch T, Paumgartner G (1992) Ursodeoxycholic acid for treatment of primary sclerosing cholangitis: a placebo-controlled trial. Hepatol 16:707–714
Bortolini O, Medici A, Poli S (1997) Biotransformations on steroid nucleus of bile acids. Steroids 62:564–577
Braun M, Link H, Liu L, Schmid RD, Weuster-Botz D (2011) Biocatalytic process optimization based on mechanistic modeling of cholic acid oxidation with cofactor regeneration. Biotechnol Bioeng 108:1307–1317
Carballeira JD, Quezada MA, Hoyos P, Simeó Y, Hernaiz MJ, Alcantara AR, Sinisterra JV (2009) Microbial cells as catalysts for stereoselective red-ox reactions. Biotechnol Adv 27:686–714
Carrea G, Pilotti A, Riva S, Canzi E, Ferrari A (1992) Enzymatic synthesis of 12-ketoursodeoxycholic acid from dehydrocholic acid in a membrane reactor. Biotechnol Lett 14:1131–1134
Duetz WA, van Beilen JB, Witholt B (2001) Using proteins in their natural environment: potential and limitations of microbial whole-cell hydroxylations in applied biocatalysis. Curr Opin Biotechnol 12:419–425
Edenharder R, Pfützner A, Hammann R (1989a) Characterization of NAD-dependent 3 alpha- and 3 beta-hydroxysteroid dehydrogenase and of NADP-dependent 7 beta-hydroxysteroid dehydrogenase from Peptostreptococcus productus. Biochim Biophys Acta 1004:230–238
Edenharder R, Pfützner M, Hammann R (1989b) NADP-dependent 3 beta-, 7 alpha- and 7 beta-hydroxysteroid dehydrogenase activities from a lecithinase-lipase-negative Clostridium species 25.11.c. Biochim Biophys Acta 1002:37–44
Gleeson D, Ruppin DC, Saunders A, Murphy GM, Dowling RH (1990) Final outcome of ursodeoxycholic acid treatment in 126 patients with radiolucent gallstones. Q J Med 76:711–729
Grimm C, Maser E, Möbus E, Klebe G, Reuter K, Ficner R (2000) The crystal structure of 3alpha-hydroxysteroid dehydrogenase/carbonyl reductase from Comamonas testosteroni shows a novel oligomerization pattern within the short chain dehydrogenase/reductase family. J Biol Chem 275:41333–41339
Hirano S, Masuda N (1982) Characterization of NADP-dependent 7 beta-hydroxysteroid dehydrogenases from Peptostreptococcus productus and Eubacterium aerofaciens. Appl Environ Microbiol 43:1057–1063
Hofmann AF (1963) The preparation of chenodeoxycholic acid and its glycine and taurine conjugates. Acta Chem Scand 17:173–186
Hofmann AF, Sjövall J, Kurz G, Radominska A, Schteingart CD, Tint GS, Vlahcevic ZR, Setchell KD (1992) A proposed nomenclature for bile acids. J Lipid Res 33:599–604
Hölsch K, Weuster-Botz D (2010) Enantioselective reduction of prochiral ketones by engineered bifunctional fusion proteins. Biotechnol Appl Biochem 56:131–140
Igimi H, Carey MC (1981) Cholesterol gallstone dissolution in bile: dissolution kinetics of crystalline (anhydrate and monohydrate) cholesterol with chenodeoxycholate, ursodeoxycholate, and their glycine and taurine conjugates. J Lipid Res 22:254–270
Koeller KM, Wong CH (2001) Enzymes for chemical synthesis. Nature 409:232–240
Liu L, Aigner A, Schmid RD (2011) Identification, cloning, heterologous expression, and characterization of a NADPH-dependent 7β-hydroxysteroid dehydrogenase from Collinsella aerofaciens. Appl Microbiol Biotechnol 90:127–135
Macdonald IA, Meier EC, Mahony DE, Costain GA (1976) 3alpha-, 7alpha- and 12alpha-hydroxysteroid dehydrogenase activities from Clostridium perfringens. Biochim Biophys Acta 450:142–153
Macdonald IA, Rochon YP, Hutchison DM, Holdeman LV (1982) Formation of ursodeoxycholic acid from chenodeoxycholic acid by a 7 beta-hydroxysteroid dehydrogenase-elaborating Eubacterium aerofaciens strain cocultured with 7 alpha-hydroxysteroid dehydrogenase-elaborating organisms. Appl Environ Microbiol 44:1187–1195
Maser E, Möbus E, Xiong G (2000) Functional expression, purification, and characterization of 3alpha-hydroxysteroid dehydrogenase/carbonyl reductase from Comamonas testosteroni. Biochem Biophys Res Commun 272:622–628
Medici A, Pedrini P, Bianchini E, Fantin G, Guerrini A, Natalini B, Pellicciari R (2002) 7alpha-OH epimerisation of bile acids via oxido-reduction with Xanthomonas maltophilia. Steroids 67:51–56
Möbus E, Maser E (1998) Molecular cloning, overexpression, and characterization of steroid-inducible 3alpha-hydroxysteroid dehydrogenase/carbonyl reductase from Comamonas testosteroni. A novel member of the short-chain dehydrogenase/reductase superfamily. J Biol Chem 273:30888–30896
Monti D, Ferrandi EE, Zanellato I, Hua L, Polentini F, Carrea G, Riva S (2009) One-pot multienzymatic synthesis of 12-ketoursodeoxycholic acid: subtle cofactor specificities rule the reaction equilibria of five biocatalysts working in a row. Adv Synth Catal 351:1303–1311
Pedrini P, Andreotti E, Guerrini A, Dean M, Fantin G, Giovannini PP (2006) Xanthomonas maltophilia CBS 897.97 as a source of new 7beta- and 7alpha-hydroxysteroid dehydrogenases and cholylglycine hydrolase: improved biotransformations of bile acids. Steroids 71:189–198
Ridlon JM, Kang DJ, Hylemon PB (2006) Bile salt biotransformations by human intestinal bacteria. J Lipid Res 47:241–259
Sambrook J, Russell DW (2001) Molecular cloning: a laboratory manual, 3rd edn. Cold Spring Harbor Laboratory, New York
Samuelsson B, Bergman S, Bak TA, Varde E, Westin G (1960) Preparation of ursodeoxycholic acid and 3alpha,7beta,12alpha-trihydroxycholanic acid. Bile acids and steroids 94. Acta Chem Scand 14:17–20
Sawada H, Kulprecha S, Nilubol N, Yoshida T, Kinoshita S, Taguchi H (1982) Microbial production of ursodeoxycholic acid from lithocholic acid by Fusarium equiseti M41. Appl Environ Microbiol 44:1249–1252
Schmid A, Dordick JS, Hauer B, Kiener A, Wubbolts M, Witholt B (2001) Industrial biocatalysis today and tomorrow. Nature 409:258–268
Serov AE, Popova AS, Fedorchuk VV, Tishkov VI (2002) Engineering of coenzyme specificity of formate dehydrogenase from Saccharomyces cerevisiae. Biochem J 367:841–847
Skålhegg BA (1974) On the 3 alpha-hydroxysteroid dehydrogenase from Pseudomonas testosteroni. Purification and properties. Eur J Biochem 46:117–125
Skålhegg BA (1975) 3alpha-Hydroxysteroid dehydrogenase from Pseudomonas testosteroni: kinetic properties with NAD and its thionicotinamide analogue. Eur J Biochem 50:603–609
Sutherland JD, Macdonald IA (1982) The metabolism of primary, 7-oxo, and 7 beta-hydroxy bile acids by Clostridium absonum. J Lipid Res 23:726–732
Sutherland JD, Macdonald IA, Forrest TP (1982) The enzymic and chemical synthesis of ursodeoxycholic and chenodeoxycholic acid from cholic acid. Prep Biochem 12:307–321
Tishkov VI, Galkin AG, Fedorchuk VV, Savitsky PA, Rojkova AM, Gieren H, Kula MR (1999) Pilot scale production and isolation of recombinant NAD+- and NADP+-specific formate dehydrogenases. Biotechnol Bioeng 64:187–193
Van der Donk WA, Zhao H (2003) Recent developments in pyridine nucleotide regeneration. Curr Opin Biotechnol 14:421–426
Acknowledgments
This work was supported by the Bayerische Forschungsstiftung within the project “Enzyme Catalyzed Steroid Oxidation” (AZ-777-07) and the company PharmaZell GmbH, Raubling, Germany. The authors gratefully acknowledge the support of the TUM Graduate School at the Technische Universität München.
Author information
Authors and Affiliations
Corresponding author
Appendix
Appendix
Rights and permissions
About this article
Cite this article
Braun, M., Sun, B., Anselment, B. et al. Novel whole-cell biocatalysts with recombinant hydroxysteroid dehydrogenases for the asymmetric reduction of dehydrocholic acid. Appl Microbiol Biotechnol 95, 1457–1468 (2012). https://doi.org/10.1007/s00253-012-4072-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-012-4072-6