
Abstract
Anticoagulant heparin has been shown to possess important biological functions that vary according to its fine structure. Variability within heparin’s structure occurs owing to its biosynthesis and animal tissue-based recovery and adds another dimension to its complex polymeric structure. The structural variations in chain length and sulfation patterns mediate its interaction with many heparin-binding proteins, thereby eliciting complex biological responses. The advent of novel chemical and enzymatic approaches for polysaccharide synthesis coupled with high throughput combinatorial approaches for drug discovery have facilitated an increased effort to understand heparin’s structure–activity relationships. An improved understanding would offer potential for new therapeutic development through the engineering of polysaccharides. Such a bioengineering approach requires the amalgamation of several different disciplines, including carbohydrate synthesis, applied enzymology, metabolic engineering, and process biochemistry.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Heparin (Fig. 1a) was discovered in 1916 and entered early clinical trials as the first biopolymeric drug during 1930s before the establishment of the US Food and Drug Administration (FDA) (Linhardt and Gunay 1999). It is still in widespread clinical use as an intravenous anticoagulant with more than 100,000 kilograms produced annually worldwide (Liu et al. 2009). Nearly a century after its discovery, heparin is still derived from animal sources such as porcine intestine and bovine lung. Heparin has a complex and diverse fine structure and is a polydisperse mixture of varying polysaccharide chain lengths. The heparin polysaccharide is comprised primarily of a trisulfated disaccharide repeating unit, 2-O-sulfo-α-l-iduronic acid 1–4 linked to 6-O-sulfo-N-sulfo-α-d-glucosamine (Pervin et al. 1995). Heparin polydispersity, defined as the ratio of weight average molecular weight to number average molecular weight, varies from 1.1 to 1.6 (Liu et al. 2009; Gray et al. 2008). This high polydispersity suggests the presence of a high mole percentage of smaller length chains within certain pharmaceutical heparin preparations. This is consistent with a molecular weight range that varies between 5,000 and 40,000 Da for pharmaceutical heparin (Linhardt and Gunay 1999; Gray et al. 2008). In addition to the trisulfated disaccharide, heparin also contains disaccharides with lesser degrees of sulfation, leading to its structural heterogeneity. Chains composed mainly of undersulfated (unsulfated or monosulfated) disaccharide are typically classified as heparan sulfate (HS). Together with heparin, HS comprises one family of glycosaminoglycans (GAGs) and shares the same biosynthetic pathway.

Structure of heparin, LMWH, and ULMWH fondaparinux. a A representative sequence of heparin comprising a major trisulfated disaccharide sequence, an AT-binding site, and a minor disulfated disaccharide sequence. b LMWH structures generated through peroxide oxidation, deaminative degradation, and chemical or enzymatic β-elimination. c ULMWH, a commercial synthetic pentasaccharide AT-binding domain stabilized as the methyl glycoside called “fondaparinux”
In addition to their anticoagulant activity, heparin and HS are known to possess a wide variety of other biological functions. This family of GAGs plays critical roles in embryonic development, inflammatory response, viral/bacterial infections, and cell differentiation. These biological activities are primarily associated with charge interactions between heparin/HS and heparin-binding proteins, thereby, making control of sulfation level and distribution, through controlled biosynthesis, of critical importance. The advent of low molecular weight heparin (LMWH) (Fig. 1b), derived from heparin through its controlled depolymerization, along with identification of serine protease inhibitor, antithrombin III (AT), and heparin-binding coagulation factors, like thrombin and factor Xa, have led to an improved understanding of the blood coagulation cascade. Development of LMWH was initially aimed at the control of heparin’s hemorrhagic effect by exploiting heparin chain size-dependent differences in binding to antithrombin and factor Xa (Johnson et al. 1976). The ensuing research led to the establishment of LMWHs as new generation of anticoagulant drugs. Anticoagulant activity associated with heparin and HS is primarily attributed to this highly specific interaction with an AT-binding pentasaccharide sequence containing a central 3,6 di-O-sulfo, 2-N-sulfo glucosamine residue (Van Boeckel et al. 1995).When this pentasaccharide sequence is bound to AT, AT undergoes a conformational change thereby accelerating its inhibition of factor Xa in the blood coagulation cascade. AT and thrombin both need to bind to the same heparin chain in a ternary bridging complex to inhibit thrombin and prevent conversion of fibrinogen into an insoluble fibrin clot. This pentasaccharide sequence was first identified through AT-based affinity chromatography following the digestion of heparin. This was followed by efforts to synthesize an ultra LMWH (ULMWH) containing this unique AT pentasaccharide sequence in the 1980s, resulting in the first pentasaccharide analogue, with the aldehyde group at the reducing-end permanently protected as the methyl glycoside (Fig. 1c). The synthesis and purification of such derivatives became simpler, resulting in their industrial-scale production (Petitou et al. 1987). In 2001, a stable methyl glycoside derivative of AT-binding pentasaccharide “fondaparinux” passed through clinical development as the first ULMWH drug leading to Arixtra® and its registration as a new antithrombotic drug in the USA and Europe (Petitou and van Boeckel 2004). Recently, a generic version of fondaparinux has been approved by the US FDA. With an estimated 110 metric tons/year of heparin produced worldwide, its annual market is estimated to be around $3–4 billion. This production volume surpasses other such anticoagulants with sales for Arixtra nearing 500 million dollars annually in the USA and Europe.
An international health crisis, associated with contamination of several heparin batches, began early 2008, reportedly resulting in nearly 100 deaths alone in the USA. The patients displayed minor symptoms like rash, fainting, racing heart, and other more severe symptoms, including hypotension leading to death. Since there was no history of such crisis related to heparin use, it led to suspicions about possible heparin contamination. These contaminated batches, however, had cleared existing quality control and quality assurance tests designed to detect proteins, lipids, DNA, dioxins, and other contaminants. These adverse side effects resulted in the withdrawal of a number of heparin batches from US markets in March 2008 followed by an investigation for the presence of contaminants in these batches. Oversulfated chondroitin sulfate (OSCS), an 18-kDa semisynthetic polysaccharide having a high charge density of −5 (sulfo + carboxyl groups)/disaccharide compared to −3.7/disaccharide for heparin), was discovered to be the contaminating agent (Guerrini et al. 2008). This contaminant was also found to be carried through the production process of certain LMWH products (Zhang et al. 2008a). The rapid and acute response elicited by this OSCS was associated with an anaphylactoid response generated due to activation of kinin–kallikrein pathway in human plasma, leading to formation of vasoactive mediator bradykinin, which caused vasodilation and a sometimes lethal drop in blood pressure (Kishimoto et al. 2008; Li et al. 2009).
Significant efforts have been directed towards elucidating the biological and functional properties of heparin and its related poly-/oligosaccharides to provide for a better understanding of this complex biopolymer. Interdisciplinary studies involving several branches of science have provided a better understanding of heparin’s complex structure and have resulted in novel synthetic approaches and structure–activity relationship (SAR) studies (Fig. 2). The health crisis in 2008 brought to light flaws within animal tissue based heparin production, which begins at slaughterhouses. This mini-review examines the various aspects of polysaccharide development including research currently underway to develop a biologically and structurally similar, bioengineered heparin that can provide a clinically safe option replacing the currently used animal-derived heparin.

SAR characterization of heparin
Current production process for heparin and heparin-like polysaccharides
Commercial-scale heparin production from animal tissues has moved from dog liver to beef lung and finally to porcine intestine. While being similar, these GAGs derived from animal sources have variation in fine structure that relates to difference in functional activities, such as AT- and thrombin-binding affinities (Liu et al. 2009). Additional variation into an already complex structure is added by the variability inherent in raising domesticated animals, including their diet and breed. For example, approximately 30,000–50,000 U (∼300 mg per animal) of USP heparin can be derived from American pigs, while this number is slightly higher for Chinese pigs (Liu et al. 2009).
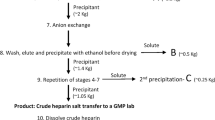
A general outline for heparin processing is presented in Fig. 3. Initial processing is carried out at slaughterhouse without current good manufacturing practices (cGMP) facilities providing an opportunity for contamination. Salting of freed intestines is first carried out using sodium metabisulfite or sodium chloride to preserve the tissues from degradation and dewatering (Liu et al. 2009; Mozen and Evans 1962; Vidic 1981). Tissues are next solubilized using proteases, which leads to a decrease in protein content, followed by heat deactivation for protease removal (Panasyuk 2011). The heparin capture step is either a precipitation step using a hydrophobic quaternary ammonium salt or resin-based chromatographic step using a strong anion exchange resin. After resolubilizing heparin in saline, it can then be precipitated using ethanol or methanol. All these steps are often performed at or near slaughterhouses generating raw heparin, which can be consolidated and shipped for purification at cGMP facilities. Quaternary ammonium salts can often selectively precipitate heparin from tissue extracts without the need for a separate protein removal step. This involves isolation of the insoluble complex followed by resolubilization of the same complex with highly concentrated sodium chloride solution (Mozen and Evans 1962) or with higher alcohols. The heparin salt of Hyamine 1622 flocculates easily at low pH, thereby allowing filtration, and the precipitate can be resolubilized in butanol followed by metathesis, leading to aqueous phase separation (Nominé and Barthelemy 1961). This can then be separated by salting out or through precipitation by miscible solvents like ethanol (Nominé and Barthelemy 1961), leading to purified heparin in high yields and in high potency. The brine formed due to salting of extracted tissues contains heparin with low impurity levels, thereby requiring milder treatments (Vidic 1981). In case of ion-exchange, the negative charge on heparin (−3.7/disaccharide unit) is used for adsorption onto macroreticular anion exchange resin with elution carried out under basic or high salt conditions depending upon the resin. Affinity purification techniques based on affinity for AT has been developed (Rosenberg 1981, 1985); however, the scalability of these approaches poses major problems.

Process flow diagram for production of heparin and LMWHs
At cGMP facilities, raw heparin is resolubilized and filtered at low pH for the removal of protein followed by bleaching step. A microreticular cation exchange resin can be employed for the removal of unwanted cations and the use of ethanol precipitation for nucleotide removal. Remaining salt present within the product is removed through membrane filtration following, which recovered heparin can be dried and analyzed.
LMWHs have a characteristic average MW of <8,000 Da (compared to 12,000–20,000 for heparin) with most of the chains weight having MW below 8,000 (Linhardt and Gunay 1999; Gray et al. 2008). LMWHs generally possess more than 70 U/mg of anti-factor Xa activity with ratio of anti-factor Xa to antifactor IIa activity ≥1.5 (Linhardt and Gunay 1999). LMWHs can be directly recovered from animal-derived heparin by size exclusion chromatography, but such a process is unsuitable for large-scale production; instead, either chemical or enzymatic depolymerization of animal-derived heparin is used to prepare LMWHs (Fig. 3).
The instability of heparin towards reactive oxygen species is utilized in hydrogen peroxide-based degradation method employed for production of ardeparin and parnaparin (Fig. 3). This proceeds by selective oxidation of susceptible nonsulfated uronic residues (Linhardt and Gunay 1999). In one such method, heparin is converted to its heparinic acid form by maintaining the pH around 3–5 followed by heating in the presence of an oxidizing agent resulting in fractions with molecular weights between 4,000 and 12,000 Da (Smith et al. 1984). Compared to other methods of depolymerization, oxidative treatment based on OH radical does not result in any residual structural modification in the LMWH chains (Gray et al. 2008) (Fig. 1b). Oxidative depolymerization using deaminative cleavage with nitrous acid or isoamyl nitrite generates anhydromannose residue at the reducing end of LMWH chains, leading to anhydromannitol residue (Fig. 1b). The presence of N-nitroso compounds obtained as contaminants with LMWHs derived from this degradation has been removed through UV irradiation (Branellec et al. 1997). In a separate strategy, the degrading enzyme heparinase (Liu et al. 2008; Linhardt 1996) can be employed for degradation via a β-elimination cleavage mechanism. Enzymatic action can be mimicked through chemical steps in which the carboxyl group of uronic acid is first esterified, and base treatment leads to selective β-elimination cleavage (Linhardt 1996; Gray et al. 2008).The products generated using enzymatic/chemical β-elimination tends to have a characteristic unsaturated uronic acid residue at the non-reducing end (Mardiguian 1984).
The introduction of Arixtra® (Fig. 1c) as a new anticoagulant drug in 2001 paved the way for industrial production of this AT-binding pentasaccharide. Multi-step chemical synthesis of Arixtra® leads to high production costs, making it more than 1,000-fold costlier than heparin. Expiration of patent protecting Arixtra® has led to development of a generic version at lower costs. Levulinate protected 2-glucuronic acid-anhydro sugar coupling methodology with deprotection and sequential sulfonation reactions leads to generation of fondaparinux (Nadji et al. 2011). This strategy offers reduced reaction time and high coupling yields of β-isomer with increased selectivity making it an efficient and scalable process for industrial-scale production.
Structural and biological activity of heparin
The biological activity of heparin is closely related to its structure and depends upon the interaction of heparin with heparin-binding proteins (Fig. 2). Heparin-binding proteins generally have patches of basic amino acid residues on their surface that interact with the negative sulfo and carboxyl groups present (−3.7/disaccharide unit) throughout the heparin chain length (Liu et al. 2009).
Disaccharide compositional and oligosaccharide mapping analysis are often used to study heparin and LMWH structure. In these methods, disaccharide and/or oligosaccharide fragments of heparin are prepared through enzymatic or chemical digestion and subsequently analyzed to provide detailed structural information (Ly et al. 2010; Yang et al. 2011; Turnbull 1993). Fourier transform ion cyclotron resonance mass spectroscopy (MS), a high resolution technique, can be used to directly determine the chain length and composition of GAGs (Chi et al. 2008; Laremore et al. 2010). Quadupole mass filter leads to removal of ions, thereby leading to an improved signal to noise ratio in Fourier transform MS (Laremore et al. 2010). Disaccharides derived from heparin have been analyzed using liquid chromatography under reverse phased ion-pairing mode coupled to electrospray ionization mass spectrometry with ion trap mass analyzer (Yang et al. 2011). Capillary electrophoresis (CE) coupled with ultraviolet or laser-induced fluorescence detection has been routinely used earlier for detection of heparin/heparan sulfate disaccharides (Pervin et al. 1993; Mao et al. 2002). Multi-dimensional NMR has been employed for assignment of 13C–15N labeled bioengineered heparin (Zhang et al. 2008b). These structural evaluation tools were critical towards determination of contaminant in adulterated heparin batches. 1D NMR and CE showed distinct variation between control lots and contaminated batches with differences in acetyl region (2.0–2.2 ppm) along with a leading peak before heparin in CE (Liu et al. 2009). NMR was employed for determination of oversulfated chondroitin sulfate as the contaminant present in heparin batches (Guerrini et al. 2008).
Bioassays are routinely used for activity studies of heparin (Linhardt et al. 1992). Surface plasmon resonance can also be used to determine kinetics and thermodynamics of heparin interaction with heparin-binding proteins affording binding kinetics and thermodynamics (Muñoz et al. 2005; Beaudet et al. 2011). Pharmacokinetic and pharmacodynamic studies have been performed on heparin delivered through oral, subcutaneous, and intravenous routes (Mousa et al. 2007).
Biosyntheis of heparin/HS
Heparin and HS share the same eukaryotic biosynthetic pathway (Fig. 4), beginning in the endoplasmic reticulum with most modifications occurring within the Golgi (Fig. 5). Different sulfation patterns in heparin and HS provide for diverse biological activities with their SAR (Fig. 2) controlled through the action of genome-encoded biosynthetic enzymes. Heparin is biosynthesized as a proteoglycan, a number of GAG chains attached to the serine residues of serglycin core protein (Sawesi et al. 2010), while HS chains are attached to a number of different core proteins (Kramer and Yost 2003). The heparin and HS GAGs are attached to their core protein through a common tetrasaccharide linker (–GlcAβ1–3Galβ1–3Galβ1–4Xylβ1–), the synthesis of which takes place in the endoplasmic reticulum (Robinson et al. 1978; Carlsson et al. 2008) (Fig. 4). A repeating GlcNAc–GlcA disaccharide unit is then attached to this tetrasaccharide linker with the sequential addition, to the non-reducing end, of uridine diphosphate (UDP) sugars through the action of EXT (Exotosin genes) glycosyltranferases (Esko and Selleck 2002). This is followed by N-deacetylation and N-sulfonation through the action of a bifunctional N-deacetylase/N-sulfotranferase (NDST) (four isoforms) (Aikawa et al. 2001). This serves as a critical step, providing substrate specificity for the appropriate action of the other modification enzymes (Kusche et al. 1991a). This N-sulfo group containing polysaccharide is then modified by C5-epimerase, converting GlcA flanked by GlcNS or GlcN to IdoA (Jacobsson et al. 1984; Gorsi and Stringer 2007).The action of 2-O-sulfotranferase then modifies the C2 position of IdoA prior to 6 and 3-O-sulfonation. The 6-O-sulfotranferase modifies the sulfated domains by placing a 6-O-sulfo group on GlcNS or GlcNAc near GlcNS/IdoA (Habuchi et al. 2003). There are three different isoforms of the 6-OST each having similar substrate specificity, but the 6-OST-1 seems to prefer the absence of 2-O-sulfo group (Habuchi et al. 2003). The 3-O-sulfotranferases add a 3-O-sulfo group to GlcNS or GlcNS6S (Gorsi and Stringer 2007). There are six different 3-OST isoforms, which mostly act on GlcNS next to GlcA/IdoA2S. The 3-OST-1 requires 6-O-sulfo GlcNS. These O-sulfo transferases and C5-epimerase are responsible for generating the specific sulfation patterns critical for the biological functions of heparin and HS. Catabolic processing of heparin and HS can take place through the action of heparanase, endo-ß-glucuronidase, and Sulf, an endo 6-O-sulfatase (Levy-Adam et al. 2005; Dhoot et al. 2001; Ai et al. 2003, 2006).

Biosynthetic pathway for heparin and HS. Synthesis of tetrasaccharide linker attached to serine residue of core protein takes place in the endoplasmic reticulum followed by enzymatic elongation and modification of polymeric chains occurring within the Golgi

Biosynthesis of HS and heparin in eukaryotic cells. Arrows in red indicate next step within the biosynthetic pathway. C5-Epi (C5-epimerase), NDST (N-deacetylase/N-sulfotranferase), 2,3,6-OST (2,3,6-O-sulfo transferases) are the enzymes involved in heparin/HS biosynthetic pathway
Most of the biosynthetic enzymatic modifications are incomplete, and epimerization can be reversible, leading to very complex sequence heterogeneity within the heparin and HS family (Bame et al. 1991). The resulting GAG chains show nearly complete sulfo group substitution in heparin (2.7/5 possible), but only partial sulfo group substitution in HS (0–2 of a total of 5 possible). The control of diversity present within HS structure is important for selective biological activity wherein many interactions within biological systems arise due to overall organization of HS domains (Kreuger et al. 2006). Mutations in the heparin HS biosynthetic enzymes have been shown to affect overall functionality. For example, EXT 1 deficient mice suffer from defects in bone growth (Lin et al. 2000), while a similar mutation in humans generates benign cartilaginous tumors (Gorsi and Stringer 2007). Mutations in NDST affect FGF, Wnt, and Hh signaling pathways in Drosophilia sulfateless (Bernfield et al. 1999), with severe phentotype showcased by NDST-1-deficient mice resulting in death (Forsberg and Kjellén 2001). In the case of 2-OST, homozygous mice with mutated 2-OST encoding genes die at birth (Ornitz 2000), while 6-OST deficient embryos of zebrafish possess abnormal muscle phenotype (Bink et al. 2003). Mice deficient in 3-OST also die at birth and display growth retardation in the womb (HajMohammadi et al. 2003). These phenotypic manifestations of mutations in biosynthetic enzymes highlight the important biological role played by heparin and HS and the critical importance of maintaining appropriate sulfation levels and domain structure.
Capsular polysaccharides from Escherichia coli K5 (ATCC 23506) and Pasteurella multicida serve the biological function of molecular camouflage (Roberts 1996; DeAngelis and White 2002) (Fig. 6). These prokaryotic GAGs have a simplified structure containing no sulfo groups. E. coli K5 derived heparosan (−4)GlcA(1–4)GlcNAc(1–) with molecular weight between 10 and 20 kDa is similar to commercial heparin, while P. multicida heparosan has a much larger molecular weight between 200 and 300 kDa (DeAngelis 2008; DeAngelis and White 2002). These bacterial capsular heparosans are shed into fermentation broth allowing their recovery and use as substrates for biosynthetic enzymes. This can result in their modification into biologically active GAGs, such as heparin or HS. Synthesis of such a capsular heparosan in E. coli K5 involves 2-keto-3-deoxyoctulosonic acid (Whitfield and Roberts 1999), leading to the formation of a starting polysaccharide in the periplasmic space, which is sequentially elongated by the glycosyltransferases, KfiA and KfiC, through addition of GlcNAc and GlcA at the non-reducing end (Arrecubieta et al. 2001). It has been suggested that synthesis of such a polysaccharide may involve a hetero-oligomeric complex bound to the membrane comprising of KpsC, KpsD, KpsE, KpsM, KpS, KpsT, and KfiA-D that aid in polymerization and translocation across plasma membrane (Whitfield and Roberts 1999; Arrecubieta et al. 2001; Silver et al. 2001).

Chemoenzymatic synthesis of heparin and neoheparin starting from E. coli capsular polysaccharide heparosan. a This pathway for the chemoenzymatic synthesis of bioengineered heparin closely resembles the heparin/HS biosynthetic pathway. The chemical de-N-acetylation/N-sulfonation reaction might be replaced by an enzymatic reaction relying on NDST. b This pathway for the synthesis of neoheparin requires selective desulfonation and resulfonation steps and affords some 3-O-sulfoglucuronic acid and iduronic acid residues not found in animal-derived heparin
Envisioning an engineered heparin based on fermentation products depends upon the success of E. coli fermentation with suitable recovery operations. Recent studies by our group have demonstrated a successful fermentation at 7-L scale with high cell density (∼85 g DCW/L) and heparosan titers up to 15 g/L with productivity of 0.4 g L−1 h−1 (Wang et al. 2010a; Wang et al. 2011). This was achieved using an exponential feeding strategy, which involved controlling the carbon source as presence of excess glucose leads to formation of toxic byproducts such as acetate resulting in cell death. In another feeding strategy employed by our group, culture pH is maintained using carbon source feeding rate. In this pH-stat feeding strategy, culture pH is known to rise with depletion of the carbon source providing for an efficient control on carbon source addition and avoiding overfeeding (Wang et al. 2011). Besides fermentation optimization, metabolic engineering of bacteria presents an exciting opportunity for increasing productivity levels.
Chemical synthesis of heparin/HS structures
The decade of the 1980s saw a huge impetus to identify and synthesize AT pentasaccharide-binding domain of heparin. As a result, Arixtra®, the stable methyl glycoside derivative of this pentasaccharide, was prepared and developed as a new anticoagulant drug in 2003 (Petitou et al. 1987; Petitou and van Boeckel 2004) (Fig. 1c). Requiring as many as 60 steps for its preparation, the structure of the Arixtra® pentasaccharide was next reduced to a structurally less complex pentasaccharide requiring fewer synthesis steps (Petitou and van Boeckel 2004) (Fig. 7). Such a structurally simplified analogue was synthesized with only O-sulfo group instead of both N-sulfo and O-sulfo groups and O-methyl esters instead of hydroxyl groups (Westerduin et al. 1994). This does not require orthogonal protecting groups for the amino sugars while reducing the number of steps to 25 from earlier 60. Thus, in order to facilitate simpler chemical synthesis, the target structure had to be modified resulting in less natural and possibly more toxic anticoagulant drug. The chemical approach is also inherently unsuitable for synthesizing longer polymeric chains like heparin because of the large number of modest yield steps and side-product formation. Furthermore, separation of products at each step provides for a challenge in scaling up these syntheses when competing against low-cost heparin and LMWHs. The flexibility of chemical synthesis for delivering unnatural oligosaccharides, however, provided an insight into the specific interactions of oligosaccharides, natural and unnatural, and helped to elucidate the specificity of heparin–protein interactions (Lee et al. 2004).

Convergent chemical synthesis of fondaparinux, which resembles the pentasaccharide AT-binding domain of heparin, stabilized as the methyl glycoside
Synthesis of heparin oligosaccharides (Lee et al. 2004) are typically carried out using a stepwise convergent synthesis as shown in the retrosynthetic scheme in Fig. 7. One pot synthesis and selective activation has recently been applied for oligosaccharide synthesis to reduce the number of synthetic steps (Polat and Wong 2007). This one pot synthesis employs rapid oligosaccharide assembly by sequential addition of thioglycoside-building blocks in reducing order of their reactivity (Polat and Wong 2007; Zhang et al. 1999). AT-binding pentasaccharide has been synthesized using one pot synthesis with starting monosaccharides followed by global deprotection and sulfonation (Zhang et al. 1999). In another such effort for synthesizing AT-binding pentasaccharide, sequential glycosylation starting with monomer building blocks was used. This involved 1-thio uronic acid building blocks, which are activated by the use of potent sulfonium activators resulting in highly stereoselective glycosylations leading towards a fully protected pentasaccharide (Codée et al. 2005). Numerous oligosaccharide containing structural variations are required for efficient SAR studies to completely understand heparin specificity with respect to a given target protein (Arungundram et al. 2009). This has been achieved through a preactivation-based one-pot combinatorial synthesis of heparin hexasaccharides (Wang et al. 2010b). In this study, thioglycosyl building blocks activated by thiophilic promoters were used with matching of donor and acceptor pairs, thereby allowing formation of stereospecific disaccharide building blocks. This resulted in a pool of 12 hexasaccharides from initial six disaccharides derived from two common intermediate disaccharides (Arungundram et al. 2009). Analysis of oligosaccharides for binding with FGF-2 showed the importance of N-sulfo groups and GlcNS–IdoA2S–GlcNS (Wang et al. 2010b). Click chemistry has also been applied to development of unnatural disaccharide and tetrasaccharide analogue of heparosan (Bera and Linhardt 2011). Protected glucosamine and glucuronic acid building blocks with azide and alkyne functional groups allow for the rapid and efficient iterative 1,3-dipolar cycloaddition, leading to formation of β(1–4) linkage as heparosan analogues. Microarrays for screening protein binding to synthetic heparin oligosaccharides or polysaccharides attached to a chip through an amine linker, compatible with protection group chemistry, have led to high throughput screening (de Paz et al. 2006; Park et al. 2008). Development of such combinatorial oligosaccharide libraries based on chemical synthesis provides for a powerful tool in SAR studies (Fig. 2) for heparin and HS through the use of such high throughput microarrays.
Chemoenzymatic synthesis of heparin, HS, and heparin oligosaccharides
Commercial-scale production of clinically safe heparin requires a robust process that retains structural and functional properties of USP heparin derived from animal sources. The capsular polysaccharide derived from E. coli K5 strain, heparosan, consisting of repeating [−4) β-d-glucuronic acid (GlcA) (1–4) N-acetyl-α-d-glucosamine (GlcNAc)(1−]n units, has been characterized as a precursor to heparin (Wang et al. 2010a; DeAngelis and White 2002). Heparosan biosynthesis has also been demonstrated in P. multicida (DeAngelis and White 2002). This capsular polysaccharide might be modified into heparin/HS using chemical or enzymatic steps involved in the biosynthetic pathway (Fig. 6).
N-Deacetylated and N-sulfonated heparosan is required for recognition by C-5 epimerase and O-sulfotransferases (Chen et al. 2007; Bame et al. 1991). While the NDST enzyme is involved in the biosynthetic pathway (Aikawa et al. 2001); hydrazine/hydrazine sulfate or NaOH can be used to chemically N-deacteylate heparosan followed by chemical N-sulfonation using trimethylamine-sulfur trioxide complex resulting in a chemically derived N-deacetylated/N-sulfonated intermediate called N-sulfo, N-acetyl heparosan (Wang et al. 2010a)
Structural constraints for formation of pentasaccharide sequence include a GlcNAc terminated GlcNAc–GlcA–GlcNS–IdoA–GlcNS substrate recognition of 3-O-sulfotransferase (Kusche et al. 1991b) for AT binding (Hricovíni et al. 2001). Lindahl and coworkers have demonstrated gram-scale production of heparin-like polysaccharide, “neoheparin,” incorporating antithrombin binding and anticoagulant activity starting from N-deacetylated/N-sulfated heparosan (Lindahl et al. 2004) (Fig. 6). In this approach, C5-epimerization of N-sulfoheparosan is carried out enzymatically using C5-epimerase derived from insect cells with higher conversion (∼60%) of GlcA–IdoA in the presence of divalent cations (Naggi et al. 2001). This is then followed by per-O-sulfonation of OH groups present in N-sulfoheparosan. Graded solvolytic desulfonation leads to removal of more of the unnatural 3-O-sulfo groups than natural 2-O-sulfo groups in IdoA residues. The 3-O sulfo groups of GlcNAc residues were resistant to this desulfonation procedure and were retained compared to 6-O sulfo groups of GlcNAc that were lost. The presence of significant number of 3-O-sulfated GlcNAc residues adjacent to unsulfated GlcA affords a pentasaccharide structurally similar to that in the natural AT-binding site. Selective O-desulfonation and re-N,6-O-sulfonation of this per-O-sulfonated product led to the generation of a low molecular weight product with anticoagulant activity. Unfortunately, the neoheparin thus generated had a high proportion of 3-O-sulfated glucuronic acid sequences, which are absent in animal-derived heparin, along with a significant amount of the unnatural 3-O-sulfo IdoA, which impede anti-factor Xa activity (Rej et al. 1991). Oversulfated chondroitin sulfate, which resulted in the 2008 contamination crisis, also possesses unnatural 3-O-sulfo glucuronic acid residues, and the presence of such unnatural sulfated patterns within neoheparin leads to concerns regarding its clinical safety.
The problems encountered using the chemoenzymatic approach to synthesize neoheparin show the challenges in controlling sulfation patterns. Enzymatic approaches lack O-sulfo groups in unnatural sites like C3 of IdoA and GlcA. Enzymes involved in heparin and HS biosynthesis have been recently cloned and over-expressed in E. coli. By scaling-up enzyme production, immobilizing these enzymes, and preparing inexpensive 3′-phosphoadenosine 5′-phosphosulfate (PAPS) cofactor, these sulfotransferases may be suitable for large-scale production (Chen et al. 2007; Chen et al. 2005). A small-scale enzymatic approach involving enzymes from heparin and HS biosynthetic pathways has been demonstrated (Zhang et al. 2008b) (Fig. 6). In this study, isotopically labeled heparosan was derived from E. coli K5, while recombinant human C5-epimerase, hamster 2-OST, hamster 6-OST-1, mouse 6-OST-3, and mouse 3-OST-1 were expressed and purified from E. coli. This approach is preceded by chemical N-deacetylation/N-sulfonation of heparosan using sodium hydroxide and trimethylamine–sulfur trioxide complex, giving N-sulfoheparosan with reduced chain length due to alkaline hydrolysis. Treatment of N-sulfoheparosan with 2-OST and C5-epimerase yields an undersulfated heparin that can further be 6-O-sulfonated by the action of 6-OST leading to heparin. The use of 3-OST-1, responsible for modification of very few residues, was the final step resulting in anticoagulant heparin. Heparin and anticoagulant heparin thus generated resembled pharmaceutical heparin with trisulfated IdoA2S(1,4)GlcNS6S, making up 86–89% of the structure. Greater degree of control over sulfation in this approach along with milligram-scale production makes it a rational approach towards preparing a bioengineered heparin comparable to the animal-derived product.
An important aspect of this chemoenzymatic approach is coupling of PAPS regeneration system in enzymatic modifications (Burkart et al. 2000). This involves the use of p-nitrophenyl sulfate as the sulfo donor in the presence of arylsulfotransferase, which can easily be expressed in E. coli, with a catalytic amount of PAPS. This and other innovative technologies (Zhou et al. 2011) reduced the impact of PAPS cost in the chemoenzymatic process, thereby improving scalability.
In addition to the potential for industrial production, a chemoenzymatic approach provides for combinatorial synthesis of oligosaccharides and polysaccharides with novel heparin-like structures. Such combinatorial libraries are essential in elucidating the substrate specificity of various heparin/HS biosynthetic pathway enzymes as well as providing novel biologically active structures. In one such approach, chemically desulfonated N-sulfo heparin was used as a starting material for modification using recombinantly produced immobilized HS sulfotransferases coupled with PAPS regeneration system (Chen et al. 2005). This resulted in heparin/HS-like structures with varying sulfation pattern generated in a block fashion. These enzymatically modified products exhibited biological activities associated with heparin-like FGF2 binding, anticoagulant activity, and binding to herpes simplex virus glycoprotein D (gD). This method can be similarly used to generate an oligosaccharide library starting from disaccharide with unnatural UDP-monosaccharide as donor (Liu et al. 2010). The generated oligosaccharides possess N-sulfo groups specifically placed along the chain and provide for critical control in enzymatic synthesis of HS oligosaccharides and polysaccharides. This combinatorial approach is further strengthened by altered substrate specificities of engineered sulfotransferases, leading to an increased diversity within such oligosaccharide/polysaccharide library.
The combinatorial approach described above can be utilized for development of HS polysaccharides having highly defined functional activity. Using N-sulfo-heparosan, the generation of differentially sulfated HS structures has shown that IdoA residue is not important for binding to AT-III as has been suggested in the literature (Chen et al. 2007). This has significant impact on reducing the complexity of engineered heparin structures as epimerization of starting GlcA is reversible, thereby resulting in significant structural heterogeneity. While IdoA has been deemed important in providing conformational flexibility, thereby leading to favorable binding to AT, it has been demonstrated that this requirement for flexibility is size dependent. This recombinant heparin version termed “Recomparin” lacks IdoA and a 2-O-sulfo group but shows anti-Xa activity at levels similar to heparin.
A recent work has shown that ultralow molecular weight heparins (ULMWHs), resembling porcine and bovine heparin’s pentasaccharide-binding domain, have been synthesized, starting from disaccharide acceptor followed by chain elongation using UDP-sugars and glycosyltransferases. Modification of this backbone chain by heparin/HS biosynthetic pathway enzymes results in constructs resembling the pentasaccharide-binding domain providing for a general chemoenzymatic method aimed at designing oligosaccharides with defined structures similar to Arixtra®. This scalable process provides for an easier method for development of oligosaccharides compared to cumbersome multistep chemical synthesis (Xu et al. 2011).
In summary, these chemoenzymatic methods involving recombinant heparin/HS biosynthetic pathway enzymes provide for scalable processes for bioengineered heparin/HS structures along with combinatorial tools for discovery of new therapeutics having simplified structures along with defined biological activity.
Metabolic engineering for heparin production
As a result of being produced as a serglycin proteoglycan in eukaryotic mast cells Golgi (Linhardt and Gunay 1999), the genetic engineering of bacteria for heparin synthesis is technologically infeasible. Such an approach would require engineering of biosynthetic pathways and enzymes along with incorporation of activities associated with Golgi within the periplasmic space in bacteria that are known to produce structurally simple GAGs like heparosan, hyaluronan, and chondroitin (DeAngelis and White 2002; Wang et al. 2010a). Owing to these problems, the biosynthesis of heparin within eukaryotic systems like yeast, insect cells, and Chinese hamster ovary (CHO) cells appear to be more achievable. Both yeast and CHO cells are widely used in the biotechnological field with protocols in place for their growth and purification of therapeutics derived from them while getting rid of the contaminating proteins (Yang et al. 2011).
Yeast strains have been shown to be capable of generating essential glycosylation patterns in mammals. Engineered yeast strains have been shown to be capable of producing antibodies (e.g., anti CD antibodies) with unique glycosylation, thereby resulting in enhancement of biological activity (Li et al. 2006). They have been shown to be capable of generating complex hybrid mammalian glycoproteins from mannose yeast glycoproteins (Hamilton and Gerngross 2007). However, yeast does not produce HS, and biosynthesis of heparin in yeast cells would be extremely challenging, entailing the expression of high levels of core protein along with controlled expression of enzymes from the entire biosynthetic pathway (Laremore et al. 2009). However, as a simpler recombinant protein expression system, efforts have been underway for expression of sulfotransferases (N-sulfotransferase, 3-OST, 2-OST, and 6-OST) and the preparation of PAPS in yeast (Zhou et al. 2011). In addition to their ease of purification, these secreted enzymes have also been shown to possess higher specific activity and thermostability and do not contain endotoxins. Yeast expression might serve as a critical optimization feature for scaling-up the chemoenzymatic production of bioengineered heparin. Glycoproteins with glycan structures similar to humans have been expressed in insect cell systems (Wolff et al. 2001). Insect cells are also capable of the biosynthesis of HS (Staatz et al. 2001; Bernfield et al. 1999) and thus might be metabolically engineered to produce heparin. One issue using insect cells is the difficulty associated with controlling baculorvirus expressing system for commercial scale production (Yin et al. 2007). Further studies of insect cells might, however, be warranted.
CHO cells are mammalian cells known to produce HS; therefore, they may be capable of producing heparin as it is produced through the same biosynthetic pathway (Bame et al. 1991; Zhang et al. 2006). CHO cells are deficient in several GAG modification enzymes and also lack granules required for the storage of the heparin proteoglycans, serglycin. They express two out of four N-sulfotransferases, one out of three 6-O-sulfotranferases, and none of the 3-O-sulfotranferases (Zhang et al. 2006). Mutant CHO cells having deficient N-sulfotransferase have been shown to possess more sparse NS distribution when compared to the wild type (Bame et al. 1991). As N-sulfonation provides for an in increase in the substrate specificity of the O-sulfotransferases, genetic engineering of CHO cells for heparin production seems promising. Mutant CHO cells have been shown to gain AT binding (Zhang et al. 2001), heparin cofactor II binding, and herpes simplex virus entry (O’Donnell et al. 2006). However, the production levels of HS in CHO cells are very low in comparison to the high levels of heparin found in (Zhang et al. 2006) mammalian mast cells. The generation of immortalized mastocytoma cell lines, a rare form of mast cell cancer, might offer another target for metabolic engineering. However, CHO cells currently provide the best alternative route for generation of heparin in eukaryotic cell lines.
The recent thrust towards development of an engineered version of heparin has focused on heparosan derived from simpler prokaryotic bacterial cells. Metabolic engineering of bacterial cells provides for an alternative approach towards increasing productivity levels of heparosan, which, when coupled with optimized fermentation strategies, can potentially lead to an economically viable engineered heparin. It has been suggested that heparosan shares its biosynthetic pathway partially with cell wall biosynthesis. Towards this goal, UDP glucose dehydrogenase, thought to be the rate-limiting step in mammalian GAG synthesis, was overexpressed in E. coli, resulting in decreased heparosan production with unaltered chain length (Roman et al. 2003), possibly due to reduction in UDP-GlcNAc, which becomes rate limiting besides interfering with polymerization (Roman et al. 2003; Wang et al. 2011). Modulating cell surface interactions with capsules through mutation of waaR gene involved in lipopolysaccharide outer-core biosynthesis has been shown to increase heparosan production (Taylor et al. 2006). In another metabolic engineering approach, controlled over-expression of glycosyltransferases, KfiA and KfiC, can direct the biosynthesis of excess heparosan, drawing sugars away from cell wall biosynthesis (Wang et al. 2011).
Conclusion and future work
Understanding complex heparin structures that show widespread involvement in diverse biological functions has generated interest in the preparation and synthesis of heparin and related poly-/oligosaccharides. Significant control over heparin’s functions might be achieved by modifying its fine structure, thereby leading to efficacious new drugs that can be targeted towards specific biological functions. Chemical synthesis will continue to drive the screening of novel and unnatural GAG sequences. Enzyme-assisted synthesis should provide greater control with regards to modification of heparin structure leading to an enhanced understanding of heparin in SAR (Fig. 2). SAR studies are currently focused on reducing the detection limits and the use of high throughput microarray and microfluidic methods (Barbulovic-Nad et al. 2008; Martin et al. 2009), which can help in deciphering these complex structures obtained from diverse sources. Evolution of a bioengineered version of heparin derived from non-animal sources having well-defined structural and functional properties with good control over its properties could lead to a clinically safer and potent version of heparin. This might also reduce side effects and health concerns around the oldest biopolymeric drug and open up new opportunities for the development of novel carbohydrate based therapeutics.
References
Ai X, Do AT, Lozynska O, Kusche-Gullberg M, Lindahl U, Emerson CP Jr (2003) QSulf1 remodels the 6-O sulfation states of cell surface heparan sulfate proteoglycans to promote Wnt signaling. J Cell Biol 162:341–351
Ai X, Do AT, Kusche-Gullberg M, Lindahl U, Lu K, Emerson CP (2006) Substrate specificity and domain functions of extracellular heparan sulfate 6-O-endosulfatases, QSulf1 and QSulf2. J Biol Chem 281:4969–4976
Aikawa J, Grobe K, Tsujimoto M, Esko JD (2001) Multiple isozymes of heparan sulfate/heparin GlcNAcN-deacetylase/GlcN N-sulfotransferase. Structure and activity of the fourth member, NDST4. J Biol Chem 276:5876–5888
Arrecubieta C, Hammarton TC, Barrett B, Chareonsudjai S, Hodson N, Rainey D, Roberts IS (2001) The transport of group 2 capsular polysaccharides across the periplasmic space in Escherichia coli. Roles for the KpsE and KpsD proteins. J Biol Chem 276:4245–4250
Arungundram S, Al-Mafraji K, Asong J, Leach FE III, Amster IJ, Venot A, Turnbull JE, Boons G-J (2009) Modular synthesis of heparan sulfate oligosaccharides for structure−activity relationship studies. J Am Chem Soc 131:17394–17405
Bame KJ, Lidholt K, Lindahl U, Esko JD (1991) Biosynthesis of heparan sulfate. Coordination of polymer-modification reactions in a Chinese hamster ovary cell mutant defective in N-sulfotransferase. J Biol Chem 266:10287–10293
Barbulovic-Nad I, Yang H, Park PS, Wheeler AR (2008) Digital microfluidics for cell-based assays. Lab Chip 8:519–526
Beaudet JM, Weyers A, Solakyildirim K, Yang B, Takieddin M, Mousa S, Zhang F, Linhardt RJ (2011) Impact of autoclave sterilization on the activity and structure of formulated heparin. J Pharm Sci 100:3396–3404
Bera S, Linhardt RJ (2011) Design and synthesis of unnatural heparosan and chondroitin building blocks. J Org Chem 76:3181–3193
Bernfield M, Götte M, Park PW, Reizes O, Fitzgerald ML, Lincecum J, Zako M (1999) Functions of cell surface heparan sulfate proteoglycans. Annu Rev Biochem 68:729–777
Bink RJ, Habuchi H, Lele Z, Dolk E, Joore J, Rauch GJ, Geisler R, Wilson SW, den Hertog J, Kimata K, Zivkovic D (2003) Heparan sulfate 6-O-sulfotransferase is essential for muscle development in zebrafish. J Biol Chem 278:31118–31127
Branellec J, Espejo J, Picart P (1997) Purified heparin fractions, method for obtaining them and pharmaceutical compositions containing them. US Patent 5,599,801
Burkart MD, Izumi M, Chapman E, Lin CH, Wong CH (2000) Regeneration of PAPS for the enzymatic synthesis of sulfated oligosaccharides. J Org Chem 65:5565–5574
Carlsson P, Presto J, Spillmann D, Lindahl U, Kjellén L (2008) Heparin/heparan sulfate biosynthesis. Processive formation of N-sulfated domains. J Biol Chem 283:20008–20014
Chen J, Avci FY, Muñoz EM, McDowell LM, Chen M, Pedersen LC, Zhang L, Linhardt RJ, Liu J (2005) Enzymatic redesigning of biologically active heparan sulfate. J Biol Chem 280:42817–42825
Chen J, Jones CL, Liu J (2007) Using an enzymatic combinatorial approach to identify anticoagulant heparan sulfate structures. Chem Biol 14:986–993
Chi L, Wolff JJ, Laremore TN, Restaino OF, Xie J, Schiraldi C, Toida T, Amster IJ, Linhardt RJ (2008) Structural analysis of bikunin glycosaminoglycan. J Am Chem Soc 130:2617–2625
Codée JDC, Stubba B, Schiattarella M, Overkleeft HS, van Boeckel CAA, van Boom JH, van der Marel GA (2005) A modular strategy toward the synthesis of heparin-like oligosaccharides using monomeric building blocks in a sequential glycosylation strategy. J Am Chem Soc 127:3767–3773
de Paz JL, Noti C, Seeberger PH (2006) Microarrays of synthetic heparin oligosaccharides. J Am Chem Soc 128:2766–2767
DeAngelis PL (2008) Heparosan-based biomaterials and coatings and methods of production and use thereof. US patent application 2008/0226690 A1
DeAngelis PL, White CL (2002) Identification and molecular cloning of a heparosan synthase from Pasteurella multocida Type D. J Biol Chem 277:7209–7213
Dhoot GK, Gustafsson MK, Ai X, Sun W, Standiford DM, Emerson CP Jr (2001) Regulation of Wnt signaling and embryo patterning by an extracellular sulfatase. Science 293:1663–1666
Esko JD, Selleck SB (2002) Order out of chaos: assembly of ligand binding sites in heparan sulfate. Annu Rev Biochem 71:435–471
Forsberg E, Kjellén L (2001) Heparan sulfate: lessons from knockout mice. J Clin Invest 108:175–180
Gorsi B, Stringer SE (2007) Tinkering with heparan sulfate sulfation to steer development. Trends Cell Biol 17:173–177
Gray E, Mulloy B, Barrowcliffe TW (2008) Heparin and low-molecular-weight heparin. Thromb Haemost 99:807–818
Guerrini M, Beccati D, Shriver Z, Naggi A, Viswanathan K, Bisio A, Capila I, Lansing JC, Guglieri S, Fraser B, Al-Hakim A, Gunay NS, Zhang Z, Robinson L, Buhse L, Nasr M, Woodcock J, Langer R, Venkataraman G, Linhardt RJ, Casu B, Torri G, Sasisekharan R (2008) Oversulfated chondroitin sulfate is a contaminant in heparin associated with adverse clinical events. Nat Biotechnol 26:669–675
Habuchi H, Miyake G, Nogami K, Kuroiwa A, Matsuda Y, Kusche-Gullberg M, Habuchi O, Tanaka M, Kimata K (2003) Biosynthesis of heparan sulphate with diverse structures and functions: two alternatively spliced forms of human heparan sulphate 6-O-sulphotransferase-2 having different expression patterns and properties. Biochem J 371:131–142
HajMohammadi S, Enjyoji K, Princivalle M, Christi P, Lech M, Beeler D, Rayburn H, Schwartz JJ, Barzegar S, de Agostini AI, Post MJ, Rosenberg RD, Shworak NW (2003) Normal levels of anticoagulant heparan sulfate are not essential for normal hemostasis. J Clin Invest 111:989–999
Hamilton SR, Gerngross TU (2007) Glycosylation engineering in yeast: the advent of fully humanized yeast. Curr Opin Biotechnol 18:387–392
Hricovíni M, Guerrini M, Bisio A, Torri G, Petitou M, Casu B (2001) Conformation of heparin pentasaccharide bound to antithrombin III. Biochem J 359:265–272
Jacobsson I, Lindahl U, Jensen J, Rodén L, Prihar H, Feingold DS (1984) Biosynthesis of heparin. Substrate specificity of heparosan N-sulfate D-glucuronosyl 5-epimerase. J Biol Chem 259:1056–1063
Johnson EA, Kirkwood TB, Stirling Y, Perez-Requejo JL, Ingram GI, Bangham DR, Brozović M (1976) Four heparin preparations: anti-Xa potentiating effect of heparin after subcutaneous injection. Thromb Haemost 35:586–591
Kishimoto TK, Viswanathan K, Ganguly T, Elankumaran S, Smith S, Pelzer K, Lansing JC, Sriranganathan N, Zhao G, Galcheva-Gargova Z, Al-Hakim A, Bailey GS, Fraser B, Roy S, Rogers-Cotrone T, Buhse L, Whary M, Fox J, Nasr M, Dal Pan GJ, Shriver Z, Langer RS, Venkataraman G, Austen KF, Woodcock J, Sasisekharan R (2008) Contaminated heparin associated with adverse clinical events and activation of the contact system. N Engl J Med 358:2457–2467
Kramer KL, Yost HJ (2003) Heparan sulfate core proteins in cell-cell signaling. Annu Rev Genet 37:461–484
Kreuger J, Spillmann D, Li JP, Lindahl U (2006) Interactions between heparan sulfate and proteins: the concept of specificity. J Cell Biol 174:323–327
Kusche M, Hannesson HH, Lindahl U (1991a) Biosynthesis of heparin. Use of Escherichia coli K5 capsular polysaccharide as a model substrate in enzymic polymer-modification reactions. Biochem J 275:151–158
Kusche M, Oscarsson LG, Reynertson R, Rodén L, Lindahl U (1991b) Biosynthesis of heparin. Enzymatic sulfation of pentasaccharides. J Biol Chem 266:7400–7409
Laremore TN, Zhang F, Dordick JS, Liu J, Linhardt RJ (2009) Recent progress and applications in glycosaminoglycan and heparin research. Curr Opin Chem Biol 13:633–640
Laremore TN, Leach FE III, Amster IJ, Linhardt RJ (2010) Electrospray ionization Fourier transform mass spectrometric analysis of intact bikunin glycosaminoglycan from normal human plasma. Int J Mass spectrom 305:109–115
Lee JC, Lu XA, Kulkarni SS, Wen YS, Hung SC (2004) Synthesis of heparin oligosaccharides. J Am Chem Soc 126:476–477
Levy-Adam F, Abboud-Jarrous G, Guerrini M, Beccati D, Vlodavsky I, Ilan N (2005) Identification and characterization of heparin/heparan sulfate binding domains of the endoglycosidase heparanase. J Biol Chem 280:20457–20466
Li H, Sethuraman N, Stadheim TA, Zha D, Prinz B, Ballew N, Bobrowicz P, Choi BK, Cook WJ, Cukan M, Houston-Cummings NR, Davidson R, Gong B, Hamilton SR, Hoopes JP, Jiang Y, Kim N, Mansfield R, Nett JH, Rios S, Strawbridge R, Wildt S, Gerngross TU (2006) Optimization of humanized IgGs in glycoengineered Pichia pastoris. Nat Biotechnol 24:210–215
Li B, Suwan J, Martin JG, Zhang F, Zhang Z, Hoppensteadt D, Clark M, Fareed J, Linhardt RJ (2009) Oversulfated chondroitin sulfate interaction with heparin-binding proteins: new insights into adverse reactions from contaminated heparins. Biochem Pharmacol 78:292–300
Lin X, Wei G, Shi Z, Dryer L, Esko JD, Wells DE, Matzuk MM (2000) Disruption of gastrulation and heparan sulfate biosynthesis in EXT1-deficient mice. Dev Biol 224:299–311
Lindahl U, Li JP, Kusche-Gullberg M, Salmivirta M, Alaranta S, Veromaa T, Emeis J, Roberts I, Taylor C, Oreste P, Zoppetti G, Naggi A, Torri G, Casu B (2004) Generation of “Neoheparin” from E. coli K5 capsular polysaccharide. J Med Chem 48:349–352
Linhardt RJ (1996) Analysis of glycosaminoglycans with polysaccharide lyases. Curr Protoc Mol Biol 17.13B.1–17.13B.16
Linhardt RJ, Gunay NS (1999) Production and chemical processing of low molecular weight heparins. Semin Thromb Hemost 25:5–16
Linhardt RJ, Ampofo SA, Fareed J, Hoppensteadt D, Mulliken JB, Folkman J (1992) Isolation and characterization of human heparin. Biochemistry 31:12441–12445
Liu D, Pojasek K, Shriver Z, Holley K, El-Shabrawi Y, Venkataraman G, Sasisekharan R (2008) Methods for preparing low molecular weight heparin with modified heparinase III. US Patent 7,390,633 B2
Liu H, Zhang Z, Linhardt RJ (2009) Lessons learned from the contamination of heparin. Nat Prod Rep 26:313–321
Liu R, Xu Y, Chen M, Weïwer M, Zhou X, Bridges AS, DeAngelis PL, Zhang Q, Linhardt RJ, Liu J (2010) Chemoenzymatic design of heparan sulfate oligosaccharides. J Biol Chem 285:34240–34249
Ly M, Laremore TN, Linhardt RJ (2010) Proteoglycomics: recent progress and future challenges. OMICS 14:389–399
Mao W, Thanawiroon C, Linhardt RJ (2002) Capillary electrophoresis for the analysis of glycosaminoglycans and glycosaminoglycan derived oligosaccharides. Biomed Chromatogr 16:77–94
Mardiguian JS (1984) Heparin esters and processes for their preparation. US Patent 4,440,926
Martin JG, Gupta M, Xu Y, Akella S, Liu J, Dordick JS, Linhardt RJ (2009) Toward an artificial Golgi: redesigning the biological activities of heparan sulfate on a digital microfluidic chip. J Am Chem Soc 131:11041–11048
Mousa SA, Zhang F, Aljada A, Chaturvedi S, Takieddin M, Zhang H, Chi L, Castelli MC, Friedman K, Goldberg MM, Linhardt RJ (2007) Pharmacokinetics and pharmacodynamics of oral heparin solid dosage form in healthy human subjects. J Clin Pharmacol 47:1508–1520
Mozen MM, Evans TD (1962) Process for purifying heparin. US Patent 3,058,884
Muñoz EM, Yu H, Hallock J, Edens RE, Linhardt RJ (2005) Poly (ethylene glycol)-based biosensor chip to study hepari protein interactions. Anal Biochem 343:176–178
Nadji S, Smoot JT, Vanartsdalen JA (2011) Process for preparaing fondaparinux sodium and intermediates useful in the synthesis thereof. US Patent Application US 2011/0105418 A1
Naggi A, Torri G, Casu B, Oreste P, Zoppetti G, Li JP, Lindahl U (2001) Toward a biotechnological heparin through combined chemical and enzymatic modification of the Escherichia coli K5 polysaccharide. Semin Thromb Hemost 27:437–443
Nominé G, Barthelemy P (1961) Process of purifying heparin US Patent 2,989,438
O’Donnell CD, Tiwari V, Oh MJ, Shukla D (2006) A role for heparan sulfate 3-O-sulfotransferase isoform 2 in herpes simplex virus type 1 entry and spread. Virology 346:452–459
Ornitz DM (2000) FGFs, heparan sulfate and FGFRs: complex interactions essential for development. Bioessays 22:108–112
Panasyuk AF (2011) Method for producing sulphated glycosaminoglycans from biological tissues. US Patent 7,943,764 B2
Park T-J, Lee M-Y, Dordick JS, Linhardt RJ (2008) Signal amplification of target protein on heparin glycan microarray. Anal Biochem 383:116–121
Pervin A, Gu K, Linhardt R (1993) Capillary electrophoresis to measure sulfoesterase activity on chondroitin sulfate and heparin derived disaccharides. Appl Theor Electrophor 3:297–303
Pervin A, Gallo C, Jandik KA, Han XJ, Linhardt RJ (1995) Preparation and structural characterization of large heparin-derived oligosaccharides. Glycobiology 5:83–95
Petitou M, van Boeckel CAA (2004) A synthetic antithrombin III binding pentasaccharide is now a drug! What comes next? Angew Chem Int Ed Engl 43:3118–3133
Petitou M, Duchaussoy P, Lederman I, Choay J, Jacquinet JC, Sinay P, Torri G (1987) Synthesis of heparin fragments: a methyl [alpha]-pentaoside with high affinity for antithrombin III. Carbohydr Res 167:67–75
Polat T, Wong CH (2007) Anomeric reactivity-based one-pot synthesis of heparin-like oligosaccharides. J Am Chem Soc 129:12795–12800
Rej RN, Ludwig-Baxter KG, Perlin AS (1991) Sulfation of some chemically-modified heparins. Formation of a 3-sulfate analog of heparin. Carbohydr Res 210:299–310
Roberts IS (1996) The biochemistry and genetics of capsular polysaccharide production in bacteria. Annu Rev Microbiol 50:285–315
Robinson H, Horner A, Höök M, Ogren S, Lindahl U (1978) A proteoglycan form of heparin and its degradation to single-chain molecules. J Biol Chem 253:6687–6693
Roman E, Roberts I, Lidholt K, Kusche-Gullberg M (2003) Overexpression of UDP-glucose dehydrogenase in Escherichia coli results in decreased biosynthesis of K5 polysaccharide. Biochem J 374:767–772
Rosenberg RD (1981) Heparin preparation. US Patent 4,301,153
Rosenberg RD (1985) Affinity fractionation of heparin on immobilized Concanavalin A. US Patent 4,539,398
Sawesi O, Spillmann D, Lundén A, Wernersson S, Åbrink M (2010) Serglycin-independent release of active mast cell proteases in response to Toxoplasma gondii infection. J Biol Chem 285:38005–38013
Silver RP, Prior K, Nsahlai C, Wright LF (2001) ABC transporters and the export of capsular polysaccharides from Gram-negative bacteria. Res Microbiol 152:357–364
Smith MR, Amaya E, Fussi F (1984) Process for manufacturing low molecular weight heparins by depolymerization of normal heparin. Eur Pat Appl EP0101141
Staatz WD, Toyoda H, Kinoshita-Toyoda A, Chhor K, Selleck SB (2001) Analysis of proteoglycans and glycosaminoglycans from Drosophila. Methods Mol Biol 171:41–52
Taylor CM, Goldrick M, Lord L, Roberts IS (2006) Mutations in the waaR gene of Escherichia coli which disrupt lipopolysaccharide outer core biosynthesis affect cell surface retention of group 2 capsular polysaccharides. J Bacteriol 188:1165–1168
Turnbull JE (1993) Oligosaccharide mapping and sequence analysis of glycosaminoglycans. Methods Mol Biol 19:253–267
van Boeckel CAA, Grootenhuis PDJ, Meuleman D, Westerduin P (1995) Glycosaminoglycans: synthetic fragments and their interaction with serine protease inhibitors. Pure Appl Chem 67:1663–1672
Vidic HJ (1981) Process for the preparation of heparin. US Patent 4,283,530
Wang Z, Ly M, Zhang F, Zhong W, Suen A, Hickey AM, Dordick JS, Linhardt RJ (2010a) E. coli K5 fermentation and the preparation of heparosan, a bioengineered heparin precursor. Biotechnol Bioeng 107:964–973
Wang Z, Xu Y, Yang B, Tiruchinapally G, Sun B, Liu R, Dulaney S, Liu J, Huang X (2010b) Preactivation based, one pot combinatorial synthesis of heparin like hexasaccharides for the analysis of heparin–protein interactions. Chemistry 16:8365–8375
Wang Z, Dordick JS, Linhardt RJ (2011) E. coli K5 heparosan fermentation and improvement by genetic engineering. Bioeng Bugs 2:63–67
Westerduin P, van Boeckel CAA, Basten JEM, Broekhoven MA, Lucas H, Rood A, van der Heijden H, van Amsterdam RGM, van Dinther TG, Meuleman DG, Visser A, Vogel GMT, Damm JBL, Overklift GT (1994) Feasible synthesis and biological properties of six ‘non-glycosamino’ glycan analogues of the antithrombin III binding heparin pentasaccharide. Bioorg Med Chem 2:1267–1280
Whitfield C, Roberts IS (1999) Structure, assembly and regulation of expression of capsules in Escherichia coli. Mol Microbiol 31:1307–1319
Wolff MW, Zhang F, Roberg JJ, Caldwell EEO, Kaul PR, Serrahn JN, Murhammer DW, Linhardt RJ, Weiler JM (2001) Expression of C1 esterase inhibitor by the baculovirus expression vector system: preparation, purification, and characterization. Protein Expr Purif 22:414–421
Xu Y, Masuko S, Takieddin M, Xu H, Liu R, Jing J, Mousa S, Linhardt RJ, Liu J, (2011) Chemoenzymatic synthesis of structurally homogeneous ultra-low molecular weight heparins. Science (in press)
Yang B, Weyers A, Baik JY, Sterner E, Sharfstein S, Mousa SA, Zhang F, Dordick JS, Linhardt RJ (2011) Ultra-performance ion-pairing liquid chromatography with on-line electrospray ion trap mass spectrometry for heparin disaccharide analysis. Anal Biochem 415:59–66
Yin J, Li G, Ren X, Herrler G (2007) Select what you need: a comparative evaluation of the advantages and limitations of frequently used expression systems for foreign genes. J Biotechnol 127:335–347
Zhang Z, Ollmann IR, Ye X-S, Wischnat R, Baasov T, Wong C-H (1999) Programmable one-pot oligosaccharide synthesis. J Am Chem Soc 121:734–753
Zhang L, Beeler DL, Lawrence R, Lech M, Liu J, Davis JC, Shriver Z, Sasisekharan R, Rosenberg RD (2001) 6-O-sulfotransferase-1 represents a critical enzyme in the anticoagulant heparan sulfate biosynthetic pathway. J Biol Chem 276:42311–42321
Zhang L, Lawrence R, Frazier BA, Esko JD (2006) CHO glycosylation mutants: proteoglycans. Methods Enzymol 416:205–221
Zhang Z, Weïwer M, Li B, Kemp MM, Daman TH, Linhardt RJ (2008a) Oversulfated chondroitin sulfate: impact of a heparin impurity, associated with adverse clinical events, on low-molecular-weight heparin preparation. J Med Chem 51:5498–5501
Zhang Z, McCallum SA, Xie J, Nieto L, Corzana F, Jiménez-Barbero J, Chen M, Liu J, Linhardt RJ (2008b) Solution structures of chemoenzymatically synthesized heparin and its precursors. J Am Chem Soc 130:12998–13007
Zhou X, Chandarajoti K, Pham TQ, Liu R, Liu J (2011) Expression of heparan sulfate sulfotransferases in Kluyveromyces lactis and preparation of 3-phosphoadenosine-5-phosphosulfate. Glycobiology 21:771–780
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Bhaskar, U., Sterner, E., Hickey, A.M. et al. Engineering of routes to heparin and related polysaccharides. Appl Microbiol Biotechnol 93, 1–16 (2012). https://doi.org/10.1007/s00253-011-3641-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-011-3641-4