
Abstract
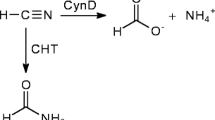
Cyanide dihydratase is an enzyme in the nitrilase family capable of transforming cyanide to formate and ammonia. This reaction has been exploited for the bioremediation of cyanide in wastewater streams, but extending the pH operating range of the enzyme would improve its utility. In this work, we describe mutants of Bacillus pumilus C1 cyanide dihydratase (CynDpum) with improved activity at higher pH. Error-prone PCR was used to construct a library of CynDpum mutants, and a high-throughput screening system was developed to screen the library for improved activity at pH 10. Two mutant alleles were identified that allowed cells to degrade cyanide in solutions at pH 10, whereas the wild-type was inactive above pH 9. The mutant alleles each encoded three different amino acid substitutions, but for one of those, a single change, E327G, accounted for the phenotype. The purified proteins containing multiple mutations were five times more active than the wild-type enzyme at pH 9, but all purified enzymes lost activity at pH 10. The mutation Q86R resulted in the formation of significantly longer fibers at low pH, and both E327G and Q86R contributed to the persistence of active oligomeric assemblies at pH 9. In addition, the mutant enzymes proved to be more thermostable than the wild type, suggesting improved physical stability rather than any change in chemistry accounts for their increased pH tolerance.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Cyanide is widely used in a variety of industrial processes, including mining, metal electroplating, plastic manufacturing, and photoengraving among others. This widespread use stems from its ability to form tight complexes with a variety of metals to make them soluble, which is especially useful in electroplating processes and leaching gold from ore. The resultant cyanide-containing wastewater must be detoxified before its release to the environment. Compared with conventional methods to detoxify cyanide (Young and Jordan 1995), microbial treatment can be inexpensive and environmentally friendly (Dubey and Holmes 1995; Knowles 1988). Enzymes capable of degrading cyanide have been found in a few bacteria and numerous fungi and plants (Jandhyala et al. 2005; Thuku et al. 2009); our work focuses on those belonging to the nitrilase superfamily (Brenner 2002).
The strain Bacillus pumilus C1 was isolated from a soil sample near a cyanide-containing wastewater dam in South Africa (Meyers et al. 1991). Its ability to degrade cyanide stems from an intracellular nitrilase that can act on the simple nitrile, cyanide, converting it to formate plus ammonia. This enzyme, a cyanide dihydratase, does not require additional cofactors or substrates during the hydrolyzing process compared with many other enzymes (Ingvorsen et al. 1991; Meyers et al. 1993; Watanabe et al. 1998). The cyanide-degrading activity of this enzyme is optimal between 37°C and 42°C and a pH of 7.0 to 7.8. The active form of the enzyme is known to be an octadecameric spiral at optimum pH and forms fibers of variable length at pH 5.4 (Jandhyala et al. 2005).
The metal plating and mining industries generate cyanide-contaminated wastewater at high pH and carrying a complex assortment of metal solutes depending on the industrial source. These conditions are generally adverse to enzyme action, and existing cyanide-degrading nitrilases have limited potential as bioremediation agents under these conditions. Therefore, engineering a cyanide-degrading enzyme with improved properties would be of significant interest.
High pH tolerance was chosen as the first target for engineering improved cyanide-degrading enzymes. Industrial cyanide waste widely exists at high pH as cyanide volatilizes easily with a low boiling point of 25.7°C under neutral conditions (Jenks 1985). Therefore, cyanide solutions are usually maintained at high pH to prevent the release of hydrogen cyanide gas. A cyanide waste sample from an electroplating plant in Houston, TX, was tested and found to be pH 11 with cyanide concentrations above 1 M (Benedik lab, unpublished observations). Enzymes capable of cyanide degradation under these alkaline conditions would be good candidates for an industrial cyanide bioremediation program.
In this study, we constructed a library of CynDpum mutants using error-prone-PCR (ePCR) (Cirino et al. 2003) and screening at two different pH values using a high-throughput strategy. Two CynDpum mutants (mutant C5 and H7) with higher pH tolerance were identified. Each of the mutants has three amino acid differences from the wild-type enzyme. Alleles were constructed carrying combinations of these mutations to study the effect of each single amino acid change on the improved activity. The activity and physical form of selected purified mutated enzymes were studied as a function of pH.
Materials and methods
Culture media and reagents
All strains were grown in Luria broth or plates supplemented as necessary with antibiotics at concentrations of 100 μg/ml for ampicillin, 30 μg/ml for chloramphenicol, and 25 μg/ml for kanamycin. X-gal was added to plates at 40 μg/ml. Protein expression was induced by the addition of IPTG at a final concentration of 1 mM to bacterial cultures. Restriction enzymes, Taq DNA polymerase, T4 ligase, and antarctic phosphatase were purchased from New England Biolabs (NEB; Ipswich, MA).
Bacterial strains and plasmids
Escherichia coli strain MB3436 (∆endA thiA hsdR17 supE44 lacI q Z∆M15) was used as the host strain for the initial screening experiments, and E. coli BL21(DE3) pLysS cells were used for bulk expression. Plasmid pBS KS+ from Stratagene was used for cloning. Plasmid pMB2890 (Jandhyala et al. 2003) is pET26b carrying the cynD gene of B. pumilus as an NdeI–XhoI insertion. Plasmids used in this study are shown in Table 1.
Error-prone PCR
Error-prone PCR was performed using pMB2890 as template, and the cynD gene was amplified with standard T7 promoter and terminator primers that anneal outside of the cloning sites. The mutagenic PCR reaction mixture consisted of 1× ThermoPol reaction buffer, 200 μM dNTPs, 2.5 U Taq DNA polymerase (NEB), and 100 ng of each primer in a 50 μl reaction volume. To establish the ePCR mutation frequency empirically, the concentration of DNA template was varied with a fixed concentration of MnCl2 and number of amplification cycles. Mutant libraries generated under these conditions were screened at pH 8.0 to determine the frequency of null mutants and conditions identified where 40–50% of the clones were inactive. Optimal ePCR conditions determined using this method were 130 ng of target DNA template in a 50-μl reaction with 0.2 mM MnCl2 and 20 cycles of amplification. The amplified product was verified for size on an agarose gel and purified using a SpinPrep® PCR clean-up Kit (Novagen, San Diego, CA).
Ligation of the PCR products
The amplified mutated PCR product was digested with XbaI and XhoI and ligated to similarly digested pBS KS+ which had been also treated with antarctic shrimp phosphatase. Ligation was performed at 16°C overnight, and the resulting ligation mixture was precipitated with ethanol before electroporation into MB3436 and plating on Luria–ampicillin agar plates containing X-gal/IPTG. Recombinant plasmids were picked from among the white colonies and screened as described below.
High-throughput screening for alkali-tolerant mutants
The cyanide-degrading activity of the mutants was tested at both pH 8.0 and pH 10.0, using a picric acid assay, as follows. White colonies were picked manually and inoculated into 96-well plates containing 150 μl Luria broth with ampicillin and cultured at 37°C overnight. The plates were replicated onto Luria agar using a 48-tooth pronger (Boekel®, Feasterville, PA), incubated at 37°C and stored at 4°C. To test the activity of the mutants at pH 8.0, 50 μl of cell culture was transferred into the corresponding wells of a new 96-well plate. Tris buffer (50 μl) (100 mM, pH 8.0) containing 8 mM KCN was added, and the 96-well plates were sealed with Parafilm and incubated at room temperature for 1 h. The reactions were terminated by adding 100 μl of alkaline picric acid reagent (0.5% picric acid in 0.25 M sodium carbonate) and incubated at 65°C for 15 min to allow color development. Active clones were identified by their yellow color. Testing at pH 10.0 was similar except that 40 μl of cells were mixed with 60 μl of Tris buffer (100 mM, pH 10.0) containing 6.7 mM KCN, conditions where the wild type fails to show activity.
Construction of single and double point mutants
Alleles of CynDpum carrying each of the six amino acid changes were constructed by site-directed mutagenesis. All mutagenesis reactions were carried out with mutagenic primers using the QuickChange® protocol (Stratagene, La Jolla, CA), and the mutations confirmed by DNA sequencing. The primers used are listed in Table 2.
Expression and purification of the wild type and mutated CynDpum
The wild-type and mutant CynDpum alleles were cloned into the expression vector pET26b as an NdeI–XhoI insertion. E. coli BL21(DE3) pLysS cells transformed with these plasmids were cultured in Nutrient Broth at 37°C to an OD600 nm of 0.4–0.6, whereafter expression was induced for 3–18 h by the addition of IPTG (1 mM). Cells were pelleted by centrifugation at 4,500 rpm and 4°C for 15 min and stored at −20°C. Pellets were thawed and resuspended on ice in 50 mM Tris pH 8.0 containing EDTA-free protease inhibitors (Roche Ltd., U.S.A.), and then lysed by sonication for 6 min on ice using a Sonicator 3,000 (Misonix, USA). Material was kept on ice or at 4°C for all subsequent steps unless stated otherwise, and fractions containing CynDpum were identified by SDS-PAGE and picric acid assay (described below).
Cell lysate was centrifuged at 20,000×g for 30 min to remove insoluble material, and ammonium sulfate precipitation was performed on the supernatant in 10% saturation increments from 20% to 50%. The sample was incubated for 20 min between increments, and fractions were recovered by centrifugation at 10,000×g for 10 min. Pellets were resuspended in 50 mM Tris, pH 8.0. Fractions containing CynDpum were filtered (0.45 μm) and separated by anion exchange chromatography using a Q Sepharose XL column (GE Healthcare) and a linear salt gradient of 100 mM to 1 M NaCl in 50 mM Tris pH 8.0. Fractions containing CynDpum were pooled, concentrated using a stirred cell ultrafiltration system with a 10-kDa cut-off membrane (Millipore Corporation, USA), and stored at −20°C in 50% glycerol. Prior to assay or microscopy, the sample was finally fractionated by HPLC gel filtration using a TSKgel PWXL4000 column (Tosoh Corporation, Tokyo) with 150 mM NaCl in 50 mM Tris pH 8.0, and A 280 nm peak fractions selected.
pH activity profiles
The activity of purified enzymes was tested over the pH range 5.0–12.0 using a picric acid assay as follows. Citrate/ Na2HPO4 buffer was used for pH 5.0 and 6.0, phosphate buffer for pH 7.0 and 8.0, glycine/NaOH for pH 9.0, sodium carbonate for pH 10.0, NaH2PO4 /NaOH for pH 11.0 and KCl/NaOH for pH 12.0. All buffers were used at a concentration of 50 mM and contained 150 mM NaCl.
The mutants were assayed in parallel using the wild-type enzyme as an internal control, and reactions were carried out in triplicate. Protein concentration was determined in triplicate using the Bradford method and protein samples were diluted to 0.057 mg/ml. Of this sample, 10 μl was mixed with 70 μl of buffer and pre-incubated for 1 h at 37°C, prior to addition of 20 μl of 25 mM KCN (in appropriate buffer). The reaction was allowed to continue for 20 min at 37°C in a water bath, and then stopped by addition of 80 μl of alkaline picric acid (0.5% picric acid in 0.25 M sodium carbonate). Color was developed by heating samples at 95°C for 5 min in a heating block, then removing them to room temperature. Room temperature distilled water (1 ml) was added to stop the color reaction, and the absorbance of this solution was measured at 520 nm on a Multiskan spectrophotometer (Titertek, Helsinki). These readings were converted to KCN concentration using standard curves determined in triplicate, separately at each pH condition. One unit of activity is defined here as the degradation of 1 μmol of cyanide per minute per microgram of protein.
Thermal stability
To determine thermal stability, an appropriate concentration of purified enzyme in 100 mM MOPS (pH 7.6) was incubated at 42°C, and samples were taken at time points ranging from 0 to 72 h. Activities were measured at room temperature as described above but using 100 mM MOPS buffer pH 7.6.
Electron microscopy
Peak fractions from gel filtration HPLC were exchanged into 150 mM NaCl, 50 mM buffer (citrate pH 5.4, Tris pH 8.0, glycine pH 9.0 or sodium carbonate pH 10.0) using a spin column (Microsep Pty Ltd., South Africa), and incubated overnight at 4°C before preparation of grids for electron microscopy. The sample (3 μl) was incubated for 30 s on a glow-discharged carbon-coated copper grid, washed twice with deionized water, blotted with filter paper, and stained with three drops of 2% uranyl acetate, blotting between drops. Grids were examined using Tecnai F20 and T20 transmission electron microscopes operated at 200 kV, at magnifications of 70,156× and 69,000× and a nominal defocus of 3.0–5.0 μm. Images were recorded using Gatan US4000 or US2000 CCD cameras.
Results
Screening for mutants with improved activity at elevated pH
A high-throughput screen was developed by adapting a standard colorimetric assay for cyanide (Fisher and Brown 1952) to a micro-plate format. This assay displays a change in color (red to yellow) upon reaction with picric acid to measure the amount of remaining cyanide. The wild-type control strain is able to degrade cyanide efficiently at pH 8.0 but not at pH 10.0 (Jandhyala et al. 2005). Clones from the ePCR mutant library were grown in 96-well plates and screened at pH 10.0 for ability to degrade cyanide.
Spot sequencing of clones from a variety of mutagenesis conditions showed that a library with about 50% inactive clones was found to represent a reasonable mutation frequency of 3–5 mutations/kb, which lead to one to five amino acid changes in the target protein. Putative mutants were reconfirmed for high pH activity by repeating the reaction under more controlled conditions in microcentrifuge tubes to eliminate false positive results.
Two candidate mutants (C5 and H7) showing activity at pH 10.0 were found among approximately 2,000 clones that were screened. DNA sequencing of the plasmid-born cynD alleles in C5 and H7 revealed that each had three amino acid changes relative to the wild-type cynD gene. The C5 allele was found to carry the amino acid changes Q86R, E96G, and D254E as well as two silent mutations (A705G and A981G). Allele H7 had changes E35K, Q322R, and E327G and one silent mutation (A816G).
Analysis of C5 and H7 mutations
Each of the three amino acid changes found in the C5 and H7 alleles was tested by generating the single and double mutants. E. coli cultures expressing each of these alleles were tested for cyanide-degrading activity at pH 8.0 and pH 10.0 (Table 3). For the C5 allele, the single and double mutants all had less activity at pH 10.0 than the original triple mutant. For the H7 allele, the Q322R and E35K single mutants displayed reduced or no activity, respectively, at pH 10.0. However, the activity of the E327G single mutant at high pH was not noticeably different from the original triple mutant, suggesting that the majority of the pH enhancement of the H7 allele can be attributed to this mutation alone.
A combined mutant carrying all three changes of the C5 allele plus the single predominant change of H7 (E327G) was also created. This had similar activity to the parental mutants, but the assay was not sufficiently quantitative to note minor differences.
pH profile of CynDpum mutants
Mutant enzymes C5, E327G, Q86R and C5 + E327G were purified using ammonium sulfate precipitation followed by anion exchange and gel filtration chromatography. pH versus activity profiles of the purified mutant and wild-type enzymes were measured using the picric acid assay for cyanide (Fig. 1a–e).

Enzymatic activities of the CynDpum mutants compared to wild-type enzyme at different pH values, using a picric acid assay. The rate of cyanide degradation by mutant enzymes, expressed as micromole substrate per minute per microgram per milliliter of enzyme are shown in panels a–d. Panel e portrays the same data as activity relative to wild type for each pH value
The mutant enzymes showed similar activity to wild type at pH 5 and pH 6. At pH 7–8, the Q86R and C5 mutants displayed elevated activity with respect to the wild type. The single mutants, Q86R and E327G, displayed very similar activity to the wild type at pH 9, but the multiple C5 and C5 + E327G mutants had approximately fivefold higher activity than the wild type (Fig. 1e). No activity was observed at pH 9.5 - 12.0 for any of the enzymes, even when using 16 times more enzyme and longer reaction times (not shown).
Thermal stability of CynDpum mutants
In order to test the hypothesis that the observed increase in activity of the mutants at elevated pH stems from improved stability (slowed unfolding at unfavorable pH) of the protein, the thermal stability of the mutant proteins H7, C5, E327G, Q86R, and C5 + E327G was investigated at neutral pH.
From a previous study in which the stability of CynDpum was tested by prolonged incubation at 23°C, 37°C, 42°C, and 55°C, we identified 42°C as an appropriate condition to assess enzyme stability (Jandhyala et al. 2005). Therefore, the wild type and mutant enzymes were incubated at 42°C for between 0 and 48 h, and the residual activity was measured for each (Fig. 2). The wild-type enzyme retained approximately 20% of its activity after 8 h. In contrast, the C5 triple mutant and the C5 + E327G combined mutant retained approximately 65% of their activity after 8 h and 20% activity after 48 h. The H7 and E327G mutants behaved similarly to one another and retained about 45–50% of their activity, respectively, after 8 h, dropping to 20% after 24 h. The Q86R single mutant was more thermostable than H7 and less so than the C5 mutant, retaining nearly 60% of its activity after 8 h and 30% after 24 h. We conclude therefore that mutations contributing to the activity at high pH also contribute to the thermostability of the enzyme. Interestingly, even though E327G appears to account for all the thermostability of the H7 mutant, it adds little to the thermostability of the C5 mutant.

Thermal stabilities of wild-type CynDpum and mutants. Residual activities were tested after incubation at the stated time periods. Values are the percentage of initial activity and are expressed as the average of duplicate experiments, with error bars representing standard deviations
Oligomerization properties of the CynDpum mutants
Previous studies have shown that CynDpum exists in two different oligomerization states, forming 18-subunit spirals at pH 8.0 and longer regular helices at pH 5.4 (Jandhyala et al. 2003). In order to investigate the relationship between pH stability and quaternary structure, we examined the purified wild-type and mutant CynDpum material by negative stain transmission electron microscopy (Fig. 3 and Table 4). Samples of the wild-type enzyme showed the presence of regular helices of varying lengths at pH 5.4, short spirals at pH 8.0, and what appeared to be partly denatured short spirals and objects of dimer size at pH 9.0. In contrast, the C5 triple mutant formed extremely long (>680 nm) helices at pH 5.4 and retained its helical form at pH 8.0. Some short helices were still present at pH 9.0, giving an average length 2.5 times longer than the wild type; however, at pH 10.0, only small aggregates remained, and the spirals showed signs of separating to the constituent dimers (not shown). A similar effect was seen in the C5 + E327G multiple mutant. We also examined the Q86R single mutant and found that this formed fibers of similar length to those of the C5 triple mutant. This suggests that the fiber length phenotype seen in the C5 mutant results from this single mutation. The E327G single mutant was similar in appearance to the wild type, with a small shift (less than twofold) towards greater average lengths at all pH. While the lengths at pH 9 were similar to those measured in the wild type, the appearance of the fibers was more regular than the wild type.

The effect of pH on oligomerization state of wild type and mutant cyanide dihydratase. Peak fractions from gel filtration were incubated at 4°C overnight at pH 5.4, 8.0 and 9.0, stained with 2% uranyl acetate and examined by transmission electron microscopy
Structural role of the mutations
Oligomerization of enzymes of this family involves off-set twofold symmetric interactions between symmetric homodimers, which coil round to form a spiral (Thuku et al. 2009). While no crystal structure is available of a close sequence homolog of CynDpum, it is possible to build a homology model by making use of a multiple alignment of enzymes of the nitrilase superfamily (Sewell et al. 2005; Thuku et al. 2009). By examining this model, we could identify the positions of the C5 and H7 mutations in the structure of CynDpum. None of the mutations is close to the active site. In the H7 mutant, E35K is located in one of the two “D” surface alpha helices which lie in close proximity to the equivalent region of the dimer belonging to the next turn of the helix, while Q322R and E327G lie in the C-terminal tail region that is thought to be located in the center of the helical or spiral oligomer (Sewell et al. 2005). Since the C-terminal tail is difficult to model accurately due to very low sequence identity with any of the template structures, we cannot pinpoint the specific location of these mutations in the structure. In the C5 mutant, Q86R and E96G both lie on the other “D” surface α-helix near to E35K, while D254E is probably located on the outside of the spiral, where it can play no role in inter-subunit contacts.
Discussion
Directed evolution methods provide powerful tools to alter the properties of proteins (Bloom et al. 2005; Cherry and Fidantsef 2003) especially in situations where detailed knowledge about the relationship between molecular structure and function is not available (Chen 2001; Farinas et al. 2001). The methodology of directed evolution usually comprises the creation of a pool of mutated genes and subsequent screening for improved gene products; this process may be iterated for two or three cycles (Kaur and Sharma 2006) or various alleles genetically shuffled. In this study, we employed error-prone PCR (ePCR) (Cirino et al. 2003) to construct a library of CynDpum mutants and developed a strategy to screen the mutants for improved activity at higher pH followed by manual recombination of the alleles.
Our decision to screen for high pH tolerance stems from the fact that cyanide-contaminated water is usually maintained in solutions above pH 9 to avoid production of hydrogen cyanide gas. Therefore, we chose to screen for cells expressing enzyme that can degrade cyanide at pH 10, where the wild type shows no activity.
From the pH 10 screening, two high pH-tolerant CynDpum mutants, C5 ad H7, were identified. Both mutants have three amino acid changes from the wild-type enzyme, and each also carried silent mutations. Testing combinations of single and double point mutants revealed that Q86R, E96G, and D254E all contribute to the increased activity of mutant C5 at high pH. In contrast, for the H7 mutant one amino acid change, E327G, showed behavior similar to the parental H7.
pH activity profiles of the purified enzymes showed that all enzymes have similar activities at pH 5.0–6.0, two of the mutants (Q86R and C5) have greater activity than the wild type at pH 7.0 and 8.0, and only the multiple C5 and C5 + E327G mutants remain active at pH 9.0. The purified mutants had little activity at pH 9.5 or higher, whereas bacterial cultures remained active at pH 10. We presume that the difference observed between the in vivo analysis and that of the purified enzymes reflects the ability of the cells to maintain partial pH homeostasis, whereby the intracellular pH does not equilibrate completely with the extracellular pH 10 during the assay.
There are two different mechanisms that could explain the increased pH tolerance of these mutants. One would be an improved capacity of the enzyme to catalyze the reaction at higher pH due to effects localized to the active site. The pH activity profile discussed above argues against this. The second is a scenario in which the enzyme is normally unstable above pH 9 but the mutants improve on that stability. This second scenario would explain why little activity was detected at pH 9 in the single mutants, which might not contribute sufficient stability to prevent unfolding during the hour's pre-incubation at high pH. The enhancement of thermal stability of the mutants at 42°C confirmed this idea, and we therefore suggest that the conformational stability of the enzyme is a key factor in maintaining activity under alkaline conditions.
Previous studies have suggested that oligomer formation is essential for the activity of these enzymes and that the C-terminal tail contributes to the formation and/or stabilization of the oligomeric quaternary structure (Sewell et al. 2003; Sewell et al. 2005). Homology modeling reveals that two of the mutations identified here as contributing strongly to pH tolerance in the C5 mutant, Q86R and E96G, lie in the same region, on one of the “D” surface alpha helices which have been proposed to form interactions with a neighboring subunit during helical oligomerization (Sewell et al. 2005). Q86R introduces a positive charge which may allow the formation of a salt bridge in the region. The removal of a negative charge in E96G does not immediately suggest an explanation for stabilizing mechanism of this mutation. The remaining mutation in C5, D245E, which does not demonstrate alkali tolerance on its own, is located on the outside of the spiral in a region not thought to be specifically involved in oligomerization.
The C terminus cannot be modeled accurately due to the absence of a homologous template structure. Furthermore, it is highly variable and in specific nitrilases, changes in this region have been shown to influence both the oligomerization state and the enzyme activity (Kiziak et al. 2007; Thuku et al. 2007). It has been shown to play roles of differing importance in the two cyanide dihydratases which have been studied to date. The oligomerization state of CynDpum switches from an octadecamer at pH 8 to a long helical fiber at pH 5.4, and this change of quaternary structure has been proposed to be due to histidine residues, which have a pKa of pH 6.0, in the C-terminal domain (Jandhyala et al. 2003). The highly homologous cyanide dihydratase from Pseudomonas stutzeri undergoes no such transition and lacks these histidines (Jandhyala et al. 2003). CynDpum was also shown to tolerate C-terminal truncations up to residue 293 before activity was lost (Sewell et al. 2005), whereas CynDstut is intolerant of C-terminal truncations CynDpum (Sewell et al. 2005).
The homology modeling showed that the key amino acid change of mutant H7, E327G, is located in this part of the C-terminal extension that is not required for activity, as is the Q322R mutation that showed slight activity at pH 10. The thermostability-enhancing effect of the H7 mutations confirm our current understanding of the structural importance of this region.
Electron microscopy studies confirmed the relationship between oligomerization and the improved activity of the mutants. The full length of the functional, 18 subunit oligomer in our molecular model has a maximum dimension of 21.2 nm (Jandhyala et al. 2003). Given that microscopic images are observed from a variety of angles, the size distribution of wild-type fibers at pH 8 (22.5 nm) is entirely consistent with the model and suggests the enzyme is primarily in the octadecameric state. At pH 9, however, the size distribution of the wild-type oligomers is significantly shorter, suggesting that the majority of proteins are no longer functional octadecamers but rather shorter inactive molecules, most likely dimers (which have a maximum length of 7.6 nm). At pH 5.4, we observed long helical fibers in the wild type; this is the pH-dependent switching in oligomerization that we reported in a previous study (Jandhyala et al. 2003).
The C5 mutant showed the stabilization of these longer helical oligomers (>21.2 nm) at pH 8.0 and 9.0 and a significant (greater than sixfold) enhancement of average fiber length at pH 5.4. This observation strongly identifies oligomerization as a strategy to enhance stability and activity under alkaline conditions. The E327G mutant does not demonstrate this effect as dramatically; however, there is an increase in the average lengths of the fibers of this mutant at all pH conditions. In the C5 mutant, the single Q86R mutation in the “D-surface” appears to be largely responsible for the increased length.
Interestingly, the majority of mutations leading to enhanced activity at low pH identified in a directed evolution study of Alcaligenes faecalis nitrilase (Schreiner et al. 2010; 32% identical to CynDpum) lay on the surface of the monomers, specifically in the “A” and “C” surfaces found in the fibers. These regions are important for oligomerization, so that although no structural characterization was carried out in that study, it is highly likely that the low-pH-tolerance effects of those mutations are also due to enhancement of higher-order structure. There is no clear evidence in either study of a contribution by mutations that might alter the titration points of the catalytic residues. Our speculation arising from this is that loss of activity as a function of both high and low pH is due to dissociation of the higher-order oligomers.
Overall, this work demonstrated the successful improvement of a cyanide dihydratase with increased activity at alkaline pH through random mutagenesis. Moreover, we have demonstrated a correlation between improved fiber formation and improved stability. None of the amino acid mutations found in mutant C5 or H7, which are remote from the active site, have been previously described to have an influence on the pH activity profile or thermal stability of this enzyme.
References
Bloom JD, Meyer MM, Meinhold P, Otey CR, MacMillan D, Arnold FH (2005) Evolving strategies for enzyme engineering. Curr Opin Struct Biol 15:447–452
Brenner C (2002) Catalysis in the nitrilase superfamily. Curr Opin Struct Biol 12:775–782
Chen R (2001) Enzyme engineering: rational design versus directed evolution. Trends Biotechnol 19:13–14
Cherry JR, Fidantsef AL (2003) Directed evolution of industrial enzymes: an update. Curr Opin Biotechnol 14:438–443
Cirino PC, Mayer KM, Umeno D (2003) Generating mutant libraries using error-prone PCR. Methods Mol Biol 231:3–9
Dubey SK, Holmes DS (1995) Biological cyanide destruction mediated by microorganisms. World J Microbiol Biotechnol 11:257–265
Farinas ET, Bulter T, Arnold FH (2001) Directed enzyme evolution. Curr Opin Biotechnol 12:545–551
Fisher FB, Brown JS (1952) Colorimetric determination of cyanide in stack gas and waste water. Anal Chem 24:1440–1444
Ingvorsen K, Hojer-Pedersen B, Godtferdsen SE (1991) Novel cyanide hydrolyzing enzyme from Alcaligenes xylosoxidans subsp. denitrificans. Appl Environ Microbiol 57:1783–1789
Jandhyala D, Berman M, Meyers PR, Sewell BT, Willson RC, Benedik MJ (2003) CynD, the cyanide dihydratase from Bacillus pumilus: gene cloning and structural studies. Appl Environ Microbiol 69:4794–4805
Jandhyala DM, Willson RC, Sewell BT, Benedik MJ (2005) Comparison of cyanide-degrading nitrilases. Appl Micro Biotechnol 68:327–335
Jenks WR (1985) Cyanide: hydrogen cyanide. In: Kirk-Othmer concise encyclopedia of chemical technology. Wiley, Somerset, p 334
Kaur J, Sharma R (2006) Directed evolution: an approach to engineer enzymes. Crit Rev Biotechnol 26:165–199
Kiziak C, Klein J, Stolz A (2007) Influence of different carboxy-terminal mutations on the substrate-, reaction- and enantiospecificity of the arylacetonitrilase from Pseudomonas fluorescens EBC191. Protein Eng Des Sel 20:385–396
Knowles CJ (1988) Cyanide utilization and degradation by microorganisms. In: Evered D, Harnett S (eds) Cyanide compounds in biology. CIBA Foundation Symposium 140. John Wiler & Sons Ltd, Chichester, pp 3–15
Meyers PR, Gokool O, Rawlings DE, Wood DR (1991) An efficient cyanide-degrading Bacillus pumilus strain. J Gen Microbiol 137:1397–1400
Meyers PR, Rawlings DE, Wood DR, Lindsey GG (1993) Isolation and characterization of a cyanide dihydratase from Bacillus pumilus C1. J Bacteriol 175:105–6112
Schreiner U, Hecher B, Obrowsky S, Waich K, Klempier N, Steinkellner G, Gruber K, Rozzell JD, Glieder A, Winkler M (2010) Directed evolution of Alcaligenes faecalis nitrilase. Enzyme Microb Tech 47:140–146
Sewell BT, Berman MN, Meyers PR, Jandhyala D, Benedik MJ (2003) The cyanide degrading nitrilase from Pseudomonas stutzeri AK61 is a two-fold symmetric, 14-subunit spiral. Structure 11:1–20
Sewell BT, Thuku RN, Zhang X, Benedik MJ (2005) The oligomeric structure of nitrilases: the effect of mutating interfacial residues on activity. Ann NY Acad Sci 1056:153–159
Thuku RN, Weber BW, Varsani A, Sewell BT (2007) Post-translational cleavage of recombinantly expressed nitrilase from Rhodococcus rhodochrous J1 yields a stable, active helical form. FEBS J 274:2099–2108
Thuku RN, Brady D, Benedik MJ, Sewell BT (2009) Microbial nitrilases: versatile, spiral forming, industrial enzymes. J Appl Microbiol 106:703–727
Watanabe A, Yano K, Ikebukuro K, Karube I (1998) Cyanide hydrolysis in a cyanide-degrading bacterium, Pseudomonas stutzeri AK61, by cyanidase. Microbiology 144:1677–1682
Young CA, Jordan TS (1995) Cyanide remediation: current and past technologies. Proceedings 10th Annual Conference on Hazard Waste Research, Manhattan, pp 104–128
Acknowledgements
This work was supported by the Texas Hazardous Substance Research Center, the Robert A. Welch Foundation (A-1310), the University of Cape Town and the South African National Research Foundation. The suggestions and support of Lacy Basile and Mary Abou-Nader at Texas A&M and Robert Ndoria Thuku and Mohamed Jaffer at the University of Cape Town are gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Wang, L., Watermeyer, J.M., Mulelu, A.E. et al. Engineering pH-tolerant mutants of a cyanide dihydratase. Appl Microbiol Biotechnol 94, 131–140 (2012). https://doi.org/10.1007/s00253-011-3620-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-011-3620-9