
Abstract
1,3-1,4-β-d-Glucanase has been widely used as a feed additive to help non-ruminant animals digest plant fibers, with potential in increasing nutrition turnover rate and reducing sanitary problems. Engineering of enzymes for better thermostability is of great importance because it not only can broaden their industrial applications, but also facilitate exploring the mechanism of enzyme stability from structural point of view. To obtain enzyme with higher thermostability and specific activity, structure-based rational design was carried out in this study. Eleven mutants of Fibrobacter succinogenes 1,3-1,4-β-d-glucanase were constructed in attempt to improve the enzyme properties. In particular, the crude proteins expressed in Pichia pastoris were examined firstly to ensure that the protein productions meet the need for industrial fermentation. The crude protein of V18Y mutant showed a 2 °C increment of Tm and W203Y showed ∼30% increment of the specific activity. To further investigate the structure-function relationship, some mutants were expressed and purified from P. pastoris and Escherichia coli. Notably, the specific activity of purified W203Y which was expressed in E. coli was 63% higher than the wild-type protein. The double mutant V18Y/W203Y showed the same increments of Tm and specific activity as the single mutants did. When expressed and purified from E. coli, V18Y/W203Y showed similar pattern of thermostability increment and 75% higher specific activity. Furthermore, the apo-form and substrate complex structures of V18Y/W203Y were solved by X-ray crystallography. Analyzing protein structure of V18Y/W203Y helps elucidate how the mutations could enhance the protein stability and enzyme activity.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
1,3-1,4-β-d-glucanase (lichenase, EC 3.2.1.73) can specifically hydrolyze 1,4-β-d-glucosidic bonds adjacent to 1,3-β-linkages in lichenan or β-d-glucans. The major products of the hydrolysis reaction are cellotriose, cellotetraose, and cellopentaose (Planas 2000). The enzyme draws much attention because of its broad spectrum of industrial applications. Supplementation of 1,3-1,4-β-d-glucanase in nonruminal animal (e.g., broiler chickens and piglets) feed largely improves digestibility of barley-based diets and reduces sanitary problems (sticky droppings). It also led to largely increase the animal feed conversion efficiency and growth rate (Selinger et al. 1996). In addition to animal feed, 1,3-1,4-β-d-glucanase is also used in beer industry as the enzyme can degrade the high molecular mass β-glucans to prevent reduced yields of extracts, inefficient wort separation and beer filtration (Planas 2000). The pelleting and expansion processes in feed industry are performed at 65–90 °C, and the optimal temperature for the malting processes in beer manufacture is between 50 °C and 70 °C. Therefore, to increase thermal stability of 1,3-1,4-β-d-glucanase is highly demanded for various industrial applications. There are two approaches to obtain a highly thermostable enzyme. The first is to directly clone the enzyme-coding genes from hyperthermophiles and to express the proteins in industrial strains. For instance, our recently solved Thermotoga maritima cellulase 12A (TmCel12A) X-ray structure that belongs to the GH12 family of glycoside hydrolases shows the strongest activity at 95 °C and has a pH optimum of 5 (Bronnenmeier et al. 1995; Liebl et al. 1996; Cheng et al. 2011). These characteristics make the enzyme highly valuable in various utilizations, since industrial processes such as plant waste treatments usually involve high temperature and low pH. Nevertheless, the hyperthermophile-derived enzymes usually exhibit low activities in physiological conditions which are between 20 °C and 37 °C, and thus severely limit their applications in aquatic and nonruminal animals. These hyperthermophilic enzymes still need to be modified to meet the requirement for different industrial usages. The second approach to obtain a thermostable enzyme is to directly modify a less thermostable enzyme by genetic manipulations.
Fibrobacter succinogenes 1,3-1,4-β-d-glucanase is classified as a member of the family 16 glycosyl hydrolases and is the only naturally occurring circularly permuted β-glucanase, among bacterial glucanases with reverse protein domains. Circular permutations were generated by changing the order of amino acids in a protein sequence, resulting in a protein structure with different connectivity, but share overall similar protein structure. The C-terminal truncated F. succinogenes 1,3-1,4-β-d-glucanase (TF-glucanase; residues 1–258) exhibits a higher thermostability and about 400% increment of enzymatic activity than the full-length enzyme (Wen et al. 2005). The structures of TF-glucanase apo-form and in complex with β-1,3-1,4-cellotriose (CLTR) have been solved (Tsai et al. 2001, 2003, 2005). TF-glucanase consists mainly of two 8-stranded anti-parallel β-sheets that are arranged in a jellyroll β-sandwich structure. Residues E11, N44, E47, E56, E60, R137, N139, W141, and T204 are involved in a hydrogen bond network, and residues F40, Y42, W203, and F205 are involved in the stacking interaction between CLTR and TF-glucanase (−3, −2 and −1 subsites; Tsai et al. 2005). This enzyme has also been well studied by mutagenesis and functional analyses (Chen et al. 2001, 2010; Cheng et al. 2002; Erfle et al. 1988; Lin et al. 2009; Tsai et al. 2008a, b). More importantly, the amounts of secreted TF-glucanase from Pichia pastoris fermentation was approaching 3 g l−1 by optimizing the codon usage, making the protein production meet the level of industrial manufacturing (range from 1 to 10 g l−1) (Huang et al. 2008). Although TF-glucanase already showed 400% increment of specific activity when compared with the full-length enzyme, the desire for a better enzyme still remains. Accordingly, TF-glucanase is an excellent target for directed mutagenesis to optimize for industrial use. In this study, we directly mutated the TF-glucanase gene in an attempt to introduce more stabilizing interactions based on the known structural information. We further transformed the mutant genes into industrial strain P. pastoris to test the protein thermostability and specific activity. Finally, the mutant proteins with higher thermostability and enzyme activities were expressed in Escherichia coli and the apo- and cellotetraose-complexed structures were solved to verify the original proposed strategies.
Materials and methods
Materials
PfuTurbo DNA polymerase was obtained from Life Technologies, Inc. The plasmid mini-prep kit, DNA gel extraction kit, and Ni-NTA resin were purchased from Qiagen. Factor Xa and the protein expression kit (including the pET32Xa/LIC vector and competent JM109 and BL21 (DE3) cells) were obtained from Novagen. The QuickChange Site-Directed Mutagenesis Kit was obtained from Stratagene, Inc.
Protein expression, mutagenesis and purification in P. pastoris
The gene encoding F. succinogenes 1,3-1,4-β-d-glucanase was amplified from genomic DNA (strain ATCC 19169/S85) by PCR using a forward primer 5′-CGCCGGAATTCTTGGTTAGCGCAAAGGATT-3′, and a reverse primer, 5′-ATAGTTTAGCGGCCGCTTATGGAGCAGGTTCGTCATCTCTTG-3′. The PCR fragments were cloned into pPICZαA vectors (Invitrogen, USA) using EcoRI and NotI sites. The TF-glucanase-expressing vector was linearized by PmeI and transformed into the P. pastoris X33 strain by electroporation. Transformants were selected on YPD (1% yeast extract, 2% peptone, 2% glucose) containing 100 μg/mL zeocin and incubated at 30 °C for 2 days. The transformants were grown in 40 ml of BMGY medium at 30 °C with shaking at 200 rpm until the OD600 reached 30–40. The cells were harvested by centrifugation at 3,500 rpm for 10 min at room temperature. The cell pellet was resuspended into 20 ml of BMMY and then cultured in a 250 ml flask at 30 °C for 5 days with shaking at 250 rpm. Then, methanol was added into the flask every 24 h to the final concentration of 0.5% (v/v). The supernatant was collected to check the protein expression amount every day.
The mutants were prepared with QuikChange Site-Directed Mutagenesis Kit using TF-glucanase gene as a template. The mutagenic oligonucleotides for performing site-directed mutagenesis are listed in Supplementary Table 1. The basic procedure of mutagenesis utilizes a supercoiled double-stranded DNA (dsDNA) vector with an insert of interest and two synthetic oligonucleotide primers containing the desired mutation. The mutation was confirmed by sequencing the entire TF-glucanase mutant gene of the plasmid obtained from an overnight culture. The correct constructs were linearized and transformed into P. pastoris as above.
The wild-type, V18Y, W203Y, V18Y/W203Y mutants were produced as follows: 100 ml of cells stock grew at 30 °C in 100 ml of yeast nitrogen base (YPD) medium (1% yeast extract, 2% peptone, and 2% dextrose) containing with 100 μg/ml Zeocin for 48 h. Cells were then transferred into 900 ml of YPD medium. After another 48 h, the cells were collected by centrifugation and grown in 1 L of minimal methanol medium (1.34% yeast nitrogen base (YNB) with ammonium sulfate without amino acids and 4 × 10−5% biotin). A total of 1% methanol was added once every 24 h to induce protein expression for 4 days.
The supernatant was collected by centrifugation and dialyzed twice against 5 L of H2O. The final solution was loaded into 20 ml DEAE Sepharose Fast Flow column (GE Healthcare Life Sciences). The buffer and gradient were 25 mM Tris, pH 7.5, and 0–500 mM NaCl. The proteins were eluted at about 350 mM NaCl. The protein purity was greater than 95% pure as judged by SDS-PAGE (Supplementary Figure 1).
Protein expression and purification in E. coli
The gene encoding F. succinogenes 1,3-1,4-β-d-glucanase was amplified by polymerase chain reaction (PCR) with forward primers 5′- GGTATTGAGGGTCGCGCGGCGGCGGCGGCGATGTTGGTTAGCGCAAAGGATT-3′ and reverse primer 5′- AGAGGAGAGTTAGAGCCTTACGGAGCAGGTTCGTCATC-3′, and then cloned into the pET32Xa/LIC vector. The mutants were prepared by using the QuickChange Site-Directed Mutagenesis Kit (Stratagene) with glucanase-pET32 Xa/LIC gene as the template. The constructs were transformed to E. coli BL21 (DE3) and the protein expression was induced with 1 mM isopropyl β-thiogalactopyranoside (IPTG) at 16 °C for 24 h.
Cell paste was harvested by centrifugation at 7,000×g and resuspended in a lysis buffer containing 25 mM Tris–HCl, pH 7.5, and 150 mM NaCl. Cell lysate was prepared with a French® pressure cell press (AIM-AMINCO Spectronic Instruments), and then centrifuged at 17,000×g to remove cell debris. The cell-free extract was loaded onto a lysis buffer-equilibrated Ni-NTA column. The column was washed with 10 mM imidazole followed by 20 mM imidazole-containing buffer. The His-tagged enzyme was eluted with 100 mM imidazole, dialyzed against the lysis buffer, and then subjected to Factor Xa digestion to remove the His tag. The mixture was then passed through another Ni-NTA column and the untagged enzyme was eluted with 10 mM imidazole-containing buffer and then dialyzed twice against a buffer of 25 mM Tris–HCl, pH 7.5. Purity of the recombinant proteins was checked by SDS-PAGE analysis (Supplementary Figure 1).
Glucanase activity assay
The β-glucanase activity was determined by dinitrosalicylic acid (DNS) method using glucose as a standard (Miller 1959). Equal amounts of enzyme solution (dissolved in 0.1 M sodium acetate, pH 5.0) and substrate solution (1% β-glucan (w/v) was co-incubated at 50 °C for 10 min. The reaction was stopped by adding 3 ml of DNS-reagent and boiled for 5 min to remove residual enzyme activity. After cooling in cold water bath for 5 min, the 540 nm absorbance of reaction solution was measured. One unit of β-glucanase activity was defined as the amount of enzyme required to liberate 1 μmol reducing sugar from β-glucan per minute per mg of total soluble proteins under the assay conditions.
For the kinetic analysis, optimal protein concentration was first determined by using a series of 6–48 ng/ml protein solutions and 20 mg/ml β-glucan substrate. The purified enzymes were diluted with 0.1 M sodium acetate, pH 5.0 for proper concentrations. The enzyme activity was then measured by using the optimal level (20 ng/ml) of protein and a series of 1–20 mg/ml β-glucan solutions. Based on these data, the kinetic parameters were obtained by curve-fitting analysis with a computer.
Thermal stability tests
The mutants were pre-treated at different temperature ranging from 50 °C to 62 °C for 2 min and then incubated in cold water bath for 5 min. The enzymatic activities were then determined as mentioned above.
Crystallization and data collection
Crystals of the glucanase in apo-form were prepared by mixing 2 μl glucanase solution (10 mg/ml in 25 mM Tris–HCl, pH 7.5) with equal amounts of mixture solution and mother liquor, and equilibrating with 500 μl of the mother liquor at room temperature by using the sitting drop method from Hampton Research (Laguna Niguel, CA). The monoclinic V18Y/W203Y unbounded crystal was obtained from the condition that contained 0.1 M Tris–HCl, pH 7.5, 0.3 M calcium acetate, and 29% PEG5KMME. The orthorhombic V14Y/W199Y cellotetraose bound crystal was firstly obtained from the refined condition that contained 0.15 M Tris, pH 8.5, 0.4 M calcium acetate and 33% PEG5KMME. The crystals were then soaked with mother liquor with 5 mM cellotetraose for 1 h.
The diffraction data from these crystals were collected at beam line BL17B2 of the National Synchrotron Radiation Research Center (NSRRC, Hsinchu, Taiwan) and at beam line BL17U of SSRF (Shanghai, China), and processed using the programs of HKL2000 (Otwinowski and Minor 1997). Prior to structural refinements, 5% randomly selected reflections were set aside for calculating R free as a monitor (Brunger 1993).
Structure determination and refinement
Two crystal structures were determined by molecular replacement method using the CNS program (Brunger et al. 1998). The previously solved truncated F. succinogenes 1,3-1,4-β-d-glucanase structure in complex with β-1,3-1,4-cellotriose (PDB code 1ZM1) was used as a search model. The V18Y/W203Y apo crystal (space group P21) contained two glucanase molecules and the V18Y/W203Y crystal in complex with cellotetraose (space group P212121) contained one molecule in an asymmetric unit. The 2Fo-Fc difference Fourier map showed clear electron densities for most amino acid residues, but the loop region of apo V18Y/W203Y crystal from Pro173 to Gly179 of molecule B lacked electron density, presumably due to disorder. Subsequent incorporation of Ca2+ ions, Tris, cellobiose (only cellobiose density was seen here) and water molecules were according to 1.0 σ map level. All the structural refinements were done with Xtalview (McRee 1999) and the CNS program (Brunger et al. 1998). The statistics are summarized in Table 1. All of the structural diagrams were drawn using PyMol software (http://pymol.sourceforge.net/).
Results
Engineering of the thermostability and specific activity of TF-glucanase in P. pastoris
In this study, we attempted to improve the TF-glucanase thermostability and specific activity according to the previously solved X-ray TF-glucanase complex structures (Chen et al. 2010; Tsai et al. 2001, 2003, 2005, 2008b). In order to reach the goal, we designed several mutations to modify the protein. First is to increase the electrostatic interaction (a single mutant S84D may form salt bridge with R120, two double mutant M27R/M39D and M27D/M39R may form salt bridge between the altered residues). The second approach is to increase the hydrophobic and hydrogen bond interactions (V18Y, M39F, V61F, and S71F). In addition, some mutations were also made to enhance the enzyme’s specific activity (W203F, W203Y and the truncated mutant at 233 amino acids). All mutations, depicted in Fig. 2a and b, were introduced into the protein by site-directed mutagenesis. In our rationale, the mutants must not only have better performance in enzymatic activities (higher thermostability and higher specific activity), but also have production levels that meets industrial requirement in industrial expression system. Therefore, the mutant and wild-type TF-glucanase proteins were expressed in P. pastoris strain and the thermostability and specific activity of the crude proteins were also measured.
The mutant enzymes expressed from P. pastoris were divided into three groups according to their performance: In group 1, the protein expressions were much lower than the wild-type enzyme. S71F, S84D, M27R/M39D, and M27D/M39R were classified into this group. For group 2, the protein expressions were comparable to wild-type enzyme, but thermostability and specific activity were not improved. 233 Stop, M39F, V61F mutants belong to this group. Due to impaired protein production and enzymatic function, mutants of groups 1 and 2 were not put to further analysis. For group 3, the protein expression levels are adequate and the enzyme properties are better. V18Y has slightly elevated protein yield and relative activity (Table 2). Noteworthily, V18Y showed a 2 °C increment of temperature tolerance (Tm, changed from 57 °C to 59 °C, Fig. 1). Therefore, this mutant is defined as group 3.

Kinetic and thermostability analysis of the TF-glucanase mutants. a Wild-type and mutant proteins (V18Y, M39F, V61F, S84D, and W203Y) were subjected to the temperature tolerance test. The V18Y mutation caused 2 °C increasing in temperature tolerance (Tm changed from 57 °C to 59 °C). Other mutants did not show significant change in temperature tolerance. W203Y had increased enzyme activity, but had similar thermostability of the wild-type enzyme. b Temperature tolerance assay was conducted in (b) P. pastoris- or (c) E. coli-expressed wild-type and mutant proteins (V18Y, W203, and V18Y/W203Y). The V18Y/W203Y double mutant exhibited increased thermostability as the V18Y single mutant did. These mutants were expressed and purified from E. coli, not only to confirm the increased activity and thermostability as observed in P. pastoris, but also to prepare the pure un-glycosylated protein for X-ray structural analysis. The temperature tolerance results of the wild-type and mutants from E. coli are 2 °C lower than the same proteins from P. pastoris. However, the thermostability patterns of V18Y and V18Y/W203Y mutants from E. coli are the same as the proteins expressed in P. pastoris with 2 °C increment (Tm changed from 54.2 °C to 56.4 °C). This lower temperature tolerance for the enzymes might be a result of absence of glycosylation in E. coli expression system
In spite of the wild-type TF-glucanase showing specific activity meeting the industrial requirement, we still tried to enhance the activity by gene mutagenesis to further reduce the production cost. It is suggested that the residue W203 is crucial for full-length glucanase function, as mutant W203R shows almost no activity (Cheng et al. 2002). Previous study has also demonstrated that the full-length glucanase W203F mutant exhibited a 4.2-fold increase in specific activity (8,726 units/mg, using lichenan as the substrate) comparing to wild-type enzyme (2,065 units/mg; Cheng et al. 2002). In this study, we tried to investigate whether the W203F mutation could also enhance the specific activity of P. pastoris-expressed TF-glucanase. Unexpectedly, we did not detect improvement of enzyme activity in the W203F mutant (Table 2). Barley β-glucan rather than lichenan was used in our experiments because lichenan is not supplemented in the animal feed. We tried to rule out if the discrepancy between our results and the previous studies was possibly caused by using different substrates. Nevertheless, W203F still showed lower specific activities than wild-type TF-glucanase (76.9%) using lichenan as substrate (data not shown), and thus was classified into group 2. From the structural comparison of TF-glucanase with B. licheniformis 1,3-1,4-β-d-glucanase (Bl-glucanase), Y24 of Bl-glucanase seems to occupy the same position as W203 in TF-glucanase. Furthermore, from the modeling result of SWISS-MODEL (http://swissmodel.expasy.org/), the W203Y mutant may not only provide the stacking interaction with the sugar bound to the −3 subsite, but also provide the hydrogen bond with the sugar. Therefore, the W203Y mutant was constructed and expressed in P. pastoris to test protein yield and enzyme function. The W203Y mutant has similar protein expression amount as wild-type TF-glucanase, and 30% elevation of specific activity (Table 2). Therefore, the W203Y was classified as group 3.
We thus took the advantage of V18Y and W203Y mutations and made the double mutant V18Y/W203Y. The thermostability experiment was then conducted to test our hypothesis. As expected, the W203Y mutant had the same Tm as the wild-type enzyme, while the V18Y/W203Y double mutant had a 2 °C increase in Tm compared with the wild-type (Fig. 1b).
The wild-type, V18Y, W203Y, and V18Y/W203Y mutant proteins expressed in P. pastoris were purified to test the enzyme specific activities. The specific activities against Barley β-glucan of the wild-type and V18Y, W203Y, and V18Y/W203Y mutant proteins are 5,291, 5,459, 6,705 and 7,237 U/mg (100%, 103.2%, 126.7%, and 136.8%, relative to the wild-type protein), respectively (Table 3). The specific activity increment ratios for the crude proteins and purified proteins in P. pastoris were similar (Tables 2 and 3).
Examination of thermostability and specific activity of TF-glucanase in E. coli
The thermostability tests and the total enzyme activity obtained from the crude and unpurified proteins in P. pastoris may not be sufficient to elucidate if the mutations of V18Y and W203Y can really improve TF-glucanase to become a better industrial enzyme. Therefore, we constructed the TF-glucanase wild-type, V18Y, W203Y and V18Y/W203Y mutants coding genes into pET32 Xa/LIC vector and expressed in E. coli BL21 (DE3) for further functional and crystallographic studies. Interestingly, the Tm of the wild-type and mutants are about 2 °C lower than those expressed from P. pastoris. One of the major differences between these two systems lies in protein glycosylation. The wild-type and mutant TF-glucanase proteins from P. pastoris did contain some glycosylations (the two major proteins M.W. were around 35–40 kDa when expressed in P. pastoris and 27 kDa in E. coli, respectively; Supplementary Figure 1). From previous studies, glycosylations could have increased temperature tolerance for some enzymes (Han and Lei 1999; Sevo et al. 2002; Koseki et al. 2006). Glycosylation might be the major reason for the Tm difference observed here. Nevertheless, the pattern of temperature tolerance among the wild-type and various mutant proteins purified from E. coli was similar to that from P. pastoris and the V18Y and V18Y/W203Y still showed 2 °C increment of the Tm (Fig. 1b and c). The specific activities against Barley β-glucan of the wild-type and mutant proteins are 5,694, 6,520, 9,263, and 9,967 U/mg (100%, 114.5%, 162.7% and 175.0%, relative to the wild type protein), respectively (Table 4). Kinetic analyses showed that the kcat and Km values are similar for the wild-type enzyme and the V18Y mutant. The kcat and Km values for W203Y and V18Y/W203Y are both increased by about the same scale, but W203Y appears to have a higher Km than does V18Y/W203Y.
Overall structures and the active site
To further investigate the underlying mechanism of V18Y/W203Y in improving the enzyme thermostability and specific activity, we tried to solve the V18Y/W203Y mutant structures. From previous structural studies, W203 interacted with the −3 subsite sugar. Because the preparation of β-1,3-1,4-cellotriose as used in previous structural study is complicated and the β-1,4-cellobiose part binds to the −3 and −2 subsites, we decided to use β-1,4-cellobiose or β-1,4-cellotetraose to soak with the V18Y/W203Y crystals as a trial. The crystal soaked with cellobiose turned out to fail. In the end, only cellobiose electron density was seen when the crystal was soaked with β-1,4-cellotetraose (Fig. 3a). The other density extending from the −3 position is obscure and can only be modeled as two water molecules. A Tris molecule was observed recently by another group (Chen et al. 2010). It was also seen clearly in our V18Y/W203Y apo- and cellotetraose complex structures (Fig. 3a).
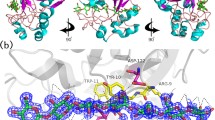
Each asymmetric unit of the V18Y/W203Y apo- and cellotetraose complex crystal unit cell (space group P21 and P212121) contains two and one enzyme monomer, respectively. There are six and two Ca2+ ions observed in molecules A and B of the V18Y/W203Y apo- crystal, respectively. There are four Ca2+ ions observed in the V18Y/W203Y cellotetraose complex crystal. Several Ca2+ ions were observed before but only Ca1 is important for the thermostability (Chen et al. 2010; Tsai et al. 2003, 2005). Here all Ca2+ ions observed in the V18Y/W203Y apo- and cellotetraose complex crystals were carefully checked. The other Ca2+ ions, except Ca1, might not worth mentioning, because they were only involved in the crystal packing or bound to the protein surface, without significant interactions with other protein residues (data not shown). The overall structures of V18Y/W203Y mutant in complex with Ca2+ ions, cellotetraose and Tris molecules are shown in Fig. 2a and b. The mutation sites of V18Y and W203Y are depicted in purple. The cellobiose from V18Y/W203Y cellotetraose complex structure superimposes well with the −3 and −2 subsites of the previous β-1,3-1,4-cellotriose structure (PDB code 1ZM1) (Tsai et al. 2005). The Tris molecule was observed in the −1 site as reported before (Chen et al. 2010). The root mean square deviation was 0.37 Å for 904 atoms. The active site residues around the W203Y position that might influence the specific activity are shown in Fig. 3c. The residue Y203, as the original W203, can still provide the stacking interaction with the sugar in the −3 subsite. There are slight conformational changes for the E11 and R137 residues. The W203Y mutation adds two hydrogen bonds to E11 and R137, so that these two residues are held in position to better recognize the sugar in the −3 and −2 subsites (Fig. 3d). The information obtained from the structural analysis might explain the significant improvement in enzyme specific activity of the W203Y mutant.

Overall fold of the V18Y/W203Y mutant bound to cellobiose. a Top view and b side view of the overall TF-glucanase V18Y/W203Y structure in complex with cellobiose (CLB) and Tris ion. Four Ca2+ ions are shown as balls and colored in blue. The C atoms of CLB and Tris in the stick models are shown in cyan and yellow, respectively. Mutants used in this study are shown in purple. Three more Ca2+ ions (Ca 2–4) were observed in this study, but they were involved in crystal packing or hardly interacted with protein residues. These Ca2+ ions may be unimportant for catalysis and thermostability

Electron density map and the substrate-binding sites. The 2Fo-Fc maps are contoured at 1.0 σ and 3.0 σ level, and are colored in green and red. a The density map of the substrates binding site from the 1.53 Å resolution crystal of the TF-glucanase V18Y/W203Y structure in complex with CLB. Cellotetraose was used in preparing the complex crystal, but only the density of a cellobiose that occupied the −3 and −2 positions was seen. Some density extended from the −3 position could not be modeled but was represented by two water molecules here. b The CLB and Tris ion observed in this study are superimposed on the previously solved TF-glucanase structure in complex with β-1,3-1,4-cellotriose (CLTR; PDB code 1ZM1). The same color styles are used for CLB and Tris ion, whereas CLTR is shown in gray. The Tris molecule superimposes well on the −1 sugar moiety of CLTR. c Three residues E11, R137 and W203 (Y203 in our study) from the previously solved TF-glucanase-CLTR complex structure (colored in gray) and our V18Y/W203Y TF-glucanase-CLB complex structure (colored in green) are shown. Either W203 or Y203 can provide stacking interaction with the −3 sugar moiety. d In our study, Y203 mutant has higher enzyme activity. Judged by the structure, E11 and R137 are both important in substrate recognition via hydrogen bonding to the OH groups of the carbohydrate substrate. Y203 can form hydrogen bonds with E11 and R137, and thus stabilizes them to maintain better orientation to interact with the substrate
Structural basis of the increased thermostability of V18Y
The hydrophobic core around the V18Y mutation was shown in Fig. 4a and b. In the beginning, we speculated that there might be space in the hydrophobic core to accommodate the larger amino acid in the V18 position. We reckoned that V18Y mutant might not only increase the hydrophobic interaction with some other hydrophobic amino acids, including L136, V168, W186 and W198 residues, but also increase the hydrogen bonds with T14 and W186 side chains. From the V18Y/W203Y apo- and cellotetraose complex structures, Y18 seemed also to increase the hydrophobic interactions with other residues. There were two water molecules observed in this region that were not observed in the previously solved TF-glucanase structures. Y18 turned out to form hydrogen bonds with T14 and W186 and interact with R197 via these two water molecules (Fig. 4c).

Overall fold and detailed structure of the V18Y hydrophobic core. a Side view of the overall TF-glucanase V18Y hydrophobic core structure was shown. Some residues (T14, F23, L136, V168, W186, R197, and W198) around V18Y were shown in cyan, and V18Y residue was shown in red. b Residues around V18 from previously solved wild-type structure TF-glucanase structure in complex with β-1,3-1,4-cellotriose (CLTR) (PDB code 1ZM1; shown in gray) were superimposed with the residues from our recently solved V18Y/W203Y cellotetraose structure (PDB code 3AXE; shown in green). The V18Y residue was shown in red. c From our V18Y/W203Y cellotetraose complex structure, Y18 seemed not only to increase the hydrophobic interactions with other residues but also to form hydrogen bonds with T14 and W186 directly. Y18 also interacts with R197 via two water molecules (Wa1 and Wa2)
Discussion
Higher thermostability is an important merit for industrial enzymes, because enzyme reaction rates, reactants solubility and sanitization can be significantly improved at higher temperature. In addition, increasing enzyme activity is also highly valued since it helps to reduce the cost for protein production. Two major genetic approaches have been conducted to improve the enzyme thermostability and specific activity: random mutagenesis and rational engineering. The random mutagenesis technologies, such as directed evolution, error-prone PCR and gene shuffling have created many new enzymes with significantly improved stability and activity (van den Burg and Eijsink 2002; Maki et al. 2009). On the other hand, rational design requires knowledge about the detailed protein structural information, especially high-resolution crystal structures. For the increment of enzyme thermostability, several rational strategies have been applied. These approaches include improvement of the packing of the hydrophobic core, introduction of disulfide bridges, stabilization of α-helix dipoles, and engineering of surface salt bridges, just to name a few (van den Burg and Eijsink 2002; Sterner and Liebl 2001; Vieille and Zeikus 2001).
In this study, we attempted to obtain higher thermostability and specific activity of glucanase by the rational design strategy. One of the major problems in selecting mutants is that the process of molecular engineering might impair protein expression in industrially used strains. TF-glucanase is an excellent 1,3-1,4-β-d-glucanase with very high specific activity. Furthermore, the production level of secreted TF-glucanase in P. pastoris fermentation reaches 3 g l−1 and is suitable for industrial manufacturing (Huang et al. 2008). Thus we used this enzyme as a template and performed rational design to produce mutants with higher thermostability and specific activity. For this purpose, 11 mutants were constructed for P. pastoris expression tests (Table 2 and Fig. 1a). These mutants, including S84D (may form salt bridge with R120), M27R/M39D and M27D/M39R, were designed for increased salt bridge to improve the thermostability based on previous β-1,3-1,4-cellotriose structure (PDB code 1ZM1). The criteria for the proposed interacting atoms should be less than 3–4 Å so that the salt bridge or H-bond could be formed. Unfortunately, the S84D mutant exhibited low protein yield than wild-type enzyme and it did not show higher thermostability. The expressions of M27R/M39D and M27D/M39R mutants were severely impaired with the protein concentration (80–90 μg/ml) indistinguishable from the vector control (data not shown). For mutants designed to increase hydrophobic and hydrogen bond interaction (V18Y, M39F, V61F, and S71F), only V18Y showed better thermostability and will be discussed later. Protein expression for M39F in P. pastoris seems similar to the wild-type enzyme, but the thermostability and specific activity are worse (Table 2 and Fig. 1a). The thermostability of V61F mutant was not enhanced and the protein expression is elevated. Interestingly, the side chain of V61F is beneath of the active site β-sheets, not face to the substrate-binding site. However, the specific activity of V61F dramatically decreases to 20%, indicating that slight conformational change might occur to interfere the substrate-binding or enzyme catalysis.
Previous studies showed that the C-terminal truncated enzyme (TF-glucanase; residues 1–258) exhibits a higher thermostability and enzymatic activity (Wen et al. 2005). From the solved β-1,3-1,4-cellotriose structure (PDB code 1ZM1; Tsai et al. 2005), the secondary structure after residue 233 is a random coil which is expected to be dispensable for enzyme function. Accordingly, the shorter C-terminal truncated enzyme was constructed (233 stop; residues 1–233) to test the hypothesis. However, the 233 stop mutant only showed 28% activity comparing to TF-glucanase in P. pastoris expression system. The random coil (residues 233–258) might still play a role in stabilizing the protein folding or enzyme catalysis.
Among these mutants, only two mutants with better performance were obtained (V18Y with increasing thermostability, W203Y with higher specificity activity). This indicates that gene modification of a nature-evolved enzyme is not easy, even with the knowledge of 3D protein structures. Finally, the V18Y/W203Y double mutant with the same amount of protein expression and better characteristics (higher thermostability and higher specific activity) was made. The characteristics of the purified V18Y, W203Y, and V18Y/W203Y mutant proteins that were expressed in P. pastoris (with glycosylations) and E. coli were further examined. The pattern of temperature tolerance of the enzymes in the two systems was essentially similar. The crude protein from P. pastoris showed lower specific activity than that expressed in E. coli. The lower specific activity of crude protein from P. pastoris could be attributed to the impurity of the target protein. The wild-type enzyme expressed in P. pastoris (5,291 U/mg) showed similar specific activity with the enzyme in E. coli (5,694 U/mg). Interestingly, the specific activities of three mutants, V18Y, W203Y, and V18Y/W203Y, expressed in P. pastoris showed lower increment of specific activities (3.2%, 26.7% and 36.8%, respectively) than the enzymes expressed in E. coli (14.5%, 62.7%, and 75.0%, respectively; Tables 3 and 4). These results indicate that the glycosylated-enzymes produced in P. pastoris still have some effect to the enzyme specific activities.
Further analysis of the kinetic parameters for the wild-type protein, the single mutants V18Y and W203Y, and the double mutant V18Y/W203Y indicates that the kcat and Km values are both increased for all mutants. Those of V18Y are only slightly different from those of the wild type, probably due to the fact that the location of V18 is away from the substrate-binding cleft and active site. Although V18Y has improved thermostability, it has little effect on the catalytic process. The other single mutant W203Y showed significant increases in both kcat and Km values, which can be translated as faster catalysis and lower substrate affinity. In the wild-type enzyme the side chain of W203 stacks with the −3 sugar. Replacement by a smaller tyrosine side chain can make the stacking interactions weaker and result in a lower substrate affinity. On the other hand, despite the additional hydrogen bonds to the phenolic oxygen, compared with the bulky asymmetric indole group of W203, the partially polar Y203 side chain can rotate more freely to facilitate product release and exchange for a new substrate molecule.
The optimal pH for the wild-type, V18Y, W203Y and V18Y/W203Y was determined to make sure if these mutant still suitable for using in animal feed (the enzyme need to work in more acidic environment) or some other industrials. Surprisingly, the optimal pH of W203Y and V18Y/W203Y mutants was changed from pH 8.0 to pH 6.0 and the working pH range also become narrower (pH 3–10 for the wild-type; pH 3–8.5 for W203Y and V18Y/W203Y mutants; Supplementary Figure 2). The characteristic of optimal pH and reaction pH range of V18Y/W203Y mutant still met our goal to be used in animal feed. For some other industrials which need more basic reaction conditions, as mentioned before, the desire for a better enzyme will never stop and that will be another story.
In conclusion, we have successfully applied structure-based rational design to endow the enzyme with improved activity and thermostability by directly mutating some important residues. The resulting V18Y/W203Y mutant had better thermostability and specific activity. Our preliminary fermentation test showed that the protein amount can reach 10 g l−1 (data not shown) and the level of expression amount makes this enzyme suitable for industrial production. In addition, similar strategy can be applied to other industrial enzymes that expressed in P. pastoris or other industrially used microbial strains.
Accession numbers
The atomic coordinates and structure factors for the truncated F. succinogenes 1,3-1,4-β-d-glucanase V18Y/W203Y in apo-form (code: 3AXD) and in complex with cellotetraose (code: 3AXE) have been deposited in the RCSB Protein Data Bank.
References
Bronnenmeier K, Kern A, Liebl W, Staudenbauer WL (1995) Purification of Thermotoga maritima enzymes for the degradation of cellulosic materials. Appl Environ Microbiol 61(4):1399–1407
Brunger AT (1993) Assessment of phase accuracy by cross validation: the free R value. Methods and applications. Acta Crystallogr D Biol Crystallogr 49(Pt 1):24–36. doi:10.1107/S0907444992007352
Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL (1998) Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr 54(Pt 5):905–921
Chen JL, Tsai LC, Wen TN, Tang JB, Yuan HS, Shyur LF (2001) Directed mutagenesis of apecific active site residues on Fibrobacter succinogenes 1,3-1,4-beta-d-glucanase significantly affects catalysis and enzyme structural stability. J Biol Chem 276(21):17895–17901. doi:10.1074/jbc.M100843200
Chen JH, Tsai LC, Huang HC, Shyur LF (2010) Structural and catalytic roles of amino acid residues located at substrate-binding pocket in Fibrobacter succinogenes 1,3-1,4-beta-d-glucanase. Proteins 78(13):2820–2830. doi:10.1002/prot.22798
Cheng HL, Tsai LC, Lin SS, Yuan HS, Yang NS, Lee SH, Shyur LF (2002) Mutagenesis of Trp(54) and Trp(203) residues on Fibrobacter succinogenes 1,3-1,4-beta-d-glucanase significantly affects catalytic activities of the enzyme. Biochemistry 41(27):8759–8766. doi:bi025766l
Cheng YS, Ko TP, Wu TH, Ma Y, Huang CH, Lai HL, Wang AH, Liu JR, Guo RT (2011) Crystal structure and substrate-binding mode of cellulase 12A from Thermotoga maritima. Proteins 79(4):1193–1204. doi:10.1002/prot.22953
Erfle JD, Teather RM, Wood PJ, Irvin JE (1988) Purification and properties of a 1,3-1,4-beta-d-glucanase (lichenase, 1,3-1,4-beta-d-glucan 4-glucanohydrolase, EC 3.2.1.73) from Bacteroides succinogenes cloned in Escherichia coli. Biochem J 255(3):833–841
Han Y, Lei XG (1999) Role of glycosylation in the functional expression of an Aspergillus niger phytase (phyA) in Pichia pastoris. Arch Biochem Biophys 364(1):83–90. doi:S0003-9861(99)91115-3
Huang H, Yang P, Luo H, Tang H, Shao N, Yuan T, Wang Y, Bai Y, Yao B (2008) High-level expression of a truncated 1,3-1,4-beta-d-glucanase from Fibrobacter succinogenes in Pichia pastoris by optimization of codons and fermentation. Appl Microbiol Biotechnol 78(1):95–103. doi:10.1007/s00253-007-1290-4
Koseki T, Miwa Y, Mese Y, Miyanaga A, Fushinobu S, Wakagi T, Shoun H, Matsuzawa H, Hashizume K (2006) Mutational analysis of N-glycosylation recognition sites on the biochemical properties of Aspergillus kawachii alpha-l-arabinofuranosidase 54. Biochim Biophys Acta 1760(9):1458–1464. doi:S0304-4165(06)00129-2
Liebl W, Ruile P, Bronnenmeier K, Riedel K, Lottspeich F, Greif I (1996) Analysis of a Thermotoga maritima DNA fragment encoding two similar thermostable cellulases, CelA and CelB, and characterization of the recombinant enzymes. Microbiology 142(Pt 9):2533–2542
Lin YS, Tsai LC, Lee SH, Yuan HS, Shyur LF (2009) Structural and catalytic roles of residues located in beta13 strand and the following beta-turn loop in Fibrobacter succinogenes 1,3-1,4-beta-d-glucanase. Biochim Biophys Acta 1790(4):231–239
Maki M, Leung KT, Qin W (2009) The prospects of cellulase-producing bacteria for the bioconversion of lignocellulosic biomass. Int J Biol Sci 5(5):500–516
McRee DE (1999) XtalView/Xfit—a versatile program for manipulating atomic coordinates and electron density. J Struct Biol 125(2–3):156–165. doi:S1047-8477(99)94094-7
Miller GL (1959) Use of dinitrosalicylic acid reagent for determination of reducing sugar. Anal Chem 31(3):426–428
Otwinowski Z, Minor W (1997) Processing of X-ray diffraction data collected in oscillation mode methods in enzymology 276 (Macromolecular Crystallography, part A):307–326
Planas A (2000) Bacterial 1,3-1,4-beta-glucanases: structure, function and protein engineering. Biochim Biophys Acta 1543(2):361–382. doi:S0167-4838(00)00231-4
Selinger LB, Forsberg CW, Cheng KJ (1996) The rumen: a unique source of enzymes for enhancing livestock production. Anaerobe 2(5):263–284. doi:S1075-9964(96)90036-0
Sevo M, Degrassi G, Skoko N, Venturi V, Ljubijankic G (2002) Production of glycosylated thermostable Providencia rettgeri penicillin G amidase in Pichia pastoris. FEMS Yeast Res 1(4):271–277. doi:S156713560100040X
Sterner R, Liebl W (2001) Thermophilic adaptation of proteins. Crit Rev Biochem Mol Biol 36(1):39–106. doi:10.1080/20014091074174
Tsai LC, Shyur LF, Lin SS, Yuan HS (2001) Crystallization and preliminary X-ray diffraction analysis of the 1,3-1,4-beta-d-glucanase from Fibrobacter succinogenes. Acta Crystallogr D Biol Crystallogr 57(Pt 9):1303–1306. doi:S0907444901010381
Tsai LC, Shyur LF, Lee SH, Lin SS, Yuan HS (2003) Crystal structure of a natural circularly permuted jellyroll protein: 1,3-1,4-beta-d-glucanase from Fibrobacter succinogenes. J Mol Biol 330(3):607–620. doi:S0022283603006302
Tsai LC, Shyur LF, Cheng YS, Lee SH (2005) Crystal structure of truncated Fibrobacter succinogenes 1,3-1,4-beta-d-glucanase in complex with beta-1,3-1,4-cellotriose. J Mol Biol 354(3):642–651. doi:S0022-2836(05)01107-1
Tsai LC, Chen YN, Shyur LF (2008a) Structural modeling of glucanase-substrate complexes suggests a conserved tyrosine is involved in carbohydrate recognition in plant 1,3-1,4-beta-d-glucanases. J Comput Aided Mol Des 22(12):915–923. doi:10.1007/s10822-008-9228-1
Tsai LC, Huang HC, Hsiao CH, Chiang YN, Shyur LF, Lin YS, Lee SH (2008b) Mutational and structural studies of the active-site residues in truncated Fibrobacter succinogenes 1,3-1,4-beta-d-glucanase. Acta Crystallogr D Biol Crystallogr 64(Pt 12):1259–1266. doi:S0907444908033428
van den Burg B, Eijsink VG (2002) Selection of mutations for increased protein stability. Curr Opin Biotechnol 13(4):333–337. doi:S0958166902003257
Vieille C, Zeikus GJ (2001) Hyperthermophilic enzymes: sources, uses, and molecular mechanisms for thermostability. Microbiol Mol Biol Rev 65(1):1–43. doi:10.1128/MMBR.65.1.1-43.2001
Wen TN, Chen JL, Lee SH, Yang NS, Shyur LF (2005) A truncated Fibrobacter succinogenes 1,3-1,4-beta-d-glucanase with improved enzymatic activity and thermotolerance. Biochemistry 44(25):9197–9205. doi:10.1021/bi0500630
Acknowledgments
The synchrotron data collection was conducted at beam line BL13B1 of NSRRC (National Synchrotron Radiation Research Center, Taiwan, R.O.C.) supported by the National Science Council (NSC) of Taiwan, and at beam line BL17U of SSRF (Shanghai, China). This work was supported by grants from National Science Council of Taiwan (NSC 98-2313-B-002-033-MY3 to JRL), National Basic Research Program of China (2011CB710800 to RTG) and Tianjin Municipal Science and Technology Commission (10ZCKFSY06000 to RTG).
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Figure 1
(DOC 701 kb)
Supplementary Figure 2
(DOC 632 kb)
Supplementary Table 1
(DOC 32 kb)
Rights and permissions
About this article
Cite this article
Huang, JW., Cheng, YS., Ko, TP. et al. Rational design to improve thermostability and specific activity of the truncated Fibrobacter succinogenes 1,3-1,4-β-d-glucanase. Appl Microbiol Biotechnol 94, 111–121 (2012). https://doi.org/10.1007/s00253-011-3586-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-011-3586-7