
Abstract
The use of cytochromes P450 for the regio- and stereoselective hydroxylation of non-activated carbon atoms in biotechnological applications reflects an efficient and cost-effective alternative in comparison to classical organic chemistry. The prokaryotic cytochrome P450 CYP106A2 from Bacillus megaterium ATCC 13368 hydroxylates a variety of 3-oxo-Δ4 steroids and recently it was identified to carry out a one-step regioselective allylic hydroxylation of the diterpene abietic acid. The anti-inflammatory pentacyclic triterpene 11-Keto-β-boswellic acid (KBA) was found to be a further substrate of CYP106A2, being the first report of a pentacyclic triterpene conversion by a prokaryotic P450. The reaction products were analyzed by HPLC and the corresponding kinetic parameters were investigated. Structure determination of the main product by NMR revealed a 15α-hydroxylation of this substrate. In order to overcome the inability of a recombinant P450 whole-cell system in E. coli for the uptake of acids with terpene structure, we developed for the first time an expression system for cytochromes P450 in B. megaterium (strains MS941 and ATCC 13368). Interestingly, CYP106A2 was only successfully expressed in the plasmid-less B. megaterium strain MS941 but not in ATCC13368. This recombinant system, with the co-expressed heterologous redox chain of the P450, bovine adrenodoxin reductase (AdR), and bovine adrenodoxin (Adx), was applied for the whole-cell conversion of KBA. The formation of 15α-hydroxy-KBA was increased 15-fold in comparison with the naturally CYP106A2-expressing B. megaterium strain ATCC 13368.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cytochrome P450 monooxygenase systems play an important role in the metabolism of a variety of hydrophobic compounds. They are involved in the synthesis of steroids, fatty acids, vitamins, and other biological processes like the detoxification of xenobiotics. Their reaction spectrum includes hydroxylations; N-oxidations; N-, O-, and S-dealkylations; sulfoxidations; epoxidations; peroxidations; deaminations; desulfurations; dehalogenations; N-oxide reductions; and even C–C bond cleavage (Bernhardt 1996, 2006). As the regio- and stereospecific oxidation of non-activated carbons is difficult to perform by means of classical organic synthesis, the biocatalysis by monooxygenases is a promising tool, especially with regard to the production of fine chemicals or pharmaceuticals (Maurer et al. 2003).
The prokaryotic cytochrome P450 CYP106A2 from Bacillus megaterium ATCC 13368 is one of only a few cloned bacterial steroid hydroxylases. It catalyzes the hydroxylation of 3-oxo-Δ4 steroids mainly in 15β-position (Berg et al. 1976; Lisurek et al. 2008; Virus and Bernhardt 2008; Zehentgruber et al. 2010). Recently, it was identified as the first reported bacterial P450 diterpene hydroxylase, which is able to carry out a one-step regioselective allylic hydroxylation of abietic acid (Bleif et al. 2011). To date, its natural function in B. megaterium is still unknown and its homologous electron transfer partners have never been cloned (Berg et al. 1979). But the electron supply can be realized with different redox partners (Berg et al. 1975; Lisurek et al. 2004; Goni et al. 2009).
The key challenges for the in vitro application of isolated external monooxygenases in biotechnology is the need for the expensive cofactor NADP(H), which has to be added in stoichiometric amounts, and the provision of redox partner(s) (Chefson and Auclair 2006). Several approaches exist to overcome the problem of cofactor supply, for example, the use of peroxides in the so-called peroxide shunt pathway, but this method leads to a fast inactivation of the P450 (Coon et al. 1996). Therefore, direct evolution efforts towards a higher stability of the enzyme had to be made (Cirino and Arnold 2003). The direct electrochemistry of the monooxygenases immobilized on a cathode is also difficult because of the instability of the P450 upon interaction with the electrode surface, so mediators which assist in the catalytic cycle had to be added (Urlacher et al. 2004). Furthermore, the electrochemical regeneration of NADP(H) leads to undesired side reactions due to high overpotentials, which can only be avoided by the addition of organometallic complexes (Hollmann et al. 2002). The most popular method for NADP(H) regeneration is the coupling of a second enzyme, in most cases a dehydrogenase, but for this approach a second substrate is required (Maurer et al. 2003).
Currently, the use of P450 monooxygenases in biocatalysis is limited to fermentation processes (Szczebara et al. 2003). The main advantages of whole-cell systems in contrast to in vitro approaches are thereby the stabilization of the P450 systems itself and the simultaneous regeneration of the cofactor in the cellular metabolism (Maurer et al. 2005). In these procedures, the utilization of bacterial P450s is extremely desirable because they are not membrane associated and exhibit a higher stability and activity than monooxygenases from plants and animals (Urlacher et al. 2004).
To date, several approaches exist for the application of the bacterial 15β-steroid hydroxylase CYP106A2 in whole-cell conversions. The B. megaterium strain ATCC 13368, carrying the endogenous CYP106A2 and its natural electron transfer partners, has been utilized for the biotransformation of steroids (Berg et al. 1976; Rauschenbach et al. 1993) and the diterpene resin acid, abietic acid (Bleif et al. 2011). But in this system, the P450 is built in the stationary growth phase and its expression is not inducible. In order to overcome these problems, a recombinant Escherichia coli whole-cell catalyst was created in which the P450 can be over-expressed together with its heterologous redox partners (Hannemann et al. 2006) and was successfully applied for the conversion of 11-deoxycorticosterone (DOC) to 15β-hydroxy-DOC (Hannemann et al. 2006). But this system was not suitable for the hydroxylation of acids, which is probably due to inability of hydrophobic acids to cross the outer membrane of the E. coli cells (Bleif et al. 2011). Therefore, a CYP106A2-dependent recombinant expression system in B. megaterium would be highly desirable, in order to overcome the existing problems like low expression levels in the natural B. megaterium strain ATCC 13368 and the inability of substrate uptake using the recombinant E. coli system.
Furthermore, B. megaterium has several advantages in comparison to other microorganisms: (1) similar to E. coli it is able to use a variety of different carbon sources, which allows its growth on low cost substances (Vary et al. 2007); (2) it does not produce the endotoxins associated with the outer membrane of gram-negative E. coli; (3) B. megaterium strains have highly stable, freely replicating plasmids, and the lack of alkaline proteases compared to industrially employed Bacillus subtilis strains (Vary 1994; Stammen et al. 2010). Thus, as a GRAS organism it is well suited for whole-cell applications.
In this work, a xylose-inducible protein production system for B. megaterium was created, in which CYP106A2 is co-expressed with its heterologous redox partners AdR and Adx and this system was successfully applied for the hydroxylation of the anti-inflammatory pentacyclic triterpene 11-keto-boswellic acid (KBA) (Jauch and Bergmann 2003).
Material and methods
Reagents and chemicals
11-keto-β-boswellic acid was synthesized as described elsewhere (Jauch and Bergmann 2003). All other chemicals were from standard sources and were of highest purity available.
Bacterial strains
All strains used in this work are listed in Table 1. Experiments for plasmid construction were performed with E. coli Top10 (Invitrogen, San Diego, CA, USA). All expression experiments were carried out using B. megaterium MS941 (Wittchen and Meinhardt 1995; Stammen et al. 2010), which was a kind gift from Prof. Dr. D. Jahn (Institute of Microbiology, TU Braunschweig, Germany). B. megaterium ATCC 13368, carrying the endogenous CYP106A2 and its natural electron transfer partners and a knockout strain of B. megaterium ATCC 13368 lacking the capacity to synthesize CYP106A2 were a kind gift from Dr. R. Rauschenbach (Schering AG, Berlin, Germany) (Rauschenbach et al. 1993). All B. megaterium experiments were carried out in baffled shake flasks in complex medium consisting of 24 g yeast extract (Difco, Detroit, MI), 4 ml glycerine, 12 g soytone (Difco, Detroit, MI), 2.31 g KH2PO4, 12.5 g K2HPO4, and 1,000 ml of distilled H2O. B. megaterium strains were stored on nutrient agar (Difco, Detroit, MI) plates at 4°C, if necessary supplemented with 10 mg/l tetracycline.
Construction of plasmids
All plasmids and synthetic oligonucleotides (MWG Biotech AG, Ebersberg, Germany) used in this work are listed in Table 1. Employed molecular biology methods were published previously (Sambrook and Russell 2001).
Site-directed mutagenesis of pKMBm4 using the primer pair KO_PacI_1 and KO_PacI_2 led to the PacI-lacking plasmid pSMF2, because this restriction site had to be used in the new multiple cloning site (mcs) in pSMF2.1. The new mcs of pSMF2.1 was constructed by denaturing 5 pmol of primer MCS1a and MCS1b for 5 min at 95°C and slowly cooling them down to room temperature. The resulting double-stranded DNA was ligated into pSMF2 digested with NheI and SphI resulting in pSMF2.1, in which the flanking SphI site was deleted during the ligation process, because it had to be used as a restriction site in the new mcs. The CYP106A2 gene was amplified by PCR from the plasmid pACYC_FHH2_8 (Hannemann et al. 2006) using the forward primer CYP106A2_BM_a, with a restriction site (SpeI) immediately upstream of the ribosome binding site and the coding region and the reverse primer CYP106A2_BM_b containing a stop codon followed by a MluI restriction site. The resulting PCR product was cloned into the pCR4-TOPO vector (Invitrogen, San Diego, CA, USA) resulting in pCR4-TOPO_CYP106A2. The pCR4-TOPO_CYP106A2 vector and the plasmid pSMF2.1 were digested with the restriction enzymes SpeI and MluI and ligated to generate pSMF2.1C. The AdR codon usage was optimized using the program JCat (Grote et al. 2005). The corresponding gene sequence including upstream the ribosome binding site was synthesized by Geneart GmbH (Regensburg, Germany). The plasmids 0838499_AdR_Bmeg_pMK-RQ (Geneart GmbH) and pSMF2.1C were digested with KpnI and SacI. The resulting AdR fragment and the digested pSMF2.1C vector were ligated to generate pSMF2.1CA. The Adx gene was amplified by PCR from the plasmid pBar_Twin (Hannemann et al. 2006) using the forward primer Adx_BM_a2, with a SacI site followed by a BsrGI restriction site immediately upstream of the ribosome binding site and the coding region and the reverse primer Adx_BM_b containing a stop codon followed by a SphI restriction site. The resulting PCR product was cloned into the pCR4-TOPO vector (Invitrogen, San Diego, CA, USA) resulting in pCR4-TOPO_Adx. The pCR4-TOPO_Adx vector and the plasmid pSMF2.1CA were digested with the restriction enzymes SacI and SphI and ligated to generate pSMF2.1CAA. The desired structures of all constructed plasmids were verified by DNA sequence analysis (MWG Biotech AG, Ebersberg, Germany).
Enzyme expression in E. coli and purification
CYP106A2, a truncated form of bovine Adx (Adx4-108) and bovine AdR were expressed and purified as described elsewhere (Sagara et al. 1993; Uhlmann et al. 1992; Simgen et al. 2000; Lisurek et al. 2004).
Spectroscopic binding assay
The binding assay was performed by using two tandem cuvettes. One chamber of each cuvette contained 10 μM CYP106A2 in 50 mM potassium phosphate buffer, pH 7.4, whereas the second chamber contained only 50 mM potassium phosphate buffer, pH 7.4. The 11-keto-β-boswellic acid was dissolved in DMSO at a stock concentration of 5 mM and was added to the CYP106A2 containing chamber of the sample cuvette giving final concentrations ranging from 0–100 μM, the same amount of KBA was also added to the buffer containing chamber of the reference cuvette (Jefcoate 1978; Schenkman and Jansson 1998) and followed by recording difference spectra (355–480 nm). The measurement was performed three times. The data were analyzed by plotting ΔΑ (386–417 nm) against c 0, where ΔΑ is the peak-to-trough difference and c 0 the total concentration of the ligand and the data were fitted by hyperbolic regression (y = (P 1 x/P 2 + x)) using the Origin version 8.1 (OriginLab Corporation, Massachusetts, USA), for the determination of the equilibrium dissociation constant (K D).
In vitro conversion and enzyme activity
The in vitro conversion of KBA was carried out using a reconstituted system in a final volume of 250 μl at 30°C for 10 min in 50 mM HEPES buffer, pH 7.4. The reconstituted system contained 10 μM Adx4-108, 1 μM AdR, 0.5 μM CYP106A2, a nicotinamide adenine dinucleotide phosphate hydrogen (NADPH)-regenerating system (1 mM MgCl2, 5 mM glucose-6-phosphate, 1 U glucose-6-phosphate dehydrogenase, and 0.1 mM NADPH), and 150 μM KBA. The reaction was stopped and the compounds were extracted twice with 250 μl chloroform and the organic phases were combined prior to solvent evaporation. The residuum was dissolved in 200 μl methanol and applied to high-performance liquid chromatography (HPLC) analysis.
The kinetics of substrate conversion with KBA was measured in a reconstituted system in a final volume of 250 μl at 30°C for 3 min in 50 mM HEPES buffer, pH 7.4. The reconstituted system contained 10 μM Adx4-108, 1 μM AdR, 0.5 μM CYP106A2, a NADPH-regenerating system (1 mM MgCl2, 5 mM glucose-6-phosphate, 1 U glucose-6-phosphate dehydrogenase, and 0.1 mM NADPH), and 10–250 μM KBA. The reaction was stopped and the compounds were extracted with 250 μl chloroform. After a second extraction with 250 μl chloroform the chloroform phases were combined prior to solvent evaporation. The residuum was dissolved in 200 μl methanol and applied to HPLC analysis. The relative amounts of the products were determined using the relative peak area and the respective activity was calculated. Each reaction was performed a minimum of three times. Data were fitted by hyperbolic regression (y = (P 1 x/P 2 + x)) using Origin version 8.1 (OriginLab Corporation, Massachusetts, USA).
HPLC analysis
HPLC experiments were carried out on a Jasco system consisting of a Pu-980 HPLC pump, an AS-950 sampler, an UV-975 UV/visible detector, and a LG-980-02 gradient unit (Jasco, Gross-Umstadt, Germany). The mobile phase methanol/water 75:25 (0.1% TFA) was degassed before use. A reversed phase ec MN Nucleodor C18 (5 μm, 4.0 × 125 mm) column (Macherey-Nagel, Bethlehem, PA, USA) was used for these experiments. The flow rate was 1.0 ml/min. Fifteen microliters of compound solution was injected each time. The column temperature was maintained at 40°C using the column thermostat BFO-04 (W.O. Electronics, Langenzersdorf, Austria). The absorbance was measured at 254 nm and the chromatogram was monitored for 20 min.
Whole-cell conversion using B. megaterium ATCC 13368
Fifty milliliters of complex medium was inoculated with a single colony of B. megaterium ATCC 13368 and incubated overnight at 100 rpm at 30°C. The main culture (50 ml) was inoculated with 500 μl of the preculture and incubated at 160 rpm at 30°C. After 24 h of incubation KBA (100 μM) was added. Samples (500 μl) were taken at time points indicated in the corresponding figure legends and extracted twice with 500 μl chloroform. The chloroform phases were combined prior to solvent evaporation. The residuum was dissolved in 200 μl methanol and applied to HPLC analysis. The relative amounts of the products were determined using the relative peak areas. Each reaction was performed a minimum of three times and the average value was calculated.
Whole-cell conversion using B. megaterium MS941
The B. megaterium strain MS941, a mutant of DSM319, was used as protein production host for all recombinant expression experiments (Wittchen and Meinhardt 1995). Protoplasted B. megaterium MS941 cells were transformed with the appropriate expression plasmids using a polyethylene glycol-mediated procedure as described before (Barg et al. 2005). B. megaterium plasmid strains were stored on nutrient agar (Difco, Detroit, MI) plates at 4°C, containing 10 mg/l tetracycline. All B. megaterium strains containing recombinant plasmids were grown in baffled shake flasks at 160 rpm at 30°C in 50 ml complex medium. The medium was supplemented with tetracycline to a final concentration of 10 mg/l. The recombinant expression of genes under transcriptional control of the xylose-inducible promoter was induced by the addition of 0.5% (w/v) xylose at an optical density measured at 578 nm (OD578) of 0.4. After a 24 h expression period KBA (100 μM) was added. Samples (500 μl) were taken at time points indicated in the corresponding figure legends and extracted twice with 500 μl chloroform. The chloroform phases were combined prior to solvent evaporation. The amounts of the products were determined using the relative peak areas. Each reaction was performed a minimum of three times and the average value was calculated.
Large-scale KBA conversion using B. megaterium MS941
Fifty milliliters of complex medium, supplemented with the appropriate amount of tetracycline was inoculated with a single colony of B. megaterium MS941 carrying the plasmid pSMF2.1CAA and incubated overnight at 100 rpm and 30°C. The main culture (1,000 ml) was inoculated with 10 ml of the preculture and incubated at 160 rpm and 30°C. The expression was induced by the addition of 0.5% (w/v) xylose at an optical density measured at 578 nm (OD578) of 0.4. After 24 h of incubation KBA (100 μM) was added. Following a 3-h incubation period, the cultures were extracted with chloroform. The organic phase was dried over anhydrous MgSO4 and evaporated to dryness. The yellowish residue was chromatographed on a silica gel column by using hexane-diethylether (1% HAc; 1:3) as an eluent, which resulted in homogeneous fractions. The fractions were evaporated to dryness and analyzed by NMR spectroscopy.
NMR measurements
All NMR experiments were performed on a Bruker DRX 500 NMR spectrometer. NMR spectra were recorded in methanol-d4. Chemical shifts are given in parts per million (ppm) on the δ scale relative to the solvent peaks CD3OD at δH = 4.84 and CD3OD at δC = 49.05. 13C multiplicities were determined using DEPT-135 pulse sequence. All assignments of 15α-hydroxy-KBA were confirmed by 2D NMR 1H,1H-COSY, HSQC, HMBC, and NOESY spectra.
SDS-PAGE and Western blot analysis
Aliquots of the cell suspension with similar cell numbers were taken, adjusted to an OD578 of 1 (1 × 109 cells) and centrifuged to pellet the cells. B. megaterium pellets were suspended in 1 ml lysis buffer (5 mg/ml lysozyme, 20 mM EDTA, and 0.1 μg/ml PSMF in a 100 mM Tris–Cl buffer pH 8.0) and incubated for 30 min at 37°C. Cell debris were removed by centrifugation. One hundred microliters of the lysate was diluted with 100 μl SDS-PAGE loading buffer and 10 μl of the proteins containing solutions were separated on 15% acrylamide gels (Laemmli 1970). The proteins were visualized by Coomassie Blue staining.
For Western blot analysis, gels were blotted onto nitrocellulose membranes and blotted membranes were blocked overnight in 30 ml TBST (50 mM Tris–Cl pH 7.5, 200 mM NaCl, 0.1% Tween 20) with 3% milk powder. The membranes were washed three times with 30 ml TBST. For immunodetection, membranes were incubated with the diluted primary antibody (1:1,000) in 30 ml TBST for 2 h. Prior to addition of the secondary antibody, the blots were rinsed three times with 30 ml TBST. The membranes were then incubated with the horseradish peroxidase linked goat antirabbit IgG secondary antibody (Dako, Glostrup, Denmark), diluted (1:2,000) in 30 ml PBS (10 mM potassium phosphate buffer pH 7.4, 150 mM NaCl) for 1 h. After that, the blots were rinsed three times with 30 ml PBS. The antigen–antibody conjugates were visualized by a color reaction, which was initiated by adding a solution consisting of 4-chloro-1-naphthol (2 ml; 3 mg/ml in ethanol), 25 ml PBS, and 10 μl 30% H2O2
Results
Cloning strategy and construction of expression vectors
Recently, we demonstrated that B. megaterium ATCC 13368 carrying the endogenous CYP106A2 and its natural redox partners is able to catalyze the hydroxylation of terpene acids (Bleif et al. 2011). Based on these promising results, our aim in this study was to improve the existing whole-cell system with regard to higher conversion rates by over-expressing CYP106A2 with its heterologous redox partners, the bovine AdR and bovine Adx in B. megaterium.
The outlined expression vectors for heterologous xylose-inducible protein production in B. megaterium are based on the shuttle vector pKMBm4, previously described by Stammen et al. (Stammen et al. 2010). However, the multiple cloning site (mcs) of pKMBm4 was not suitable for our cloning strategy and had to be replaced. For this purpose, the PacI site in the starting plasmid pKMBm4 had to be deleted first, in order to use it as a restriction site in the new mcs. This was performed by QuickChange® mutagenesis, which resulted in the plasmid pSMF2. The new multiple cloning site for B. megaterium consisting of seven unique restriction sites was introduced by insertion of a 81-bp oligonucleotide pair into the NheI and SphI sites of pSMF2 to create pSMF2.1. For the further construction of the desired expression vectors, the genes of interest were prior checked upon their codon adaptation index (CAI) using the program JCat (Grote et al. 2005). The parameter CAI describes how well the codons match the codon usage preference of the target organism. Thus, a CAI of one represents optimal codon usage. The average CAI for all 4.814 open reading frames of B. megaterium is 0.35 (Yang et al. 2007), meaning all genes having a lower CAI value should be optimized. The codons used in the AdR gene differ largely from the codon bias of B. megaterium evident by a CAI of 0.12. The sequence was optimized (CAI of 0.96) and synthesized by Geneart. The genes of CYP106A2 and Adx reached CAI values of 0.33 and 0.35, respectively. Consequently, they did not require improvement. The CYP106A2-encoding gene was cloned in pSMF2.1, resulting in pSMF2.1C. After this, the cDNAs of AdR and Adx were integrated in pSMF2.1C downstream to the P450, with the reductase preceding the ferredoxin, giving pSMF2.1CAA (Fig. 1).

Coexpression plasmid for recombinant protein production in B. megaterium. The 9,525 bp vector pSMF2.1CAA contains the cDNAs of CYP106A2, AdR, and Adx. Elements for inducible recombinant gene expression in B. megaterium are the xylose-inducible promotor (PxylA) and the gene encoding the xylose repressor (xylR). Elements for plasmid replication in Bacillus are the origin of replication oriU, RepU, and the tetracycline resistance gene. Elements for plasmid replication in E. coli are the origin of replication (ori E.coli) and the ampicillin resistance gene
Heterologous protein production in B. megaterium
After protoplast transformation of B. megaterium strain ATCC 133368 with each of the above described plasmids, cells were grown in complex medium at 30°C and gene expression was induced with 0.5% (w/v) xylose at an OD578nm of 0.4. Samples were taken 24 h after induction of gene expression, but no obvious protein production was observed using SDS-PAGE and Western blot analysis (data not shown). Therefore, we chose subsequently the B. megaterium strain MS941 for our further expression experiments. The genome of this plasmidless strain has been sequenced and proved to be useful for the expression of heterologous proteins (Stammen et al. 2010). After protoplast transformation of B. megaterium MS941 with the plasmids of interest and subsequent recombinant protein production, SDS-PAGE analysis revealed the expression of CYP106A2 and AdR (Fig. 2).

SDS gel of recombinant protein production in B. megaterium MS941. B. megaterium MS941 transformed with pSMF2.1 (lane 1), pSMF2.1C (lane 3), and pSMF2.1CAA (lane 6) were cultivated in complex medium at 30°C. Expression of the proteins was induced at an OD578 of 0.4 by the addition of 0.5% (w/v) xylose. Samples were taken 24 h after induction of gene expression. Cell-free extracts prepared from approximately 109 cells were analyzed by 15% SDS-PAGE. Proteins were visualized by Coomassie Blue staining. Lane 5 bands of the molecular weight marker. The black boxes highlight produced CYP106A2 (∼47.5 kDa; lane 3) as well as CYP106A2 and AdR (∼51.5 kDa; lane 6). No samples were applied to lanes 2 and 4
However, recombinant Adx (M r ∼ 12 kDa) could not be detected via SDS-PAGE gel analysis, because it has nearly the same size as lysozyme, which has been used for disrupting the cells (Fig. 2).
In order to show that Adx is co-expressed with CYP106A2 and AdR, Western blot analyses were performed. The immunostaining using primary antibodies raised against Adx, AdR, and CYP106A2 showed that all three proteins appear at the same size as the corresponding purified recombinant proteins from E. coli (Fig. 3).

Western blot analysis performed with polyclonal antibodies raised against Adx4-108 (∼12 kDa) (a), AdR (∼51.5 kDa) (b), and CYP106A2 (∼47.5 kDa) (c). a Lane 1 represents the sample of the purified recombinant Adx4-108 from E. coli. Lysate of B. megaterium containing pSMF2.1 24 h after induction (negative control) is shown in lane 2. Lane 3 shows the bands of the prestained molecular weight marker. Lysate of B. megaterium containing pSMF2.1CAA 24 h after induction is shown in lane 6. No samples were applied to lanes 4 and 5. b Lane 1 represents the sample of the purified recombinant AdR from E. coli. Lysate of B. megaterium containing pSMF2.1 24 h after induction (negative control) is shown in lane 2. Lane 4 shows the bands of the prestained molecular weight marker. Lysate of B. megaterium containing pSMF2.1CAA 24 h after induction is shown in lane 6. No samples were applied to lanes 3 and 5. (c) Lane 1 represents the sample of the purified recombinant CYP106A2 from E. coli. Lysate of B. megaterium containing pSMF2.1 24 h after induction (negative control) is shown in lane 2. Lane 4 shows the bands of the prestained molecular weight marker. Lysate of B. megaterium containing pSMF2.1CAA 24 h after induction is shown in lane 5. No sample was applied to lane 3
In vitro substrate characterization
Before starting with whole-cell conversions, we investigated whether CYP106A2 can convert triterpenes and accept the pentacyclic triterpene KBA as substrate. For these experiments recombinant proteins purified from E. coli were used. First, the binding of KBA to the active site of CYP106A2 was investigated by performing difference spectroscopy. Binding of KBA to the P450 induced a so-called type I spectral shift, which is due to the replacement of an axial water molecule from the heme iron coordination sphere and characterized by a maximum at ∼390 nm and a minimum at ∼420 nm. Titration of the enzyme with KBA leads to an equilibrium dissociation constant (K D) of 33.34 ± 2.87 μM (supplementary material).
Binding of a substrate is a necessary but not sufficient prerequisite for substrate conversion, therefore in the next step the catalytic activity of CYP106A2 towards KBA had to be studied. CYP106A2 was reconstituted with its heterologous redox partners, AdR and Adx, together with NADPH and the substrate conversion was analyzed by reversed phase HPLC. KBA was converted by the CYP106A2-dependent system into one main and three minor products (Fig. 4).

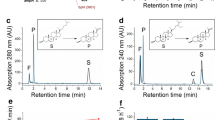
HPLC chromatogram of the in vitro conversion of KBA by CYP106A2. The reaction was carried out in a final volume of 250 μl in HEPES buffer (50 mM, pH 7.4) at 30°C for 10 min in the presence of a NADPH-regenerating system. The used concentrations were 0.5 μM CYP106A2, 1 μM AdR, and 10 μM Adx4-108. P1 (t R = 2.292 min), 15α-hydroxy-KBA (t R = 3.508 min), P2 (t R = 5.033 min), P3 (t R = 6.467 min), and KBA (t R = 17.358 min) are shown. The negative control without addition of CYP106A2 is shown in gray
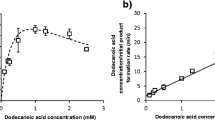
The conversion of the pentacyclic triterpene KBA by CYP106A2 was further investigated by kinetic analyses. The enzyme performed the reaction to the corresponding products with a V max value of 97.4 nmol products per minute per nmol CYP106A2 and a K M of 106.3 μM (Fig. 5). The achieved K M value is comparable to the K M values described for abietic acid hydroxylation by this enzyme (Bleif et al. 2011).

Determination of kinetic parameters for KBA conversion catalyzed by CYP106A2. Reactions were carried out in a final volume of 250 μl in HEPES buffer (50 mM, pH 7.4) at 30°C over 3 min in the presence of a NADPH-regenerating system. The used concentrations were 0.5 μM CYP106A2, 1 μM AdR, and 10 μM Adx4-108 with differing concentrations of KBA (10–250 μM)
Whole-cell conversion with B. megaterium ATCC 13368
After showing that KBA can be converted by CYP106A2 in vitro, we aimed to develop a whole-cell approach. In our first attempt, we investigated whether the B. megaterium strain ATCC 13368, carrying the endogenous CYP106A2 and its natural redox partners shows the same product pattern after incubation with KBA as demonstrated in vitro. A knock out strain of B. megaterium ATCC 13368 lacking the capacity to synthesize CYP106A2 served as a negative control. Both strains were incubated for 24 h prior to the addition of KBA (100 μM). Samples were taken after 8 h, extracted with chloroform, and analyzed by reversed phase HPLC (Fig. 6a). No conversion occurred with the knock out strain, indicating that the reaction was catalyzed by CYP106A2 and the HPLC elution profile shows the same product pattern as in the reconstituted in vitro system.

A HPLC chromatogram (after chloroform extraction) of the complex medium containing B. megaterium ATCC 13368 without adding KBA (a). HPLC chromatogram of the knock out strain after incubation with KBA (b). HPLC chromatogram of the in vivo conversion of KBA by B. megaterium ATCC 13368 (c). Reactions were carried out in 50 ml complex medium at 30°C for 8 h. P1 (t R = 2.317 min), 15α-hydroxy-KBA (t R = 3.492 min), P2 (t R = 4.992 min), P3 (t R = 6.475 min), and KBA (t R = 17.275 min) are shown. B HPLC chromatogram of CYP106A2-dependent substrate conversion. Chromatograms were obtained from extracted samples with B. megaterium MS941 cells containing pSMF2.1 without adding KBA (a). pSMF2.1 with KBA (b), and pSMF2.1CAA with KBA. Reactions were carried out in 50 ml complex medium at 30°C for 8 h. P1 (t R = 2.258 min), 15α-hydroxy-KBA (t R = 3.442 min), P2 (t R = 4.942 min), P3 (t R = 6.550 min), and KBA (17.280 min) are shown
Whole-cell conversion with B. megaterium MS941
In the next step, we focused on the B. megaterium strain MS941 for further improvement of the whole-cell biocatalysis. CYP106A2 was successfully co-expressed with its heterologous redox chain (Fig. 3), so it had to be explored whether the proteins are functionally active. Expression cultures of B. megaterium MS941 carrying either the plasmid pSMF2.1CAA or pSMF2.1 (negative control) were supplemented with inducer and after a 24-h expression period the substrate KBA (100 μM) was added. The KBA intermediates have been extracted after an 8-h incubation period and were applied to HPLC analyses. All MS941 strains, which do not contain the plasmid pSMF2.1CAA showed no conversion, whereas cells expressing the complete electron transfer chain showed the same HPLC elution profile as demonstrated with the B. megaterium strain ATCC 13368 (Fig. 6b) and the reconstituted in vitro system.
A large-scale conversion of KBA and silica gel purification of the reaction mixture was performed to obtain sufficient amounts of the main reaction product for structure determination. Thirty milligrams of the purified main product were obtained after purification and could be identified as 15α-hydroxy-KBA by NMR spectroscopy (Scheme 1). The synthesis and NMR analysis of this compound has not been published so far.

CYP106A2-catalyzed hydroxylation of KBA at the 15 position
1H NMR (CD3OD; 500 MHz): δ = 5.55 (s, 1 H; H-12), 4.24 (dd, J = 11.5, 6 Hz, 1 H; H-15β), 3.96 (dd, J = 3, 3 Hz, 1 H; H-3), 2.53 (s, 1 H; H-9), 2.37 (ddd, J = 13, 4.5, 3, 1 H; H-1β), 2.24 (dddd, J = 14.5, 14.5, 4.5, 3, 1 H; H-2β), 2.11 (dd, J = 13, 11.5, 1 H; H-16β), 1.89 (m, 2 H; H-6β, H-7α), 1.79 (dd, J = 10.5, 3.5, 1 H, H-7β), 1.72 (m, 1 H; H-6α), 1.56 (dd, J = 11, 2, 1 H; H-18), 1.51 (m, 1 H; H-22α), 1.49 (m, 1 H; H-2α), 1.48 (m, 1 H; H-5), 1.46 (m, 1 H; H-21β), 1.36 (m, 4 H; H-1α, H-27), 1.34 (m, 1 H; H-21α), 1.33 (m, 1 H; H-22β), 1.26 (m, 1 H; H-16α), 1.25 (s, 3 H; H-23), 1.23 (s, 3 H; H-26), 1.13 (s, 3 H; H-25), 0.97 (m, 5 H; H-19, H-20, H-30), 0.87 (s, 3 H, H-28), 0.85 ppm (d, J = 6.5, 3 H; H-29); 13 C NMR (CD3OD; 125 MHz): δ = 201.97 (C-11), 181.01 (C-24), 167. 72 (C-13), 131.68 (C-12), 71.41 (C-3), 68.52 (C-15), 61.80 (C-9), 60.88 (C-18), 50.79 (C-14), 49.49 (C-5), 48.33 (C-4), 47.69 (C-8), 41.70 (C-22), 40.40 (C-20), 40.25 (C-19), 39.35 (C-16), 38.82 (C-10), 37.08 (C-7), 35.15 (C-1), 35.07 (C-17), 31.84 (C-21), 30.01 (C-28), 27.06 (C-2), 25.01 (C-23), 21.41 (C-30), 20.34 (C-6), 19.37 (C-26), 17.78 (C-29), 15.16 (C-27), 14.09 ppm (C-25).
Time-dependent in vivo conversion of KBA
In order to investigate whether an enhancement of product formation can be achieved by over-expressing CYP106A2 and its heterologous redox partners in comparison to the native B. megaterium strain ATCC 13368, the time-dependent KBA conversion in both strains was compared.
Experiments were performed on a 50-ml scale and 100 μM substrate was added. As shown in Fig. 7, after 24 h the KBA conversion with the B. megaterium strain ATCC 13368 was 100% and a selectivity for 15α-hydroxylation of 80% was achieved (Fig. 7a). The conversion of KBA using Bacillus megaterium MS941, which expressed CYP106A2 and its heterologous redox chain, achieved a complete conversion after 100 min, the main product 15α-hydroxy-KBA was also formed to 80% (Fig. 7b). In both experiments around 20% minor products were produced. This demonstrates that an increase in the relative productivity by a factor of 15 was obtained leading to 560.7 mg 15α-hydroxy-KBA L−1 day−1 when using the heterologous expression system in B. megaterium MS941 in comparison to a relative productivity of 37.6 mg 15α-hydroxy-KBA L−1 day−1 when using the natural converting B. megaterium strain ATCC 13368. It can be expected that a further optimization of this process will significantly increase the yield of the hydroxylated product.

A Conversion of KBA by B. megaterium ATCC 13368. Reaction conditions: T = 30°C, 50 ml complex medium, 100 μM KBA. Samples (500 μl) were taken after 2, 4, 6, 8, and 24 h. B Conversion of KBA by B. megaterium MS941 expressing CYP106A2, AdR, and Adx. Reaction conditions: T = 30°C, 50 ml complex medium, 100 μM KBA. Samples (500 μl) were taken after 20, 40, 60, 80, and 100 min
Discussion
The aim of this study was the construction of a xylose-inducible protein expression system for B. megaterium, in which CYP106A2 will be co-expressed with its heterologous redox partners, bovine AdR and Adx, in order to improve the natural CYP106A2 expressing whole-cell system towards higher conversion rates. The cytochrome P450 monooxygenase was, together with its heterologous redox chain, successfully cloned into the B. megaterium shuttle vector pSMF2.1. Surprisingly, no obvious protein production was observed after protoplast transformation of B. megaterium ATCC 13368 and subsequent induction of gene expression with the plasmid of interest. A possible explanation might be that most isolated B. megaterium strains contain more than four plasmids (Vary et al. 2007). The best studied strain QM B1551 even carries 11% of its cellular DNA on seven plasmids, emphasizing the ability of the bacterium to stably maintain extra-chromosomal DNA elements in parallel (Gamer et al. 2009). Therefore, the plasmidless B. megaterium strain MS941 was chosen for further transformation and expression experiments. CYP106A2 could successfully be co-expressed together with its heterologous electron transfer proteins. This is to the best of our knowledge the first report of a recombinant B. megaterium, expressing a cytochrome P450. We further investigated whether CYP106A2 is able to hydroxylate triterpenes and to accept the pentacyclic triterpene KBA as substrate. KBA shares some structural similarities with steroids and includes an acid moiety like the recently identified substrate of CYP106A2, abietic acid. Pentacyclic triterpenes have gained significant importance because their chemical structures resemble those of steroids. Boswellic acids (Bas), the main active components of the gum resin extract of Boswellia serrata (Salei guggal), are used in Indian ayurvedic medicine for the treatment of inflammatory and arthritic diseases (Krüger et al. 2008; Gupta et al. 2001). BAs possess potent anti-inflammatory properties by inhibiting 5-lipoxygenase, human leukocyte elastase, and the nuclear factor-κB pathway, without exerting the adverse effects known of steroids (Krüger et al. 2008; Poeckel and Werz 2006). Pure β-boswellic acids were also found to act as efficient inhibitors of the topoisomerases I and IIα (Syrovets et al. 2000). Boswellic acids are therefore very fascinating molecules and there is an increasing demand for these substances for pharmaceutical and medicinal studies (Jauch and Bergmann 2003). Thus, KBA is an optimal target for structural modifications with regard to possible enhancement of its biological activity.
At first it was studied whether CYP106A2 can bind and convert KBA. It was demonstrated that KBA induced a type I spectrum during binding to CYP106A2 with an equilibrium dissociation constant (K D) of 33.34 ± 2.87 μM and an in vitro conversion of KBA with CYP106A2 led to one main and three minor products. The main product was analyzed by NMR and identified as 15-α-hydroxy-KBA. This hydroxylation position is extremely interesting, because it is not accessible via synthetic chemical methods. In order to further characterize the catalytic activity of the reaction, the K M and V max values of the KBA conversion were determined and resulted in a K M of 106.3 μM and a V max of 97.4 nmol products per minute per nmol CYP106A2. The achieved values are in the same range as described for abietic acid hydroxylation by CYP106A2 (Bleif et al. 2011).
After the successful identification of KBA as substrate, we were able to demonstrate that the B. megaterium strain ATCC 13368 carrying the endogenous CYP106A2 and its natural redox partners showed the same product pattern with KBA as shown in the in vitro reaction. To improve the efficiency of the in vivo biocatalyst, we focused on the B. megaterium strain MS941. It was demonstrated that CYP106A2 as well as its redox partners, Adx and AdR, could be expressed in this strain and that the expressed proteins were functionally active. They converted KBA to the same products as demonstrated with the B. megaterium strain ATCC 13368 and the reconstituted in vitro system.
To further characterize the new expression system, time-dependent in vivo conversions of KBA were performed. It was shown that both strains converted KBA to 100% with 80% selectivity for the 15α-hydroxylation. The overall reaction time for a complete conversion could be 14-fold reduced by using the recombinant system. The relative productivity thus was increased 15-fold. It comprised 560.7 mg 15α-hydroxy-KBA L−1 day−1 when using the heterologous expression system in B. megaterium MS941 in comparison with a relative productivity of 37.6 mg 15α-hydroxy-KBA L−1 day−1 when using the natural B. megaterium strain ATCC 13368.
In summary, we developed for the first time a recombinant expression system for cytochromes P450 in B. megaterium, which was successfully applied for the whole-cell conversion of the pentacyclic triterpene 11-keto-β-boswellic acid.
References
Barg H, Malten M, Jahn M, Jahn D (eds) (2005) Protein and vitamin production in Bacillus megaterium vol 18. Microbial processes and products, 1st edn. Humana Press, Inc, Totowa
Berg A, Carlstrom K, Gustafsson JA, Ingelman-Sundberg M (1975) Demonstration of a cytochrome P-450-dependent steroid 15beta-hydroxylase in Bacillus megaterium. Biochem Biophys Res Commun 66(4):1414–1423
Berg A, Gustafsson JA, Ingelman-Sundberg M (1976) Characterization of a cytochrome P-450-dependent steroid hydroxylase system present in Bacillus megaterium. J Biol Chem 251(9):2831–2838
Berg A, Ingelman-Sundberg M, Gustafsson JA (1979) Isolation and characterization of cytochrome P-450meg. Acta Biol Med Ger 38(2–3):333–344
Bernhardt R (1996) Cytochrome P450: structure, function, and generation of reactive oxygen species. Rev Physiol Biochem Pharmacol 127:137–221
Bernhardt R (2006) Cytochromes P450 as versatile biocatalysts. J Biotechnol 124(1):128–145
Bleif S, Hannemann F, Lisurek M, von Kries JP, Zapp J, Dietzen M, Antes I, Bernhardt R (2011) Identification of CYP106A2 as a regioselective allylic bacterial diterpene hydroxylase. Chembiochem. doi:10.1002/cbic.201000404
Chefson A, Auclair K (2006) Progress towards the easier use of P450 enzymes. Mol Biosyst 2(10):462–469
Cirino PC, Arnold FH (2003) A self-sufficient peroxide-driven hydroxylation biocatalyst. Angew Chem Int Ed Engl 42(28):3299–3301
Coon MJ, Vaz AD, Bestervelt LL (1996) Cytochrome P450 2: peroxidative reactions of diversozymes. FASEB J 10(4):428–434
Gamer M, Frode D, Biedendieck R, Stammen S, Jahn D (2009) A T7 RNA polymerase-dependent gene expression system for Bacillus megaterium. Appl Microbiol Biotechnol 82(6):1195–1203
Goni G, Zollner A, Lisurek M, Velazquez-Campoy A, Pinto S, Gomez-Moreno C, Hannemann F, Bernhardt R, Medina M (2009) Cyanobacterial electron carrier proteins as electron donors to CYP106A2 from Bacillus megaterium ATCC 13368. Biochim Biophys Acta 1794(11):1635–1642
Grote A, Hiller K, Scheer M, Munch R, Nortemann B, Hempel DC, Jahn D (2005) JCat: a novel tool to adapt codon usage of a target gene to its potential expression host. Nucleic Acids Res 33 (Web Server issue):W526-531
Gupta I, Parihar A, Malhotra P, Gupta S, Ludtke R, Safayhi H, Ammon HP (2001) Effects of gum resin of Boswellia serrata in patients with chronic colitis. Planta Med 67(5):391–395
Hannemann F, Virus C, Bernhardt R (2006) Design of an Escherichia coli system for whole cell mediated steroid synthesis and molecular evolution of steroid hydroxylases. J Biotechnol 25:25
Hollmann F, Withold B, Schmid A (2002) [cp*Rh(bpy)(H2O)]2+: a versatile tool for efficient and non-enzymatic regeneration of nicotinamide and flavin coenzymes. J Mol Cat B Enzym 791:1–10
Jauch J, Bergmann J (2003) An efficient method for large-scale preparation of 3-O-Acetyl-11-oxo-β-boswellic acid and other boswellic acids. Eur J Org Chem 2003:4752–4756
Jefcoate CR (1978) Measurement of substrate and inhibitor binding to microsomal cytochrome P-450 by optical-difference spectroscopy. Methods Enzymol 52:258–279
Krüger P, Daneshfar R, Eckert GP, Klein J, Volmer DA, Bahr U, Muller WE, Karas M, Schubert-Zsilavecz M, Abdel-Tawab M (2008) Metabolism of boswellic acids in vitro and in vivo. Drug Metab Dispos 36(6):1135–1142
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227(5259):680–685
Lisurek M, Kang MJ, Hartmann RW, Bernhardt R (2004) Identification of monohydroxy progesterones produced by CYP106A2 using comparative HPLC and electrospray ionisation collision-induced dissociation mass spectrometry. Biochem Biophys Res Commun 319(2):677–682
Lisurek M, Simgen B, Antes I, Bernhardt R (2008) Theoretical and experimental evaluation of a CYP106A2 low homology model and production of mutants with changed activity and selectivity of hydroxylation. Chembiochem 9(9):1439–1449
Maurer SC, Kühnel K, Kaysser LA, Eiben S, Schmid RD, Urlacher VB (2005) Catalytic hydroxylation in biphasic systems using CYP102A1 mutants. Adv Synth Catal 347:1090–1098
Maurer SC, Schulze H, Schmid RD, Urlacher V (2003) Immobilisation of P450 BM-3 and an NADP+ cofactor recycling system: towards a technical application of heme-containing monooxygenaes in fine chemical synthesis. Adv Synth Catal 345:802–810
Poeckel D, Werz O (2006) Boswellic acids: biological actions and molecular targets. Curr Med Chem 13(28):3359–3369
Rauschenbach R, Isernhagen M, Noeske-Jungblut C, Boidol W, Siewert G (1993) Cloning sequencing and expression of the gene for cytochrome P450meg, the steroid-15 beta-monooxygenase from Bacillus megaterium ATCC 13368. Mol Gen Genet 241(1–2):170–176
Sagara Y, Wada A, Takata Y, Waterman MR, Sekimizu K, Horiuchi T (1993) Direct expression of adrenodoxin reductase in Escherichia coli and the functional characterization. Biol Pharm Bull 16(7):627–630
Sambrook J, Russell DW (2001) Molecular cloning: a laboratory manual, vol 3. CSHL, Cold Spring Harbor
Schenkman JB, Jansson I (1998) Spectral analyses of cytochromes P450. Methods Mol Biol 107:25–33
Simgen B, Contzen J, Schwarzer R, Bernhardt R, Jung C (2000) Substrate binding to 15beta-hydroxylase (CYP106A2) probed by FT infrared spectroscopic studies of the iron ligand CO stretch vibration. Biochem Biophys Res Commun 269(3):737–742
Stammen S, Muller BK, Korneli C, Biedendieck R, Gamer M, Franco-Lara E, Jahn D (2010) High-yield intra- and extracellular protein production using Bacillus megaterium. Appl Environ Microbiol 76(12):4037–4046
Syrovets T, Buchele B, Gedig E, Slupsky JR, Simmet T (2000) Acetyl-boswellic acids are novel catalytic inhibitors of human topoisomerases I and IIalpha. Mol Pharmacol 58(1):71–81
Szczebara FM, Chandelier C, Villeret C, Masurel A, Bourot S, Duport C, Blanchard S, Groisillier A, Testet E, Costaglioli P, Cauet G, Degryse E, Balbuena D, Winter J, Achstetter T, Spagnoli R, Pompon D, Dumas B (2003) Total biosynthesis of hydrocortisone from a simple carbon source in yeast. Nat Biotechnol 21(2):143–149
Uhlmann H, Beckert V, Schwarz D, Bernhardt R (1992) Expression of bovine adrenodoxin in E. coli and site-directed mutagenesis of /2 Fe-2S/ cluster ligands. Biochem Biophys Res Commun 188(3):1131–1138
Urlacher VB, Lutz-Wahl S, Schmid RD (2004) Microbial P450 enzymes in biotechnology. Appl Microbiol Biotechnol 64(3):317–325
Vary PS (1994) Prime time for Bacillus megaterium. Microbiology 140(Pt 5):1001–1013
Vary PS, Biedendieck R, Fuerch T, Meinhardt F, Rohde M, Deckwer WD, Jahn D (2007) Bacillus megaterium—from simple soil bacterium to industrial protein production host. Appl Microbiol Biotechnol 76(5):957–967
Virus C, Bernhardt R (2008) Molecular evolution of a steroid hydroxylating cytochrome P450 using a versatile steroid detection system for screening. Lipids 43(12):1133–41
Wittchen KD, Meinhardt F (1995) Inactivation of the major extracellular protease from Bacillus megaterium DSM319 by gene replacement. Appl Microbiol Biotechnol 42(6):871–877
Yang Y, Malten M, Grote A, Jahn D, Deckwer WD (2007) Codon optimized Thermobifida fusca hydrolase secreted by Bacillus megaterium. Biotechnol Bioeng 96(4):780–794
Zehentgruber D, Hannemann F, Bleif S, Bernhardt R, Lutz S (2010) Towards preparative scale steroid hydroxylation with cytochrome P450 monooxygenase CYP106A2. Chembiochem 11(5):713–721
Acknowledgment
The authors thank Prof. Dr. Dieter Jahn (TU Braunschweig) for the kind provision of the B. megaterium strain MS941 and the plasmid pKMBm4 and Dr. Simon Stammen (TU Braunschweig) for constructive discussions. We are thankful to Katharina Bompais and Wolfgang Reinle for the expression and excellent purification of CYP106A2, AdR, and Adx.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOC 68 kb)
Rights and permissions
About this article
Cite this article
Bleif, S., Hannemann, F., Zapp, J. et al. A new Bacillus megaterium whole-cell catalyst for the hydroxylation of the pentacyclic triterpene 11-keto-β-boswellic acid (KBA) based on a recombinant cytochrome P450 system. Appl Microbiol Biotechnol 93, 1135–1146 (2012). https://doi.org/10.1007/s00253-011-3467-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-011-3467-0