
Abstract
Ethical and technical difficulties for in vivo studies on gut microbiotas argue for the development of alternative in vitro models: here, we describe a system simulating the proximal part of a human colon both nutritionally and physico-chemically with a procedure aimed to limit experimental variations over the time (Proximal Environmental Control System For Intestinal Microbiota—P-ECSIM). The continuous culture system P-ECSIM is first inoculated by a −20°C glycerol stock established from the batch culture of a stool-inoculated medium. The anaerobic atmosphere is self-maintained by the gases produced in the ordinary metabolism of fermentations. The monitoring of metabolic activities and microbial constitutions indicates that different steady states are obtained according to the dilution rate. Finally, the glycerol conservation of the batch culture-derived inoculum gives a similar differential response between the two dilution rates (D = 0.08 h−1 and D = 0.04 h−1) after a 1-year storage time as well for their metabolism and constitution in steady states, but with a lower abundance. Molecular fingerprints of the microbiota reveal however alterations over the time. Further efforts are needed concerning the preservation of standardized inoculums in order to improve the process for intra- and inter-lab comparison. Combined with appropriate analytical techniques, this system provides an efficient alternative means of studying functionally human microbiota in its constitution, metabolism and adaptation to environmental changes, particularly nutritional.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Our understanding of human microbiota and its metabolic impact on host health has considerably evolved in recent years, partly due to new molecular techniques (Zoetendal et al. 2008; Brugere et al. 2009). These tools permit the description of gut microbial diversity (Eckburg et al. 2005; Tap et al. 2009) in various situations (Dethlefsen et al. 2008; Zhang et al. 2009; Claesson et al. 2009). The potent metabolic behaviour of microbiota can also be inferred from these studies, especially when using metagenomics (Qin et al. 2010). Functional studies (metabolism, host–microbes and microbes–microbes interactions,…), however, are still difficult for in vivo ethical and technical reasons. The development of an in vitro system able to simulate environmental conditions of the human gut can partially facilitate such studies. Such in vitro O2-free models are principally based on either batch or chemostats cultures. Batch cultures have the advantage of providing results within a relatively short time period of about 24–48 h (Gibson and Fuller 2000) but do not reflect the near stable in vivo conditions, even for such short incubation times (Rumney and Rowland 1992). Continuous culture systems are, therefore, preferred, formed either of one or of more vessels depending on complexity and/or purpose. A single-stage chemostat simulating the human colon was developed 20 years ago, mimicking the nutrient availability and environmental conditions extrapolated from our knowledge of the in vivo conditions (Allison et al. 1989). This was further developed into a three-stage continuous system to simulate the three different functional parts of the human colon, i.e. the proximal, transversal and distal parts (Gibson et al. 1988; Macfarlane et al. 1998). In this type of system, the growth medium was continuously sparged with O2-free N2 and each reactor maintained anaerobically under CO2. A different system called “simulator of the human intestinal microbial ecosystem” (SHIME) was developed (Molly et al. 1993), which allows for the assessment of the dynamics of microbial ecology in the gastrointestinal tract. The stomach and small intestine are simulated in one vessel of 0.3 L each and the large intestine by three vessels of different volumes. The anaerobic conditions are obtained by a continuous N2 flux. An evolution of this system is the Twin-SHIME, which consists of two identical SHIME units (Van den Abbeele et al. 2010). Another computer-controlled system (called TIM-2) integrates a simulation of metabolites and water absorption in the proximal human colon (Minekus et al. 1999). Microbial diversity was assessed using phylogenetic DNA chips during the first 88 h (Rajilić-Stojanović et al. 2010). It was shown to vary throughout the duration of the experiment, with the most similar time (compared to the stool) obtained for the last experimental point. It is suggested that immobilized faecal microbiotas be used in continuous fermentations to solve the problems of long stabilization delay coupled with washing effects (Cinquin et al. 2004). This phenomenon is particularly important when simulating the digestive system of an infant. Immobilization could also be applied to pathogens in order to allow their implantation (Le Blay et al. 2009). In this model, anaerobiosis is guaranteed by a continuous flow of CO2. Here, we propose an alternative continuous culture system called “Environmental Control System for Intestinal Microbiota” (ECSIM), which differs from others by the maintaining of an anaerobic atmosphere initially established with a N2 flush and then kept by the fermentative activity of the microbiota itself. Furthermore, attempt for limiting inoculum variations over the time were realized by preparing and storing at −20°C batch-derived inoculums from an initial stool sample.
Materials and methods
Bacterial media composition
For the Proximal ECSIM (P-ECSIM) procedure, the so-called artificial gut medium was used as the fermentative medium. It is derived from the one used by Molly et al. (1993) and Macfarlane et al. (1998) and is a mix of three solutions: the trace elements solution, the vitamin solution (1 mL of each for 1 L of artificial gut medium) and the core medium (all compounds are expressed in grams per litre): mucin, 4.0; starch, 5.0; pectin, 2.0; guar gum, 1.0; xylan, 2.0; arabinogalactan, 2.0; inulin, 1.0; L-cystein-HCl, 0.8; casein, 3.0; peptone, 5.0; tryptone, 5.0; yeast extract, 4.5; bile salts, 0.4; tween 80, 1.0; FeSO4·7H2O, 0.005; NaCl, 4.5; KCl, 4.5; KH2PO4, 0.5; MgSO4·7H2O, 1.25; CaCl2·2H2O, 0.15; NaHCO3, 1.5 and Hemin, 0.05, pH adjusted to 6.0. The solution of trace elements contains (in grams per litre): MnSO4·2H2O, 0.5; FeSO4·7H2O, 0.1; CoSO4, 0.1; ZnSO4, 0.1; CuSO4·5H2O, 0.01; AlK(SO4), 0.01; H3BO3, 0.01; Na2MoO4·2H2O, 0.01; NiCl2·6H2O, 0.1 and Na2SeO3, 0.01. The vitamin solution contains (in milligrams per litre): menadione, 1; d-biotin, 2; pantothenate, 10; nicotinamide, 5; vitamin B12, 0.5; thiamin, 4 and para-aminobenzoic acid, 5.
All the microbiological procedures (cultivation, inoculation,…) were performed under strict anaerobic conditions. For bacterial enumeration, samples were anaerobically diluted under CO2 flux (serial 10-fold dilution). The reductive dilution medium was composed of (in grams per litre) K2HPO4·3H2O, 0.3; KH2PO4, 0.3; (NH4)2SO4, 0.6; NaCl, 0.6; MgSO4·7H2O, 0.06; CaCl2·2H2O, 0.06; cysteine HCl, 0.5; Na2S·9H2O, 0.3; NaHCO3, 5.0 and resazurin, 0.001. The solid bacterial media were Wilkins–Chalgren agar for total anaerobes and total facultative anaerobes (Bearne et al. 1990), Bacteroides mineral salt for Bacteroides spp. (Allison et al. 1989), Beerens agar for bifidobacteria (Beerens 1991), Reinforced Clostridial Medium for Clostridium spp., MacConkey agar for enterobacteria and Rogosa for lactobacilli (Mitsuoka and Hayakawa 1973). For each counting, triplicate plates were inoculated with 0.1 mL of each dilution and incubated at 37°C for 5 (facultative anaerobes) or 7 days (anaerobes).
Faeces, batch cultures, glycerol stocks and ECSIM procedure
Fresh faeces were obtained from a healthy female volunteer (29 years old), without recent treatment with antibiotics and known not to be a carrier of methanogens. Preculture of the faecal slurry at 20% (w/v) of medium was carried out on a 5-mL tube of medium at 37°C for 10 h. The culture in growth phase was transferred into a 1-L Erlenmeyer flask containing 95 mL of medium at pH 5.75. After 15 h of growth at 37°C, 100 mL of the broth was used to inoculate 900 mL of the same medium in a 2-L bioreactor (pH = 5.75, 400 rpm, 37°C) and stored after a new 8-h growth at −20°C, in aliquots of 2 mL of 20% (v/v) glycerol stock. A correlation of measures of turbidity and dry weight was established (R = 0.994, data not shown) in order to facilitate the rapid follow-up of the culture (see below). For ECSIM experiments, one of these aliquots was unfrozen on ice and used to inoculate a preculture of 5 mL of artificial gut medium, grown at 37°C for 10 h. It was then transferred for 15 h into a 1-L Erlenmeyer flask containing 95 mL of artificial gut medium. This was further transferred into 900 mL of complete artificial gut medium in a stirred tank 2-L bioreactor, previously N2 flushed (GPC—Global Process Concept Inc., France), for a 24-h batch culture. Then, a continuous cultivation was launched (Table 1) with in initial dilution rate of D = 0.08 h−1 (experiment P1), followed by D = 0.04 h−1 (P2) then back to D = 0.08 h−1 (P3). The same procedure was used 1 year later with a dilution rate of D = 0.08 h−1 (experiment P4), followed by D = 0.04 h−1 (P5). The acquisition and control software C-BIO (GPC—Global Process Concept Inc.) was used for the batch culture and continuous conditions. The stirring rate was adjusted at 400 rpm, the temperature maintained at 37°C and the pH at 5.75 by the automatic addition of 2 M NaOH. The oxidation/reduction potential (ORP) was monitored every 5 min using an Argenthal reference probe from Mettler Toledo (Inpro® 3253i/SG/225 probe). Measured redox potentials E were corrected into an E h value, at 37°C, by adding +199 mV. Steady states were expected to be obtained after five residence times (i.e. after more than 62.5 h for P1, P3 and P4 and 125 h for P2 and P5). Each one was maintained at least during 2 days during which samples were collected for microbial and biochemical analyses. Microbiota growth was evaluated by spectrophotometric (620 nm, Beckman Coulter® DU 640B spectrophometer) and dry weight measurements. The biomass was measured on 5 mL of culture that was first centrifuged (13,000×g, 10 min), then the pellet was washed three times with distilled water before being deposited under vacuum and dried at 104°C onto a pre-weighted membrane (Polyamid 0.45 μm, Sartorius). Anaerobic conditions were continuously checked during the experimentation (at least three times a day), by off-gas analysis of the atmosphere using gas chromatography (HP 6890 series columns: Molecular Sieve 5A and Porapack Q, Agilent Technologies, USA; coupled with an FID detector) permitting the detection of O2, CO2, CH4, and H2.
Biochemical analyses of P-ECSIM reactors
For short-chain fatty acids (SCFA) measurements, a bioreactor volume of about 200 μL was quickly sampled, deproteinized using 400 μL of cold methanol and centrifuged (8,000×g, 10 min). Supernatant allowed the assay of acetate, propionate, butyrate, isobutyrate, isovalerate, valerate, caproate, isocaproate and heptanoate using gas chromatography (HP 6890 series, column HP-INNOWAX 30 m × 250 μm × 0.25 μm, split ratio = 25:1, Agilent Technologies). 2-Ethyl-butyrate was used as an internal standard.
Molecular fingerprints
Molecular fingerprints of the microbiota were performed by ribosomal intergenic spacer amplifications (RISA). Total DNA was extracted from 1-mL samples (Yu and Morrison 2004) and RISA performed using conditions and primers ITSF/ITSReub described by Cardinale et al. (2004). After electrophoresis (20 h run on a 30-cm-long 2% (m/v) agarose gel, at 35 V, 0.5× TBE), fingerprints were analysed using the Bio-RAD Quantity One software. The DICE method was used for similarity matrix calculation and the clustering was done by the neighbour-joining method.
Statistics
Data was analysed using KaleidaGraph 4.03 (Synergy Software). Results were expressed as mean±standard deviations. Means were further analysed using one-way ANOVA variance analysis. Multiple comparison tests were further performed using Tukey's test, and differences were considered significant for p values less than 0.05.
Results
Technical principles, operating features and course of the P-ECSIM experiments
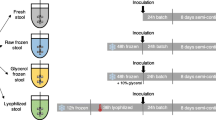
A procedure ending with a continuous culture system (the P-ECSIM) was developed in order to maintain the diversity of a gut microbiota and its metabolic properties (Fig. 1). Conceptually, the process was standardized as much as possible. It was performed using a microbiological engineering approach simulating the environmental conditions encountered in the proximal part of a human colon. Firstly, this concerns the inoculation of the P-ECSIM realized after an anaerobic culture regeneration of a permanent stock stored at −20°C and originating from a human faecal sample (see “Materials and methods” section for further explanations). The different experiments were initiated the month after the realization of the inoculum stock (experiments P1, P2 and P3) or 1 year later (P4 and P5). Secondly, this was achieved with a culture medium derived from those previously described (Macfarlane et al. 1998; Molly et al. 1993) which mimics the terminal ileal chyme from a common western diet. As mentioned in Table 1, this was also achieved by choosing a fermentation parameters/components ratio simulating the proximal colon environment (volume, retention time, stirring, pressure, temperature, pH,…). Moreover, pH was maintained at 5.75 using a volume-controlled alkaline solution. This indirectly evaluated the acidification of the medium, being the metabolic activity of the microbiota. The experiment was also technically designed to lower variations during sampling (liquid and gaseous parts). There was no alteration of the sterility, nor of the anaerobic atmosphere, and modifications of volume due to sampling were transient and inferior to 2% of the total volume. Therefore, dilution effects due to a sustained constant volume were limited. Two different retention times were used: a short one of 12.5 h (P1, P3 and P4) and a longer one (25 h, experiments P2 and P5) to simulate the part of the transit time in a proximal colon for a total 48 or 96-h transit time, i.e. a physiological transit time or a slow transit time (Table 1). Also, no N2 (or any other gas/gases mix) was flushed during the course of the continuous culture, as is usually the case to maintain a controlled anoxic environment. In this case, it was obtained from an initial N2 atmosphere which was progressively and dynamically enriched by the gases originating directly from the microbiota metabolism (H2 and CO2, no detectable O2 and CH4 in all our experiments, Table 1). H2 was very low all over the experiments, while CO2 was about twice lower 1 year after (experiments P4 and P5). Redox potential values were recorded as stable near −200 mV among the different experimental conditions (Table 1).

Schematic representation of the different stages leading to maintaining a microbiota in P-ECSIM. A fresh stool sample was progressively cultured (three-stage procedure) and stored as glycerol stocks at −20°C for future use, either 1 month later (simulations P1, P2 and P3) or 1 year later (simulations P4 and P5). At that time, a preculture was performed with a two-stage procedure in batch and used for inoculating the bioreactor, first with a batch culture without substrates limitation then continued as a chemostat in conditions simulating the proximal human colon. This continuous culture was considered to have reached a steady-state after a five-residence time delay. This marked the beginning of an experimental condition (called P1 to P5) in the P-ECSIM. The “Materials and Methods” section provides more details
Facultative and total anaerobes monitoring
Microbiological enumeration of total anaerobes and facultative anaerobes were performed on Wilkins–Chalgren agar plates during the entire process (from fresh stools to the various steady states in P-ECSIM). Data are reported in Fig. 2a. In all the samples (faeces, batch before storage and P-ECSIM cultures), there were between 109 and 1010 total anaerobes per mL, without any significant differences from one stage to another. A more contrasted view was obtained with the enumeration of facultative anaerobes, with a significant increase in the batch culture vs faeces (about 109 vs 106 mL−1, p < 0.0001) and vs experiments in the P-ECSIM (about 107 mL−1, p < 0.006). Differences between faeces and several P-ECSIM presented a p value of between 0.0012 and 0.0062. Differences were not significant among the diverse P-ECSIM steady-states analyses.

Enumeration of anaerobes (a) and RISA fingerprints (b) from the faecal sample, the culture batch for inoculum storage and the different P-ECSIM experiments. The enumeration of total anaerobes (left part of a) and of facultative anaerobes (right part of a) are indicated in log10 of CFU per millilitre (or per gram for the faecal sample). Mean values ± SD are obtained from four independent samples collected along the supposed “steady-state” condition of an experiment. Asterisk indicates a significant difference (p < 0.05) between ex vivo (the faecal sample) and the in vitro values
Evolution of molecular bacterial fingerprints during the pre-inoculation stages and the steady states in the P-ECSIM
A molecular fingerprint was obtained by RISA in each stage of our procedure (Fig. 2). The lack of enough differentiable bands on gels (less than 20) prevented any robust statistic comparisons of similarities. When compared to the faeces sample, however, the fingerprints from both the glycerol stock and the P-ECSIM looked different. Among the P-ECSIM experiments, samples taken at two different times during a steady-state looked highly similar (compare lanes P1a and P1b, Fig. 2b, corresponding to two samplings separated for ~24 h). This was also the case for P1 and P3, two experiments conducted similarly just after a retention time change. However, experiments conducted identically 1 year apart gave different fingerprints (compare P1 and P4, or P2 and P5 fingerprints, Fig. 2b). This indicated probable shifts between bacterial populations in these samples due to retention time modifications (P2, P5 vs P1, P3 and P4) and inoculum preservation over a 1-year period.
Bacterial variations in P-ECSIM experiments
Bacteroides spp., Bifidobacterium spp., Clostridium spp., Enterobacteriaceae and Lactobacillus spp. were enumerated using selective media (Fig. 3). No significant differences were observed over all the experiments concerning the number of Bacteroides, Clostridium and Enterobacteria. They were all present at about 108.5, 109.5 and 106.5 per mL, respectively (Fig. 3a, c, d). Some significant differences were detected among P-ECSIM experiments for bifidobacteria and lactobacilli: values ranging from 106 to 108.5 per mL for bifidobacteria (respectively experiment P2 and P4 Fig. 3b) and from 106 to 107.3 per mL for lactobacilli (respectively experiment P2 and P3, Fig. 3e). The experiment P2 was different to P1, as was P5 with P4 for bifidobacteria and lactobacilli (p < 0.002). This was also the case between P2 and P3, markedly so for lactobacilli (p < 0.0001), less for bifidobacteria (p < 0.026). Similarly, significant differences (p < 0.0001) could be detected in the counts of lactobacilli between P1 or P3 compared to P4, and between P2 compared to P5. This effect was present but attenuated for bifidobacteria (p values ranging from less of 0.0001 to 0.0155)

Counts of aBacteroides spp., bBifidobacterium spp., cClostridium spp., d enterobacteria and e lactobacilli. Results in log10 of CFU per millilitre correspond to the mean values ± SD of four independent samples. Significant differences among points are indicated in the text
SCFA production in P-ECSIM experiments
In the P-ECSIM, total SCFA concentration was around 134 mM for P1, P2 and P3 (range 131–138) or 103 mM for P4 and P5 (range 96–110 mM), indicating a high fermentative activity devoted to SCFA production (Table 2). Major SCFAs were respectively acetate, propionate and butyrate, while the others (isobutyrate, isovalerate and valerate) represented around 10% (Table 2). The amount of each major SCFA displayed significant differences between experiments. Concerning the ratio level between major SCFAs, percentages were relatively homogenous between all the experiments, with acetate values representing 58 ± 2%, propionate 25 ± 2% and butyrate 17 ± 1%.
Discussion
The aim of this work was to propose an alternative method for studying the metabolism and constitution of human gut microbiotas in an in vitro model, specifically the proximal part, where studies are made difficult due to ethical and technical limitations. Ideally, conditions have to be rigorously standardized to allow for reproducibility, and the model has to be stable over a long period to allow testing of abiotic or biotic factors (for example responses to nutritional, physical or microbial modifications). Here we have tried to control several important factors (such as pressure, volume, pH, residence time, sterility, medium, temperature, stirring, redox potential, gas flow, gas condensation) in order to obtain a simulation of the proximal human colon environment. The atmosphere during the steady states was composed of N2, CO2 and H2, with no detectable O2 or CH4. This at least partly reflected the metabolic activity of the microbiota and probably exerted feedback controls on microbial populations and their metabolisms. In other in vitro models (Cinquin et al. 2004; Gibson et al. 1988; Macfarlane et al. 1998; Minekus et al. 1999; Molly et al. 1993), N2 is continuously flushed during the experiments. By this method, we believe to be closer to in vivo conditions and to facilitate the action of hydrogenotrophs (by keeping H2 be disposable for them) and, in turn, the action of fermentative microbiota, by decreasing the H2 pressure. High levels of bacteria in the bioreactor (around 109.5 anaerobes) agree with this hypothesis, together with the high level of SCFAs [around 130 mM for the three first experiment part, i.e. quite similar in quality, quantity and proportion with in vivo data (Cummings et al. 1987)]. Moreover, by facilitating hydrogenotrophs, the P-ECSIM model could help the study of methanogens, sulphate-reducing prokaryotes and reductive acetogens on the gut microbiota. This could be of particular interest for studying the microbiota of elderly people and the role of methanogens, as they are more prevalent with age (Mihajlovski et al. 2010) or the original lineage of intestinal Thermoplasmatales (Mihajlovski et al. 2008) for which no in vitro culture has been described to date.
One of the main difficulties for simulating a proximal part is the initial inoculums. Ideally it should be a luminal content from a human proximal colon, limiting microbes exclusively to the habitants of this part of the gastrointestinal tract and allowing for an efficient comparison between in vivo and in vitro measurements. On the other hand, we found it important to be able to reproduce an in vitro model of the proximal part with a faecal sample in order to have a more convenient process. It was postulated, therefore, that faecal bacteria encompassed qualitatively the proximal ones, at least for the most part. As stools may contribute to modify the in vitro cultures by adding uncontrolled substrates, we decided to initially amplify the faecal bacteria using a batch reactor. This led to the use of a small inoculum and reduced the impact of substrate contamination, thereby obtaining larger homogenous amounts. This was useful for storage and for addressing long-term reproducibility. This stage clearly lowered the apparent diversity, but this seemed transient (see the number of total facultative anaerobes and the RISA fingerprints, between the batch culture and the stools/the ECSIM experiments, Fig. 2). The RISA fingerprints also demonstrated an apparent lower diversity between stools and P-ECSIMs. The protocol used, therefore, was able to reveal the diversity and the richness of its components, from a faecal inoculum (compare lane 1 with lanes from P-ECSIM experiments in Fig. 2). The loss of species within the batch reactor stage seemed limited. More exhaustive techniques like sequencing or phylogenetic chips would confirm this vision. However, reproducibility tested with similar experiments performed 1 year later was partly obtained using metabolic and bacterial indicators: total anaerobes and total facultative anaerobes seemed not to be modified (Fig. 2) but RISA fingerprints showed some alterations (compare lanes P1/P3 and P4, lanes P2 and P5, Fig. 2b). Based on the few bands on gels, P4 and P5 were more closely related together than with those obtained a year before (data not shown). This illustrates the need for further efforts to determine the best conditions to preserve (store and/or regenerate) gut microbiota for long periods. This must be done using stools and biopsies so that microbiotas can be shared between labs for a more efficient comparative analysis. Once again, molecular techniques with a higher resolution (16S sequencing, phylogenetic DNA chips) will greatly improve our knowledge of lost bacteria if any during the preservation steps, in order to apply selective corrective processes.
Another important feature of the P-ECSIM model is the efficient control of several parameters which could disturb a steady-state continuous culture. One such feature is sampling. The subtracted volume should be kept as low as possible in order not to influence fermentation (dilution effect due to the maintenance of a constant volume). Here, we managed to obtain a stable microbiota for a few days, as shown by the bacterial constitution (enumerations of anaerobes and of some genera, molecular fingerprints) and some final metabolites (SCFAs). This was also indicated by the stability of indirect factors, such as dry weight, ORP or the linear addition of NaOH for stabilizing pH. Even when taken over a long period in the same steady-state (more than 4 days), the P-ECSIM experiments were characterized by relatively low standard deviations. Tests have to be made over a longer period for this observation to be generalised.
As an example of the adaptability of the P-ECSIM to various conditions, we easily modified the retention time by changing the dilution rate from 0.08 to 0.04 h−1. This corresponded to a retention time of 12.5 and 25 h respectively, and, if compared to the length of the different parts of the colon, to a global transit time of ~48 and ~96 h. For western populations, this is supposed to represent a standard transit time and a very slow one (Cummings et al. 1992). This modification significantly affected the quantities of bifidobacteria and lactobacilli (p < 0.05), without significantly altering the total anaerobic bacteria or SCFAs production, regardless of storage duration. Previously described in vitro studies have also revealed the importance of retention time in a three-stage continuous culture model (Child et al. 2006). Retention times were different in the two studies (corresponding to a total transit time of 20 or 60 h in a three-stage system vs 12.5 and 25 h here), together with a different bacterial counting methodology (FISH vs culture media); therefore, no comparison can be made. However, taken together, this highlights the importance of the retention time/transit time parameter that should not be eluded in in vitro and in vivo studies, particularly when studying human populations where transit time is known to have changed. For example less bifidobacteria have been reported in elderly people (Hopkins et al. 2001), a population known to have an increased transit time (Probert et al. 1993; Woodmansey 2007). This highlights the relevance of flexible in vitro models like the P-ECSIM, and a probable underestimated role of abiotic factors in the human gut. On the other hand, the consequence for the microbiota of a change in retention time was at least partly reversible in the P-ECSIM, indicating a potent resilience of the microbiota. This is further illustrated by closely related RISA fingerprints (compare P1 and P3 in Fig. 2b), although there was a stage with an alteration of the retention time between these two experiments (P-ECSIM P2). A more precise analysis of the RISA fingerprints (similarity matrix, neighbour-joining algorithm) confirms this view, although obtained from few differentiated bands (data not shown).
In summary, these studies show the potentiality of this alternative fermentation system to better understand the human proximal colon. Particularly, P-ECSIM should facilitate in vitro studies on microbiota from elderly people characterized by particular biotic (methanogens) and abiotic (transit time) components. Further developments are needed, particularly relative to long-term storage in order to improve reproducibility and inter-lab comparisons. The same approach is now used to validate a three-part continuous fermentation system. This three-stage ECSIM would provide a mean for more closely simulating the proximal, transversal and distal parts of the human colon.
References
Allison C, McFarlan C, MacFarlane GT (1989) Studies on mixed populations of human intestinal bacteria grown in single-stage and multistage continuous culture systems. Appl Environ Microbiol 55(3):672–678
Bearne CA, Mallett AK, Rowland IR, Brennan-Craddock WE (1990) Continuous culture of human faecal bacteria as an in vitro model for the colonic microflora. Toxicol In Vitro 4(4–5):522–525
Beerens H (1991) Detection of bifidobacteria by using propionic acid as a selective agent. Appl Environ Microbiol 57(8):2418–2419
Brugere JF, Mihajlovski A, Missaoui M, Peyret P (2009) Tools for stools: the challenge of assessing human intestinal microbiota using molecular diagnostics. Expert Rev Mol Diagn 9(4):353–365
Cardinale M, Brusetti L, Quatrini P, Borin S, Puglia AM, Rizzi A, Zanardini E, Sorlini C, Corselli C, Daffonchio D (2004) Comparison of different primer sets for use in automated ribosomal intergenic spacer analysis of complex bacterial communities. Appl Environ Microbiol 70(10):6147–6156
Child MW, Kennedy A, Walker AW, Bahrami B, Macfarlane S, Macfarlane GT (2006) Studies on the effect of system retention time on bacterial populations colonizing a three-stage continuous culture model of the human large gut using FISH techniques. FEMS Microbiol Ecol 55(2):299–310
Cinquin C, Le Blay G, Fliss I, Lacroix C (2004) Immobilization of infant fecal microbiota and utilization in an in vitro colonic fermentation model. Microb Ecol 48(1):128–138
Claesson MJ, O'Sullivan O, Wang Q, Nikkila J, Marchesi JR, Smidt H, de Vos WM, Ross RP, O'Toole PW (2009) Comparative analysis of pyrosequencing and a phylogenetic microarray for exploring microbial community structures in the human distal intestine. PLoS One 4(8):e6669
Cummings JH, Pomare EW, Branch WJ, Naylor CP, Macfarlane GT (1987) Short chain fatty acids in human large intestine, portal, hepatic and venous blood. Gut 28(10):1221–1227
Cummings JH, Bingham SA, Heaton KW, Eastwood MA (1992) Fecal weight, colon cancer risk, and dietary intake of nonstarch polysaccharides (dietary fiber). Gastroenterology 103(6):1783–1789
Dethlefsen L, Huse S, Sogin ML, Relman DA (2008) The pervasive effects of an antibiotic on the human gut microbiota, as revealed by deep 16S rRNA sequencing. PLoS Biol 6(11):e280
Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M, Gill SR, Nelson KE, Relman DA (2005) Diversity of the human intestinal microbial flora. Science 308(5728):1635–1638
Gibson GR, Fuller R (2000) Aspects of in vitro and in vivo research approaches directed toward identifying probiotics and prebiotics for human use. J Nutr 130(2S Suppl):391S–395S
Gibson GR, Cummings JH, Macfarlane GT (1988) Use of a three-stage continuous culture system to study the effect of mucin on dissimilatory sulfate reduction and methanogenesis by mixed populations of human gut bacteria. Appl Environ Microbiol 54(11):2750–2755
Hopkins MJ, Sharp R, Macfarlane GT (2001) Age and disease related changes in intestinal bacterial populations assessed by cell culture, 16S rRNA abundance, and community cellular fatty acid profiles. Gut 48(2):198–205
Le Blay G, Rytka J, Zihler A, Lacroix C (2009) New in vitro colonic fermentation model for Salmonella infection in the child gut. FEMS Microbiol Ecol 67(2):198–207
Macfarlane GT, Macfarlane S, Gibson GR (1998) Validation of a three-stage compound continuous culture system for investigating the effect of retention time on the ecology and metabolism of bacteria in the human colon. Microb Ecol 35(2):180–187
Mihajlovski A, Alric M, Brugere JF (2008) A putative new order of methanogenic Archaea inhabiting the human gut, as revealed by molecular analyses of the mcrA gene. Res Microbiol 159(7–8):516–521
Mihajlovski A, Doré J, Levenez F, Alric M, Brugère J-F (2010) Molecular evaluation of the human gut methanogenic archaeal microbiota reveals an age-associated increase of the diversity. Environ Microbiol Rep 2:272–280
Minekus M, Smeets-Peeters M, Bernalier A, Marol-Bonnin S, Havenaar R, Marteau P, Alric M, Fonty G, Huis in't Veld JH (1999) A computer-controlled system to simulate conditions of the large intestine with peristaltic mixing, water absorption and absorption of fermentation products. Appl Microbiol Biotechnol 53(1):108–114
Mitsuoka T, Hayakawa K (1973) The fecal flora in man. I. Composition of the fecal flora of various age groups. Zentralbl Bakteriol Orig A 223(2):333–342
Molly K, Vande Woestyne M, Verstraete W (1993) Development of a 5-step multi-chamber reactor as a simulation of the human intestinal microbial ecosystem. Appl Microbiol Biotechnol 39(2):254–258
Probert CJ, Emmett PM, Heaton KW (1993) Intestinal transit time in the population calculated from self made observations of defecation. J Epidemiol Community Health 47(4):331–333
Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, Nielsen T, Pons N, Levenez F, Yamada T, Mende DR, Li J, Xu J, Li S, Li D, Cao J, Wang B, Liang H, Zheng H, Xie Y, Tap J, Lepage P, Bertalan M, Batto JM, Hansen T, Le Paslier D, Linneberg A, Nielsen HB, Pelletier E, Renault P, Sicheritz-Ponten T, Turner K, Zhu H, Yu C, Li S, Jian M, Zhou Y, Li Y, Zhang X, Li S, Qin N, Yang H, Wang J, Brunak S, Dore J, Guarner F, Kristiansen K, Pedersen O, Parkhill J, Weissenbach J, Bork P, Ehrlich SD, Wang J (2010) A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464(7285):59–65
Rajilić-Stojanović M, Maathuis A, Heilig HG, Venema K, de Vos WM, Smidt H (2010) Evaluating the microbial diversity of an in vitro model of the human large intestine by phylogenetic microarray analysis. Microbiology 156(11):3270–3281
Rumney CJ, Rowland IR (1992) In vivo and in vitro models of the human colonic flora. Crit Rev Food Sci Nutr 31(4):299–331
Tap J, Mondot S, Levenez F, Pelletier E, Caron C, Furet JP, Ugarte E, Munoz-Tamayo R, Paslier DL, Nalin R, Dore J, Leclerc M (2009) Towards the human intestinal microbiota phylogenetic core. Environ Microbiol 11(10):2574–2584
Van den Abbeele P, Grootaert C, Marzorati M, Possemiers S, Verstraete W, Gerard P, Rabot S, Bruneau A, El Aidy S, Derrien M, Zoetendal E, Kleerebezem M, Smidt H, Van de Wiele T (2010) Microbial community development in a dynamic gut model is reproducible, colon region specific, and selective for Bacteroidetes and Clostridium cluster IX. Appl Environ Microbiol 76(15):5237–5246
Woodmansey EJ (2007) Intestinal bacteria and ageing. J Appl Microbiol 102(5):1178–1186
Yu Z, Morrison M (2004) Improved extraction of PCR-quality community DNA from digesta and fecal samples. Biotechniques 36(5):808–812
Zhang H, DiBaise JK, Zuccolo A, Kudrna D, Braidotti M, Yu Y, Parameswaran P, Crowell MD, Wing R, Rittmann BE, Krajmalnik-Brown R (2009) Human gut microbiota in obesity and after gastric bypass. Proc Natl Acad Sci U S A 106(7):2365–2370
Zoetendal EG, Rajilic-Stojanovic M, de Vos WM (2008) High-throughput diversity and functionality analysis of the gastrointestinal tract microbiota. Gut 57(11):1605–1615
Acknowledgements
This work was supported by a postdoctoral scholarship support from the European Union (UE) and the Auvergne Council, to DF-G. (FEDER). We thank Pascal Vandekerckove from Lesaffre SA and Zsolt Popse from Global Process Concept (France) for their support. J-FB thanks Céline Vidal and Claire Ardaens for their technical help. The authors are grateful of the assistance of Meghan Schaut (Michigan State University) and William Tottey for their remarks on the English version of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Feria-Gervasio, D., Denis, S., Alric, M. et al. In vitro maintenance of a human proximal colon microbiota using the continuous fermentation system P-ECSIM. Appl Microbiol Biotechnol 91, 1425–1433 (2011). https://doi.org/10.1007/s00253-011-3462-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-011-3462-5