
Abstract
We previously described five arabinanolytic enzymes secreted by Penicillium chrysogenum 31B into the culture medium. Here, we describe a sixth such enzyme, termed AbnS1. Analysis of the reaction products of debranched arabinan revealed that AbnS1 cleaved the substrate in an endo manner. The optimum temperature of AbnS1 was 60°C, which was much higher than that of a cold-adapted endo-arabinanase (Abnc) produced by this strain. The abns1 cDNA gene encoding AbnS1 was isolated by in vitro cloning. The deduced amino acid sequence of AbnS1 had 70% identity with that of Abnc. Pfam analysis revealed a Glyco_hydro_43 domain at positions 28 to 318 of AbnS1. Semi-quantitative reverse transcription-polymerase chain reaction analysis indicated that the abns1 gene was constitutively expressed in P. chrysogenum 31B at a low level, although the expression was only slightly induced with arabinose and arabinan. In contrast, expression of the abnc gene encoding Abnc was strongly induced by arabinose, arabinitol, and arabinan. Using debranched arabinan as substrate, recombinant AbnS1 (rAbnS1) accumulated arabinobiose and arabinotriose as the major products. Recombinant Abnc (rAbnc) released mainly arabinotriose and lesser amounts of arabinose and arabinobiose than did rAbnS1. Branched arabinan was completely degraded to arabinose by the action of rAbnS1 or rAbnc in combination with α-l-arabinofuranosidase.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
-
Sugar beet pulp, which is a by-product of sucrose refining, is abundantly produced in Europe, Japan, and the USA. It is mainly used as feed for ruminants, but its biotechnological conversion to value-added products such as ethanol or industrial chemicals is desirable. Sugar beet pulp is largely composed of pectins (56% of dry pulp). The abundant sugars in the pectins are d-galacturonic acid (21.1% of dry pulp), l-arabinose (20.9%), d-galactose (5.1%), and l-rhamnose (2.4%). Sugar beet pectin also contains methanol (1.8%), acetic acid (3.9%), and ferulic acid (0.8%) as side groups (Micard et al. 1996). The d-galacturonic acid residues exist in both homogalacturonan and rhamnogalacturonan regions of the pectins. The carboxyl groups of pectins are partially esterified with methanol, and their hydroxyl groups at position 2 or/and 3 of the d-galacturonic acid residues are sometimes acetylated. In contrast, the l-arabinoses are mainly present in the rhamnogalacturonan region and form branched l-arabinans, highly branched α-1,5-l-arabinans possessing 1,2- and/or 1,3-α-linked l-arabinofuranosyl residues as side chains (Tagawa and Kaji 1988). Ferulic acids are mainly linked to hydroxyl groups at C-2 of α-1,5-linked arabinofuranose residues or at C-6 of β-1,4-linked galactopyranose residues.
Among pectic components, l-arabinose is an important sugar in food industry and biofuel industry. l-Arabinose selectively inhibits intestinal sucrase in a noncompetitive manner, leading to decreased plasma glucose levels after sucrose ingestion (Seri et al. 1996). Arabinooligosaccharides that are prepared from sugar beet arabinan by treatment with a commercial pectinase (Viscozyme L) are considered as potential prebiotics (Al-Tamimi et al. 2006). In addition, two groups have constructed genetically engineered Saccharomyces cerevisiae strains to produce ethanol from l-arabinose through fermentation. One group used a prokaryotic l-arabinose pathway with an ethanol yield of 60% (Becker and Boles 2003) and another group used a eukaryotic pathway with a yield of 40% (Bera et al. 2010). Therefore, degradation of l-arabinan in sugar beet pectin can significantly improve the utilization of sugar beet pulp.
We previously isolated Penicillium chrysogenum 31B from rotten sugar beet and showed that it has high degrading activity towards sugar beet arabinan (Sakamoto and Thibault 2001). When the culture filtrate of this strain was incubated with sugar beet pulp, approximately 90% of the total arabinose in the pulp was released as the monomer to the reaction mixture (unpublished results). The arabinanolytic enzymes secreted by P. chrysogenum 31B include an exo-arabinanase (Abnx; Sakamoto and Thibault 2001; Sakamoto et al. 2004), three distinct α-l-arabinofuranosidases (AFQ1 and AFS1, Sakamoto and Kawasaki 2003; AXS5, Sakamoto et al. 2011), and an endo-arabinanase (Abnc, Sakamoto et al. 2003; 2005). Abnc has optimal activity at 30°C to 40°C, which makes it a cold-adapted enzyme compared with the endo-arabinanases of Aspergillus niger (Rombouts et al. 1988; Flipphi et al. 1993) and Aspergillus aculeatus (Beldman et al. 1993; Skjøt et al. 2001) which have optimal activities at 50°C to 60°C. We previously described five arabinanolytic enzymes produced by P. chrysogenum 31B. Recently, in the course of purifying these enzymes, we found a sixth enzyme, a mesophilic endo-arabinanase, which we call AbnS1. Here, we describe the purification, molecular cloning, and overexpression in Escherichia coli of AbnS1. We also report biochemical characterizations of AbnS1 and Abnc and their gene expression profiles in P. chrysogenum 31B.
Materials and methods
Chemicals and reagents
HiLoad 16/60 Superdex 75 and Ni Sepharose 6 Fast Flow were purchased from GE Healthcare UK Ltd. (Buckinghamshire, UK). CM-Toyopearl 650 M, DEAE-Toyopearl 650 M, and Butyl-Toyopearl 650 M were obtained from Tosoh Corp. (Tokyo, Japan). Sugar beet l-arabinan, sugar beet debranched arabinan, α-1,5-arabinooligosaccharides, wheat arabinoxylan (low viscosity), lupin galactan, and soy bean rhamnogalacturonan were obtained from Megazyme International Ireland Ltd. (Wicklow, Ireland). Na-polygalacturonic acid was from Sigma-Aldrich Co. (St. Louis, MO, USA). Sugar beet pulp was kindly provided by Nippon Beet Sugar Manufacturing Co., Ltd. (Tokyo). All other chemicals were from Wako Pure Chemical Industries, Ltd. (Osaka, Japan) unless otherwise stated and were of certified reagent grade.
Strain, media, growth conditions, and plasmids
The P. chrysogenum 31B strain used in this study is deposited in International Patent Organism Depositary, National Institute of Advanced Industrial Science and Technology (acc. no. FERM P-19163; http://unit.aist.go.jp/ipod/cie/index.html). To purify AbnS1, 20 ml of medium (2% glucose, 0.5% yeast extract, and 0.5% peptone, pH 5.0) was inoculated with 106 spores/ml and incubated at 30°C in a reciprocating shaker at 120 strokes/min for 1 day. The whole culture broth was then transferred to a 5-l Erlenmeyer flask containing 2 l of basal medium (0.2% NH4NO3, 0.1% K2HPO4, 0.05% MgSO4·7H2O, 0.05% KCl, and 0.001% FeSO4 and 0.1% peptone, pH 5.0) supplemented with 0.1% glucose and 2% sugar beet pulp and cultured at 30°C for 15 days under static conditions.
E. coli DH5α, E. coli BL21 (DE3), and the plasmid pET-30a(+) (Novagen Inc., Madison, WI, USA) were used for cloning and expression of the polymerase chain reaction (PCR) products. Transformants were grown in LBG medium (1% peptone, 0.5% yeast extract, 1% NaCl, and 2% glucose, pH 7.0) supplemented with 20 μg/ml of kanamycin.
Enzyme assay
Arabinan-degrading activity was assayed by measuring the release of reducing groups in a reaction mixture containing 190 μl of 0.1% debranched arabinan in 20 mM Na-acetate buffer (pH 5.0) and 10 μl of enzyme sample at 37°C. Reducing sugars were measured by the method of Somogyi (1952). One unit was defined as the amount of enzyme that forms reducing groups corresponding to 1 μmol of l-arabinose in 1 min under the above conditions.
Purification of AbnS1
AbnS1 was purified from the culture filtrate of P. chrysogenum 31B as follows. The culture filtrate (4 l) was concentrated to 200 ml by ultrafiltration (10-kDa cutoff) and precipitated with ammonium sulfate at 80% saturation. After centrifugation (22,000×g for 15 min at 4°C), the pellet was dissolved in 20 mM Na-acetate buffer (pH 5.0) and dialyzed against the same buffer. The enzyme solution was loaded onto a CM-Toyopearl 650 M column (4 × 25 cm) equilibrated with the dialysis buffer. The unbound fraction was put on a DEAE-Toyopearl 650 M column (4 × 25 cm) equilibrated with 20 mM Na-acetate buffer (pH 5.0). The unbound fraction was added ammonium sulfate to a 30% saturation. The enzyme solution was put on a Butyl-Toyopearl 650 M column (4 × 25 cm) equilibrated with 20 mM Na-acetate buffer (pH 5.0) containing ammonium sulfate at a concentration of 30% saturation. The adsorbed proteins were eluted by a linear gradient of ammonium sulfate (500 ml, from 30% to 0% of saturation). The active fractions were pooled, dialyzed against 20 mM Na-acetate buffer (pH 5.0), concentrated, and injected into a size-exclusion column of Superdex 75 equilibrated with 100 mM NaCl in 20 mM Na-acetate buffer (pH 5.0). Proteins were eluted with the same buffer at a flow rate of 1 ml/min.
Protein concentration was determined by a Coomassie Plus-The Better Bradford Assay Kit (Pierce Biotechnology Inc., Rockford, IL, USA) with bovine serum albumin as the standard and by measurement of absorbance at 280 nm. Protein homogeneity was evaluated, and the molecular mass of the purified enzyme was estimated by sodium dodecyl sulfate-polyacrilamide gel electrophoresis (SDS-PAGE) in a 10% gel by the method of Laemmli (1970). The proteins were stained with Coomassie Brilliant Blue R-250.
N-terminal amino acid sequence of AbnS1
To determine the N terminus of AbnS1, 5 μg of the purified protein was directly blotted on a polyvinylidene difluoride membrane (ATTO Corp., Tokyo, Japan). The N terminus of the protein was determined by automated Edman degradation using a gas phase sequencer.
Influence of temperature and pH
To determine the optimum temperature, the enzyme reaction was performed at various temperatures in 20 mM Na-acetate buffer (pH 5.0) for 10 min. The temperature stability was evaluated by measuring the residual activity after 1 h pre-incubation of the enzyme at temperatures between 30°C and 70°C in 20 mM Na-acetate buffer (pH 5.0) containing 50 μg/ml of bovine serum albumin. The optimum pH was determined by measuring the activity at 37°C over the pH range 3 to 9 using 100 mM Na-acetate-HCl buffer (pH 3), Na-acetate buffer (pH 4 and 5), Na-phosphate buffer (pH 6 to 8), and Na-carbonate buffer (pH 9). The pH stability was studied by pre-incubation of the enzyme at 30°C for 16 h at various pHs in 100 mM the above buffers described.
Determination of nucleotide sequence of gene encoding AbnS1
The nucleotide sequence of abns1 was determined by 3′-rapid amplification of cDNA ends (3′-RACE) and cassette ligation-mediated PCR (CLM-PCR), as reported previously (Sakamoto et al. 2010). In 3′-RACE, two abns1-specific degenerate primers were synthesized based on the N-terminal amino acid sequence of AbnS1 determined by Edman degradation. Total RNA was prepared from mycelia of P. chrysogenum 31B grown in basal medium supplemented with 2% sugar beet pulp for 12 days at 30°C under static conditions. Single-stranded cDNA was synthesized using total RNA with a linker adapted oligo-dT primer. In CLM-PCR, two abns1-specific primers were designed based on the nucleotide sequence of the PCR product of 3′-RACE. SalI cassette-ligated genomic DNA was used as the template for CLM-PCR. To clone the full-length abns1 cDNA and genomic DNA, two primers were synthesized based on the sequencing data of 3′-RACE and CLM-PCR. The nucleotide sequence was determined with a BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA, USA) on a DNA sequencer (ABI 3730xl; Applied Biosystems).
Sequence analysis
The deduced amino acid sequence of AbnS1 was analyzed for its similarity using the UniProtKB blast program (http://www.uniprot.org/blast/uniprot). Module sequence analysis of AbnS1 was performed using the Pfam database (http://pfam.sanger.ac.uk/search).
Expression of the abns1 and abnc gene in E. coli
The region between the N terminus (YANPG) and the C terminus (GWPVV) of the mature AbnS1 was PCR-amplified using primers abns1-NdeI-Fw (5′-TTCCCATATGTACGCAAACCCTGGC) and abns1-XhoI-Rv (5′-TTTCTCGAGAACCACTGGCCAGCC) and single-stranded cDNA synthesized from total RNA as the template. NdeI and XhoI sites (underlined) were added to the primers, respectively. The amplified fragment was digested with NdeI and XhoI, and ligated to the restriction enzyme sites of the pET-30a(+) vector, forming a new plasmid termed abns1-His-pET-30a(+). For production of the recombinant AbnS1 (rAbnS1), the overnight culture of E. coli BL21 (DE3) transformant having the constructed plasmid was inoculated to LBG medium containing kanamycin (20 μg/ml) and cultured at 37°C for 90 min. Then, isopropyl β-thiogalactopyranoside was added to 0.1 mM, and incubation was continued at 18°C for 20 h. The cells were harvested by centrifugation, suspended in column buffer (500 mM NaCl and 20 mM Na-phosphate buffer, pH 7.4) containing protease inhibitor cocktail (Nacalai Tesque, Inc, Kyoto, Japan), and disrupted by sonic treatment on ice. After centrifugation, the supernatant was loaded onto a Ni Sepharose 6 Fast Flow column equilibrated with the column buffer. The bound proteins were eluted by the column buffer containing 100 mM imidazole. The active fractions were pooled, dialyzed against 20 mM Na-acetate buffer (pH 5.0), concentrated, and injected into a size-exclusion column of Superdex 75 equilibrated with the column buffer.
Recombinant Abnc (rAbnc) was synthesized with the same host/vector system used to synthesize rAbnS1. The mature region of the abnc gene, which encodes Abnc, was PCR-amplified using primers abnc-NdeI-Fw (5′-TTCCCATATGTACGCGAATCCCGGAGCTT) and abnc-NotI-Rv (5′-TTGCGGCCGCCACCGAAGGCCAGCCGCTCGAGAAATCAAT), and the pMAL-AC1 vector containing the abnc cDNA (Sakamoto et al. 2005) as the template. NdeI and NotI sites (underlined) were added to the primers, respectively.
Analysis of the enzymatic products by high-performance anion-exchange chromatography
Hydrolysis of debranched arabinan was carried out by incubating the reaction mixture containing 1 ml of 0.5% substrate in 20 mM Na-acetate buffer (pH 5.0) and rAbnS1 (80 mU) or rAbnc (80 mU) at 50°C for rAbnS1 and 30°C for rAbnc. Aliquots were taken at intervals, and the products were analyzed by high-performance anion-exchange chromatography (HPAEC) using a Dionex DXc-500 system (Dionex Corp., Sunnyvale, CA, USA) with a CarboPac PA-1 column (4 × 250 mm; Dionex). Sugars were eluted at a flow rate of 1 ml/min with 0.1 M NaOH for 5 min followed by a linear gradient from 0 to 0.45 M sodium acetate in 0.1 M NaOH for 30 min. The effluent was monitored with pulsed amperometric detection.
Substrate specificities of rAbnS1 and rAbnc towards polysaccharides
Substrate specificity towards polysaccharides was tested by incubating 12 mU of the enzyme with 200 μl of 0.1% substrate in 20 mM Na-acetate buffer (pH 5.0) at 50°C (rAbnS1) or 30°C (rAbnc) for 30 min. Reducing sugars released into the mixtures were measured by the method of Somogyi (1952).
Degradation of l-arabinan by rAbnS1 or rAbnc with α-l-arabinofuranosidase
Enzyme activity was tested by incubating 20 mU of the enzyme with 300 μl of 0.1% l-arabinan in 20 mM Na-acetate buffer (pH 5.0) at 37°C. The mixture was withdrawn at 3 and 16 h followed by quantification of arabinose released into the reaction mixtures by HPAEC. Yields of release of arabinose (%) were calculated taking the amount of the total arabinose in the substrate as 100%. Total arabinose content in l-arabinan was determined by HPAEC after hydrolysis of the polysaccharide with 1 M sulfuric acid at 100°C for 1 h. Recombinant α-l-arabinofuranosidase (rAFQ1) used in this experiment was isolated from the cell-free extract of E. coli BL21 (DE3) transformed with the pET-32a vector (Novagen) to which the P. chrysogenum afq1 gene (acc. no. AB461442) is inserted.
Semi-quantitative RT-PCR
To study the effect of different carbon sources or different concentrations of arabinose on the expression of abns1 and abnc gene in P. chrysogenum 31B, the microorganism was cultured in 100-ml Erlenmeyer flasks containing 20 ml of a medium (2% glucose, 0.5% yeast extract, and 0.5% peptone, pH 5) at 30°C for 1 week at static conditions. After washing mycelia with 0.85% NaCl, 0.5 g of mycelia were transferred to 50-ml Erlenmeyer flasks containing 10 ml of a fresh basal medium supplemented with sugar and cultured at 30°C for 1, 2, and 3 days under static conditions. The amount of monomeric sugars remaining in the culture filtrate was quantified by HPAEC. Total RNA was prepared from mycelia, and single-stranded cDNA was synthesized using the RNA with random hexamers (Takara Bio Inc., Shiga, Japan). The partial abns1 and abnc cDNAs were PCR-amplified using the gene-specific primers (5′-AACGGCCCGTGGACTAACAT and 5′-TAGAGGACGACGAGGCAATA for abns1 and 5′-CGACCTTCGAGACCCAAATC and 5′-GGCATACCCTGATCATACTTG for abnc) followed by electrophoresis in 2% agarose gel. The PCR reaction mixtures consisted of 5 μl Go-Taq Green Master Mix (Promega Co., Madison, WI, USA), 3 μl cDNA, 1 μl sense primer (1 μM), and 1 μl antisense primer (1 μM). An initial denaturation step of 2 min at 94°C was followed by 30 s at 94°C, 90 s at 55°C, and 30 s at 72°C for 30 cycles. The conditions were determined by testing a number of cycles ranging from 20 to 35, so that none of the RNA samples reached a plateau at the end of the amplification. A part of mitochondrial small rDNA was amplified using a set of primers (5′-TGATGGCTCTAACTGAACACTG and 5′-AAACCCAATAAAGATAACTAACACT) as a control.
Nucleotide sequences
The nucleotide sequences of abns1 cDNA and genomic DNA were submitted to the DDBJ/EMBL/GenBank (acc. nos. AB186044 and AB605633, respectively).
Results
Purification of AbnS1
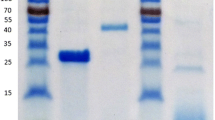
We previously demonstrated that P. chrysogenum 31B produces five arabinanolytic enzymes that include an Abnx (Sakamoto and Thibault 2001), a cold-adapted endo-arabinanase (Abnc, Sakamoto et al. 2003), three α-l-arabinofuranosidases (AFQ1 and AFS1, Sakamoto and Kawasaki 2003; AXS5, Sakamoto et al. 2011). A sixth arabinanolytic enzyme, termed AbnS1, of this strain was purified from 4 l fermentation broth of P. chrysogenum 31B. After elimination of the other five arabinanolytic enzymes by ion-exchange chromatographies using CM- and DEAE-Toyopearl columns, the enzyme solution was applied to a hydrophobic column of Butyl-Toyopearl, in which the enzyme activity was detected in bound fractions as a single peak. Finally, AbnS1 was separated from contaminating proteins with a Superdex 75 size-exclusion column. The enzyme was highly purified as judged from an N-terminal amino acid analysis and by SDS-PAGE (Fig. 1a). Specific activity of AbnS1 was 18.8 U/mg.

SDS-PAGE of native and recombinant AbnS1 and Abnc. a Lane 1, purified native AbnS1; lane 2, AbnS1 after endoglycosidase H treatment; lane 3, molecular markers. b Lane 1, purified rAbnS1; lane 2, purified rAbnc; lane 3, molecular markers
Characterization of AbnS1
The molecular mass of AbnS1 was estimated to be 31 kDa by SDS-PAGE (Fig. 1a). Size-exclusion chromatography of AbnS1 using Superdex 75 indicated a molecular mass of 27 kDa. These results confirmed the monomeric structure of the protein. The molecular mass of the enzyme was not affected by treatment with endoglycosidase H (Fig. 1a), suggesting that AbnS1 was not a glycoprotein. The N-terminal amino acid sequence of AbnS1 was determined to be YANPGSXSGAXIVHDPTLIQRSSDN, where X indicates undetermined residues.
The optimal temperature of the activity was 60°C (curve with open circles in Fig. 2). This was significantly higher than the optimal temperature for another endo-arabinanase of this strain (Abnc; closed circles in Fig. 2). However, the temperature dependency of AbnS1 is similar to the temperature dependencies of two other fungal endo-arabinanases, ABNA of A. niger (Rombouts et al. 1988; Flipphi et al. 1993) and ARA1 of A. aculeatus (Beldman et al. 1993; Skjøt et al. 2001). AbnS1 maintained 100% of the initial activity by treatment it at 50°C for 1 h. Approximately 70% of the activity was lost at 60°C. This result demonstrated that AbnS1 was much more stable than Abnc, which is unstable even at 40°C (Sakamoto et al. 2003). Arabinanase activity was highest in the pH range 6.0-7.0, dropping to about 55% and 70% of the highest activity at pH 5.0 and 8.0, respectively. The enzyme was stable between pH 4.0 and pH 8.0. When the purified enzyme was incubated with debranched arabinan, several arabinooligosaccharides were detected by HPAEC (data not shown), which indicated that AbnS1 was an endo-acting enzyme. Other properties of AbnS1 were evaluated with a recombinant enzyme to exclude the possibility of contamination with other arabinanolytic enzymes. These results are described below.

Effect of temperature on enzyme activity of AbnS1 (open circles) and Abnc (closed circles). Reactions were carried out in 20 mM Na-acetate buffer (pH 5.0). All experiments were performed in duplicate
Nucleotide sequence and amino acid sequence alignment
The coding sequence of the abns1 cDNA contains 960 bp encoding a protein of 320 amino acids. The calculated molecular mass and isoelectric point of the polypeptide consisting of residues 19 to 320 was 32059 and 5.09, respectively. The calculated molecular mass of AbnS1 was in good agreement with that of the native protein (31 kDa). No asparagine residue as a potential N-glycosylation site was found in the deduced amino acid sequence. Pfam analysis revealed a Glyco_hydro_43 domain (accession no. PF04616) at positions 28 to 318 of the enzyme. No carbohydrate-binding module was found. Determination of the genomic DNA sequence of the abns1 gene revealed that the gene was interrupted by three introns at nucleotides 265 to 334, 812 to 867, and 954 to 1006. The fungal consensus 5′ and 3′ splicing sites, GTPuNGPy and PyAG (Unkles 1992), were conserved in the three introns.
Using the deduced amino acid sequence of the abns1 gene as a query, the best hits in a UniProtKB blast search were proteins Pc13g15920 (B6H2Z7, 100% identity) and Pc20g15730 (B6HE62; 83% identity) from P. chrysogenum Wisconsin 54–1255, whose complete genome sequence was determined (Van den Berg et al. 2008). To our knowledge, previous studies have reported five genes encoding functional fungal endo-arabinanases: A. niger ABNA (Flipphi et al. 1993; acc. no. P42256), A. aculeatus ARA1 (Skjøt et al. 2001; acc. no. Q9HFS9), A. nidulans ABNB and ABNC (Bauer et al. 2006; acc. no. Q5AZC8 and Q5AUM3), and P. chrysogenum Abnc (Sakamoto et al. 2005; acc. no. Q75WE6). In an alignment of the deduced amino acid sequences of these proteins and AbnS1, A. nidulans ABNB was the least similar. The deduced amino acid sequence of AbnS1 had 77% and 72% identities with those of mesophilic endo-arabinanases from A. niger and A. aculeatus, respectively. Although P. chrysogenum Abnc is a cold-adapted endo-arabinanase, it has 70% amino acid identity to AbnS1. X-ray crystallographic studies of arabinanase 43A of Cellvibrio japonicus suggest that the Asp38 and Glu221 of this enzyme act as the catalytic base and acid, respectively, and that Asp and Glu residues serve the same functions in other enzymes belonging to glycoside hydrolase (GH) family 43 (Nurizzo et al. 2002). These amino acid residues are conserved at Asp15 and Glu181 in the mature AbnS1.
Expression of abns1 and abnc genes in P. chrysogenum 31B
The time-course transcriptional profiles of abns1 and abnc gene in P. chrysogenum 31B grown in the presence of different carbon sources were studied using semi-quantitative RT-PCR. For each of 13 carbon sources examined, the abns1 gene was constitutively expressed at a low level (Fig. 3a), although the expression was only slightly induced with arabinose (Ara) and arabinan (AN). In contrast, expression of the abnc gene was strongly induced by arabinose, arabinitol (Aol), and arabinan and less strongly induced by arabinoxylan (AX). Remaining sugar content in the culture filtrates of mycelia grown in monomeric carbon source is shown in Table 1. These data show that uptake of sugars by P. chrysogenum 31B is slow and that xylitol and rhamnose are hardly assimilated. In fact, expression of the abnc gene was scarce in the fungus grown in the presence of arabinose for 2, 4, 6, or 8 h (data not shown). To examine whether the expression of abns1 gene is repressed under the above conditions (supplementation with 3% arabinose), the expression in the presence of lower concentrations of arabinose was studied. However, little transcript of abns1 was detected even at these conditions (Fig. 3b). These results suggest that the expression of the two genes is regulated by different mechanisms in P. chrysogenum 31B.

Semi-quantitative RT-PCR analysis of the expressions of abns1 and abnc in P. chrysogenum 31B grown on different carbon sources (a) and on different concentrations of arabinose (b). The experimental conditions are described in the text. a The initial carbon source contents of the basal media were 3% for monomeric carbohydrates and 1% for polymeric carbohydrates. None: without supplementation of carbon source, Glc: d-glucose, Ara: l-arabinose, Aol: l-arabinitol, Xyl: d-xylose, Xol: d-xylitol, Gal: d-galactose, Rha: l-rhamnose, GA: d-galacturonic acid, AN: sugar beet l-arabinan, AX: wheat arabinoxylan, GT: lupin galactan, RG: soy bean rhamnogalacturonan, PGA: polygalacturonic acid. b The initial arabinose contents of the basal media were 0.003%, 0.03%, and 0.3%
Overexpression of abns1 and abnc genes in E. coli
One liter of culture medium of the E. coli BL21 (DE3) transformant having the abns1-His-pET-30a(+) vector yielded 0.5 mg of rAbnS1 tagged with (His)6 at its C terminus. Its purity was evaluated to be over 95% on SDS-PAGE (Fig. 1b). Specific activity of the recombinant enzyme was 17.0 U/mg, which is similar to that of the native enzyme. In contrast, His-tagged rAbnc was synthesized in E. coli BL21 (DE3) as inclusion bodies. Active rAbnc was obtained by refolding of the recombinant inclusion body protein. The enzyme was purified using a Ni Sepharose 6 Fast Flow column and a Superdex 75 size-exclusion column. Its molecular mass was slightly larger than that of rAbnS1 (Fig. 1b). Details on the refolding and purification conditions of rAbnc will be described elsewhere.
Characteristics of rAbnS1 and rAbnc
rAbnS1, like the native enzyme, showed optimal activity at 60°C and was stable up to 50°C. Using debranched arabinan as substrate, rAbnS1 produced several arabinooligosaccharides in the initial stage of hydrolysis, and their levels increased with time (Fig. 4), demonstrating that rAbnS1 is an endo-acting enzyme. The major products were arabinobiose and arabinotriose. rAbnc released lesser amounts of arabinose and arabinobiose than did rAbnS1. The specificities of rAbnS1 and rAbnc toward polysaccharides including debranched arabinan, l-arabinan, and wheat arabinoxylan were examined. Both enzymes strongly degraded debranched arabinan but not l-arabinan under the conditions used, indicating that the α-l-arabinofuranosyl side chains interfered with access of the enzymes to the α-1,5-arabinan backbone. Neither enzyme hydrolyzed wheat arabinoxylan. Both recombinant enzymes had higher activities towards oligosaccharides with higher degrees of polymerization (DPs; Table 2). The arabinopentaose/arabinohexaose activity ratio of rAbnS1 was 0.46, which was much higher than that of rAbnc (0.24). Neither enzyme showed degrading activity towards arabinotriose or arabinotetraose under the experimental conditions used.

HPAEC chromatograms showing enzymatic products of debranched arabinan with rAbnS1 or rAbnc after different reaction times. Ara2, arabinobiose; Ara3, arabinotriose; Ara4, arabinotetraose
Synergistics between endo-arabinanases and α-l-arabinofuranosidase
To investigate whether there are any synergistic effects among arabinanolytic enzymes including rAbnc, rAbnc, and a recombinant rAFQ1 in the degradation of branched l-arabinan, the substrate was incubated in combination with the above enzymes. As shown in Table 3, no arabinose was detected in a reaction mixture containing rAbnS1 or rAbnc alone. rAFQ1 released 34% of total arabinose in the substrate when the time of incubation was 16 h. When endo-arabinanases acted in combination with rAFQ1, l-arabinan was completely hydrolyzed to arabinose in 16 h. AFQ1 shows activities that debranch arabinose residues from side chains of l-arabinan and degrades α-1,5-arabinooligosaccharides to monomeric arabinose (Sakamoto and Kawasaki 2003). In contrast, endo-arabinanases accumulate α-1,5-arabinooligosaccharides from debranched arabinan, which is produced from branched l-arabinan by the action of AFQ1. Complete degradation of branched l-arabinan is therefore achieved by synergistic effects of their enzymatic functions.
Discussion
Although enzymes that hydrolyze α-l-arabinofuranosyl linkages including endo-arabinanases, exo-arabinanases, and α-l-arabinofuranosidases have been grouped into GH families 3, 43, 51, 54, 62, and 93 based on their amino acid sequences (http://www.cazy.org/Glycoside-Hydrolases.html), all known endo-arabinanases are in GH family 43. In this paper, we reported the identification of the second GH43 endo-arabinanase, AbnS1, produced by P. chrysogenum 31B. The P. chrysogenum Wisconsin strain genome has 14 GH family 43 genes [http://www.cazy.org/e823.html]. To estimate the functions of these 14 genes, we blasted their deduced amino acid sequences against the UniProtKB database. Seven of them (Pc12g01330, Pc12g02040, Pc12g10800, Pc12g12910, Pc13g15920, Pc20g04270, and Pc20g15730) had hits with more than 50% identities to endo-arabinanases and putative endo-arabinanases. Pc12g02040 and Pc13g15920 were endo-arabinases with 99% and 100% identities to Abnc and AbnS1, respectively. It is therefore likely that P. chrysogenum 31B also has the other five endo-arabinase genes. To our knowledge, no other microorganisms have this many endo-arabinanase genes, although it is uncertain whether all of them are functional. Even A. niger, which is known to be a potent producer of various pectinases, has four genes encoding putative endo-arabinanase (Martens-Uzunova and Schaap 2009). We are presently attempting to identify the five remaining putative endo-arabinanases in P. chrysogenum 31B to compare their enzymatic properties, expression profiles, and structures. A BLAST search also revealed that the P. chrysogenum Wisconsin 54–1255 genome has nine putative α-l-arabinofuranosidases and an exo-arabinanase, which suggests that P. chrysogenum Wisconsin 54–1255, and probably P. chrysogenum 31B as well, have high abilities to degrade arabinose-containing polysaccharides in plant cell walls.
In the present study, the abnc gene was strongly expressed in P. chrysogenum 31B grown on arabinose, arabinitol, and l-arabinan (Fig. 3). Arabinitol, an intermediate of l-arabinose catabolism, was also found to induce the expression of endo-arabinanases in A. niger (van der Veen et al. 1993; Flipphi et al. 1994) and A. nidulans (Ramón et al. 1993). These results suggest that arabinitol specifically induces the expression of the abnc gene. The finding that the abns1 gene was only slightly induced by arabinose and l-arabinan (Fig. 3) suggests that the expressions of abns1 and abnc in P. chrysogenum 31B are regulated differently. The abnc gene was much more strongly expressed than abns1 in the presence of l-arabinan, suggesting that Abnc mainly contributes to the cleavage of arabinan backbones in P. chrysogenum 31B. It is unclear why this fungus produces two different endo-arabinanases.
Debranched arabinan was the best substrate for rAbnS1 and rAbnc. However, the final major products from the substrate were different: arabinobiose and arabinotriose for rAbnS1 and arabinotriose for rAbnc. The final products of rAbnS1 and rAbnc are similar to those of A. niger ABNA and A. aculeatus ARA1, respectively (Beldman et al. 1993; Pitson et al. 1997). These results indicate that monomeric arabinose is not produced from debranched arabinan by the action of fungal endo-arabinanases. A bacterial endo-arabinanase from Bacillus subtilis liberates arabinose from α-1,5-l-arabinan (Sakai and Sakamoto 1990). Both endo-arabinanases from Aspergillus produce much greater amounts of arabinooligosaccharides with high DPs from debranched arabinan in the early stage of the reactions than do the two Penicillium endo-arabinanases. The reaction modes by which rAbnS1 and rAbnc degrade the substrate seem to be different from those of the two Aspergillus enzymes.
There has been increasing interest in arabinanolytic enzymes because l-arabinose is the second most abundant pentose in nature and is considered to be a renewable carbon and energy source (Seiboth and Metz 2011). The finding of new endo-arabinanases with different properties should expand our ability to produce arabinose and arabinooligosaccharides from industrial by-products.
References
Al-Tamimi MAHM, Palframan RJ, Cooper JM, Gibson GR, Rastall RA (2006) In vitro fermentation of sugar beet arabinan and arabino-oligosaccharides by the human gut microflora. J Appl Microbiol 100:407–414
Bauer S, Vasu P, Persson S, Mort AJ, Somerville CR (2006) Development and application of a suite of polysaccharide-degrading enzymes for analyzing plant cell walls. Proc Natl Acad Sci USA 103:11417–11422
Becker J, Boles E (2003) A modified Saccharomyces cerevisiae strain that consumes l-arabinose and produces ethanol. Appl Environ Microbiol 69:4144–4150
Beldman G, Searle-van Leeuwen MJF, De Ruiter GA, Siliha HA, Voragen AGJ (1993) Degradation of arabinans by arabinanases from Aspergillus aculeatus and Aspergillus niger. Carbohydr Polym 20:159–168
Bera AK, Sedlak M, Khan A, Ho NWY (2010) Establishment of l-arabinose fermentation in glucose/xylose co-fermenting recombinant Saccharomyces cerevisiae 424A(LNH-ST) by genetic engineering. Appl Microbiol Biotechnol 87:1803–1811
Flipphi MJA, Panneman H, van der Veen P, Visser J, de Graaff LH (1993) Molecular cloning, expression and structure of the endo-1,5-α-l-arabinase gene of Aspergillus niger. Appl Microbiol Biotechnol 40:318–326
Flipphi MJA, Visser J, van der Veen P, de Graaff LH (1994) Arabinase gene expression in Aspergillus niger: indications for coordinated regulation. Microbiology 140:2673–2682
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685
Martens-Uzunova ES, Schaap PJ (2009) Assessment of the pectin degrading enzyme network of Aspergillus niger by functional genomics. Fungal Genet Biol 46:S170–S179
Micard V, Renard CMGC, Thibault JF (1996) Enzymatic saccharification of sugar-beet pulp. Enzyme Microb Technol 19:162–170
Nurizzo D, Turkenburg JP, Charnock SJ, Roberts SM, Dodson EJ, McKie VA, Taylor EJ, Gilbert HJ, Davies GJ (2002) Cellvibrio japonicus α-l-arabinanase 43A has a novel five-blade β-propeller fold. Nat Struct Biol 9:665–668
Pitson SM, Voragen AGJ, Vincken JP, Beldman G (1997) Action patterns and mapping of the substrate-binding regions of endo-(1- > 5)-α-l-arabinanases from Aspergillus niger and Aspergillus aculeatus. Carbohydr Res 303:207–218
Ramón D, van der Veen P, Visser J (1993) Arabinan degrading enzymes from Aspergillus nidulans: induction and purification. FEMS Microbiol Lett 113:15–22
Rombouts FM, Voragen AGJ, Searle-van Leeuwen MF, Geraeds CCJM, Schols HA, Pilnik W (1988) The arabinanases of Aspergillus niger-purification and characterisation of two α-l-arabinofuranosidases and an endo-1,5-α-L-arabinanase. Carbohydr Polym 9:25–47
Sakai T, Sakamoto T (1990) Purification and some properties of a protopectin-solubilizing enzyme that has potent activity on sugar-beet protopectin. Agric Biol Chem 54:879–889
Sakamoto T, Kawasaki H (2003) Purification and properties of two type-B α-l-arabinofuranosidases produced by Penicillium chrysogenum. Biochim Biophys Acta (General subjects) 1621:204–210
Sakamoto T, Thibault JF (2001) An exo-arabinanase of Penicillium chrysogenum able to release arabinobiose from α-1,5-l-arabinan. Appl Environ Microbiol 67:3319–3321
Sakamoto T, Ihara H, Kozaki S, Kawasaki H (2003) A cold-adapted endo-arabinanase from Penicillium chrysogenum. Biochim Biophys Acta (General subjects) 1624:70–75
Sakamoto T, Ihara H, Shibano A, Kasai N, Inui H, Kawasaki H (2004) Molecular characterization of a Penicillium chrysogenum exo-1,5-α-l-arabinanase that is structurally distinct from other arabinan-degrading enzymes. FEBS Lett 560:199–204
Sakamoto T, Ihara H, Shibano A, Nagahiro H, Kawasaki H (2005) Molecular identification of a cold-adapted endo-arabinanase of Penicillium chrysogenum. J Appl Glycosci 52:369–372
Sakamoto T, Tsujitani Y, Fukamachi K, Taniguchi Y, Ihara H (2010) Identification of two GH27 bifunctional proteins with β-L-arabinopyranosidase/α-d-galactopyranosidase activities from Fusarium oxysporum. Appl Microbiol Biotechnol 86:1115–1124
Sakamoto T, Ogura A, Inui M, Tokuda S, Hosokawa S, Ihara H, Kasai N (2011) Identification of a GH62 α-l-arabinofuranosidase specific for arabinoxylan produced by Penicillium chrysogenum. Appl Microbiol Biotechnol 90:137–146
Seiboth B, Metz B (2011) Fungal arabinan and l-arabinose metabolism. Appl Microbiol Biotechnol 89:1665–1673
Seri K, Sanai K, Matsuo N, Kawakubo K, Xue C, Inoue S (1996) l-Arabinose selectively inhibits intestinal sucrase in an uncompetitive manner and suppresses glycemic response after sucrose ingestion in animals. Metabolism 45:1368–1374
Skjøt M, Kauppinen S, Kofod LV, Fuglsang C, Pauly M, Dalbøge H, Andersen LN (2001) Functional cloning of an endo-arabinanase from Aspergillus aculeatus and its heterologous expression in A. oryzae and tobacco. Mol Genet Genomics 265:913–921
Somogyi M (1952) Notes on sugar determination. J Biol Chem 195:19–23
Tagawa K, Kaji A (1988) Preparation of l-arabinan and 1,5-l-arabinan. Methods Enzymol 160:542–545
Unkles SE (1992) Gene organization in industrial filamentous fungi. In: Kinghorn JR, Turner G (eds) Applied molecular genetics of filamentous fungi, 1st edn. Blackie Academic and Professional, London Tokyo, pp 28–53
Van den Berg MA, Albang R, Albermann K, Badger JH, Daran JM, Driessen AJ, Garcia-Estrada C, Fedorova ND, Harris DM, Heijne WH, Joardar V, Kiel JA, Kovalchuk A, Martín JF, Nierman WC, Nijland JG, Pronk JT, Roubos JA, van der Klei IJ, van Peij NN, Veenhuis M, von Döhren H, Wagner C, Wortman J, Bovenberg RA (2008) Genome sequencing and analysis of the filamentous fungus Penicillium chrysogenum. Nat Biotechnol 26:1161–1168
van der Veen P, Flipphi MJA, Voragen AGJ, Visser J (1993) Induction of extracellular arabinases on monomeric substrates in Aspergillus niger. Arch Microbiol 159:66–71
Acknowledgments
The authors thank Dr. H. Ihara, Osaka Prefecture University, for skillful technical assistance. This research was financially supported in part by grants-in-aid for scientific research (22580091) from Japan Society for the Promotion of Science.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Sakamoto, T., Inui, M., Yasui, K. et al. Biochemical characterization and gene expression of two endo-arabinanases from Penicillium chrysogenum 31B. Appl Microbiol Biotechnol 93, 1087–1096 (2012). https://doi.org/10.1007/s00253-011-3452-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-011-3452-7