
Abstract
Recombinant strains of the oleaginous yeast Yarrowia lipolytica expressing the PHA synthase gene (PhaC) from Pseudomonas aeruginosa in the peroxisome were found able to produce polyhydroxyalkanoates (PHA). PHA production yield, but not the monomer composition, was dependent on POX genotype (POX genes encoding acyl-CoA oxidases) (Haddouche et al. FEMS Yeast Res 10:917–927, 2010). In this study of variants of the Y. lipolytica β-oxidation multifunctional enzyme, with deletions or inactivations of the R-3-hydroxyacyl-CoA dehydrogenase domain, we were able to produce hetero-polymers (functional MFE enzyme) or homo-polymers (with no 3-hydroxyacyl-CoA dehydrogenase activity) of PHA consisting principally of 3-hydroxyacid monomers (>80%) of the same length as the external fatty acid used for growth. The redirection of fatty acid flux towards β-oxidation, by deletion of the neutral lipid synthesis pathway (mutant strain Q4 devoid of the acyltransferases encoded by the LRO1, DGA1, DGA2 and ARE1 genes), in combination with variant expressing only the enoyl-CoA hydratase 2 domain, led to a significant increase in PHA levels, to 7.3% of cell dry weight. Finally, the presence of shorter monomers (up to 20% of the monomers) in a mutant strain lacking the peroxisomal 3-hydroxyacyl-CoA dehydrogenase domain provided evidence for the occurrence of partial mitochondrial β-oxidation in Y. lipolytica.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
There is growing concern about the environmental effects of petroleum-derived plastic waste, which takes several decades to degrade. Biodegradable polymers are thus attracting increasing attention as a potential replacement for conventional plastics (Steinbüchel and Lütke-Eversloh 2003). Furthermore, biologically produced polymers are renewable and may have a more positive carbon balance than synthetic polymers (Steinbüchel and Lütke-Eversloh 2003). Polyhydroxyalkanoates (PHAs) are polymers of 3-hydroxyacids naturally produced by many bacteria that can be completely degraded within a year by various microorganisms. PHA constitutes a family of polyesters with thermoplastic and elastomeric properties dependent on their monomer composition (Sudesh et al. 2000; Suriyamongkol et al. 2007; Steinbüchel and Hein 2001).
Many approaches have been used to improve PHA production by natural producers of these molecules, through either traditional metabolic engineering or systems metabolic engineering (Jung et al. 2010; Steinbüchel and Lütke-Eversloh 2003). Efforts have also been made to engineer PHA production in organisms that do not usually produce these molecules, including bacteria (E. coli), yeasts such as S. cerevisiae (Leaf et al. 1996; Poirier et al. 2001; Zhang et al. 2006; Marchesini et al. 2003), Pichia pastoris (Poirier et al. 2002), Yarrowia lipolytica (Haddouche et al. 2010), and Arxula adeninivorans (Terentiev et al. 2004), the filamentous fungus Aspergillus nidulans (Magliano et al. 2010), plants (Poirier and Brumbley 2010), and insect cells (Williams et al. 1996).
PHAs can essentially be assigned to two classes on the basis of their monomer composition: short-chain length (SCL) and medium-chain length (MCL) PHAs. SCL-PHAs are polymers of 3-hydroxyacid monomers with a chain length of three to five carbons, whereas MCL-PHAs contain 3-hydroxyacid monomers with six to 16 carbons (Witholt and Kessler 1999; Steinbüchel and Valentin 1995).
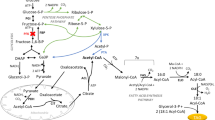
3-Hydroxy-acyl-CoAs are intermediates of the β-oxidation cycle involved in the degradation of fatty acids that can be used as substrates for PHA biosynthesis by PHA synthase (Fig. 1a). In animal cells, the β-oxidation pathway occurs in both mitochondria and peroxisomes. However, short-chain and medium-/long-chain fatty acids are almost exclusively degraded by the mitochondrial β-oxidation pathway, whereas very long-chain fatty acids are predominantly degraded by the peroxisomal β-oxidation pathway (Kunau et al. 1995; Hiltunen and Qin 2000). Unlike animals, yeasts, such as S. cerevisiae, degrade their fatty acids exclusively via the peroxisomal β-oxidation pathway. In this organism, peroxisomal β-oxidation can handle a wide variety of substrates, ranging from very long to short-chain fatty acids (Qin et al. 1999).

Peroxisomal β-oxidation of fatty acyl-CoA and multifunctional enzyme. a Schematic representation of the four reaction steps of peroxisomal β-oxidation catalyzed by an acyl-CoA oxidase, a multifunctional enzyme with a 2-enoyl-CoA hydratase and a 3-hydroxyacyl-CoA dehydrogenase and, finally, a 3-ketoacyl-CoA thiolase. b Schematic representation of the domains of the MFE. The human MFE-2 (H. sapiens) contains three domains, the N terminus domain containing the (3R)-hydroxyacyl-CoA dehydrogenase activity (gray rectangles), the 2-enoyl-CoA hydratase 2 domains (hatched rectangles) and a domain homologous to the sterol carrier protein (SCP domain, open rectangle). The oval represents the binding site for NAD+ and the moon shape, the amino acid motif Typ-X-X-X-Lys. The S. cerevisiae and Y. lipolytica MFE-2 polypeptides contain two 3-hydroxyacyl-CoA dehydrogenase domains and one 2-enoyl-CoA hydratase domain. c Schematic representation of the MFE-2 domains expressed in this study; the full-length MFE-2 (MFE-ABC), the two C-terminal domains (MFE-BC) and the single hydratase domain (MFE-C)
The first step of the peroxisomal β-oxidation pathway is catalyzed by acyl-CoA oxidase (Aox), which converts acyl-CoAs to 2-trans-enoyl-CoAs (Fig. 1a). This enzyme is encoded by a single gene in most yeasts, but Y. lipolytica has six genes (POX1-POX6) encoding isoenzymes with different substrate specificities. The principal Aox isozymes are Aox2p and Aox3p, which are specific for medium- and short-chain length acyl-CoAs, respectively (Wang et al. 1998; Luo et al. 2000, 2002), whereas Aox4p and Aox5p demonstrate only lower levels of nonspecific activity against a broad spectrum of acyl-CoAs (Wang et al. 1998).
The peroxisomal β-oxidation occurring in yeasts also involves the MFE type2 enzyme responsible for catalyzing the second and third reactions of the pathway via a 3(R)-hydroxyacyl-CoA intermediate (Fig. 1a). This enzyme is found in a broad range of organisms, from yeasts to mammals, and in each case carries three functional domains with two or three enzymatic activities. The human MFE-2 contains three domains, with two activities (Fig. 1b): the 3(R)-hydroxyacyl-CoA dehydrogenase domain catalyzes the third reaction of β-oxidation and is located at the NH2-terminus of MFE-2; it is followed by the (2E)-enoyl-CoA hydratase-2 domain, which is responsible for the second reaction. In mammalian MFE-2s, the hydratase-2 domain is followed by a sterol carrier protein type 2 (SCP-2)-like domain. In yeast, the MFE-2 enzyme contains two dehydrogenase domains (domains A and B) and a single hydratase domain (domain C). The S. cerevisiae MFE dehydrogenase spans amino acids 1–280 (domain A) and amino acids 310–600 (domain B), whereas the hydratase domain extends from amino acids 600–900 (domain C) (Qin et al. 1999). In Y. lipolytica, these domains are located from positions 1–288 for domain A, 325–758 for domain B and 758–901 for domain C (Fig. 1b).The Y. lipolytica MFE-2 enzyme is encoded by the MFE1 gene (YALI0E15378g) and contains a conserved variant of the PTS1 peroxisomal-targeting sequence (AKI) at the C terminus (Smith and Aitchison 2009). In the fourth and last reaction of β-oxidation, the 3-ketoacyl-CoA undergoes thiolytic cleavage to yield an acetyl-CoA and an acyl-CoA with a chain shortened by two carbon atoms (Fig. 1a).
Y. lipolytica can grow on various hydrophobic carbon sources, such as alkanes, fatty acids, and triglycerides (Barth and Gaillardin 1996; Fickers et al. 2005). This yeast is also an oleaginous yeast because, in defined conditions, it stores lipids, mostly as triglyceride (TAG), which may account for up to 35% of its dry weight (Ratledge 2005; Beopoulos et al. 2009). The limiting step in TAG synthesis is the final step of the Kennedy pathway, corresponding to the acylation of the diacylglycerol (DAG) by either a DAG acyltransferase or a phospholipid DAG acyltransferase, to produce TAG (Czabany et al. 2007). In S. cerevisiae, lipid accumulation in lipid bodies is completely abolished by the deletion of four genes encoding different acyltransferases (LRO1, DGA1, ARE1, and ARE2). The strain in which all four of these genes are deleted, Q4, is unable to accumulate TAG (Sandager et al. 2002). A similar Q4 strain lacking all four acyltransferases has been constructed in Y. lipolytica (Beopoulos et al., unpublished).
MCL-PHA synthesis has been demonstrated in several hemiascomycetes, including S. cerevisiae, P. pastoris, A. adeninivorans, and Y. lipolytica (Leaf et al. 1996; Poirier et al. 2001, 2002; Marchesini et al. 2003; Terentiev et al. 2004; Haddouche et al. 2010), and in the filamentous fungus A. nidulans (Magliano et al. 2010). In these organisms, PHA has been produced by polymerization of the 3-hydroxyacyl-CoA intermediates of the β-oxidation cycle, through expression of the PhaC synthase from Pseudomonas aeruginosa in the peroxisomes (Fig. 1a). However, these organisms produced only small amounts of PHA, generally corresponding to <1% of cell dry weight.
Efforts have been made to modify both the amount of PHA produced and its monomer composition in recombinant yeast cells. Marchesini et al. modified the monomer composition of MCL-PHAs in S. cerevisiae, by expressing mutated forms of the peroxisomal multifunctional enzyme (MFE-2, encoded by the FOX gene) (Marchesini et al. 2003). Zhang et al. engineered PHA monomer composition and PHA levels in S. cerevisiae, by targeting a PHA synthase to either the cytosol or the peroxisome. They demonstrated the accumulation of PHA to about 7% of cell dry weight (Zhang et al. 2006). In Y. lipolytica, against expectations, the PHA monomer composition of MCL-PHAs was not modified by altering the POX genotype background (this gene encoding acyl-CoA oxidase), whereas PHA levels were significantly affected. However, PHA levels remained low in these strains, corresponding to at most 1% of cell dry weight (Haddouche et al. 2010).
In this report, we assessed the effects on PHA production and monomer composition of modifications to the MFE-2 enzyme and the redirection of fatty acid flux through abolition of the TAG synthase pathway.
Materials and methods
Bacterial and yeast strains and growth conditions
The Y. lipolytica strains used in this study were derived from the wild-type (WT) strain Y. lipolytica W29 (ATCC 20460) (see Table 1). The auxotrophic strain Po1d (Leu−, Ura−) has been described elsewhere (Barth and Gaillardin 1996). The auxotrophic derivative of the strain with the quadruple acyltransferase deletion Q4 (Leu−, Ura−; Beopoulos et al., unpublished data) was obtained by successive gene deletions in Po1d, as previously described (Fickers et al. 2003).
Standard media and growth conditions (Sambrook et al. 1989) were used for Escherichia coli, and those described by Barth and Gaillardin (1996) were used for Y. lipolytica. Rich medium (YPD), minimal glucose medium (YNB) and minimal medium with casamino acids (YNBcas) were prepared as previously described (Mlickova et al. 2004). Minimal medium (YNB) contains a 0.17% w/v yeast nitrogen base (without amino acids and ammonium sulfate, YNBww; Difco, Paris, France), 0.5% w/v NH4Cl, 0.1% w/v yeast extract (Bacto-DB) and 50 mM phosphate buffer (pH 6.8).
General genetic techniques
Standard molecular genetics techniques were used throughout this study (Sambrook et al. 1989). Restriction enzymes were obtained from Eurogentec S.A. (Liege, Belgium). Yeast cells were transformed by the lithium acetate method (Le Dall et al. 1994). Genomic DNA from yeast transformants was prepared as described by Querol et al. (1992). PCR amplification was carried out in an Eppendorf 2720 thermal cycler with either Pyrobest™ or Ex Taq DNA polymerase (TAKARA BIO Inc.). PCR fragments were purified with the QIAgen Purification Kit (Qiagen, Hilden, Germany) and recovered from agarose gels with the QIAquick Gel Extraction Kit (Qiagen). The STADEN package of programs (Dear and Staden 1991) was used for sequence analysis.
Strain construction
Construction of the MFE deletion cassette
We deleted the MFE1 gene by replacing the coding region of this gene by a cassette containing the URA3 gene as a selectable marker, as described by Fickers et al. (2003). We first obtained the promoter (P) and terminator (T) regions of MFE1 by PCR amplification, using Y. lipolytica Po1d genomic DNA as the template and MFE-P1/MFE-P2, MFE-T1/MFE-T2 oligonucleotides (Table 2), respectively, as primer pairs. MFE-P2 and MFE-T1 were designed to introduce an IsceI restriction site at the 3′ end of the P fragment and at 5′ end of the T fragment. The corresponding P-IsceI and T-IsceI fragments were pooled and used as templates for amplification of the P-IsceI-T cassette with the MFE-P1/MFE-T2 primers. This cassette was then inserted into the pGEMT easy cloning vector. The resulting construct, JMP1072, was checked by restriction analysis with IsceI and sequenced. The selectable excisable marker URA3ex was then inserted between the P and T fragments of the P-IsceI-T cassette, at the IsceI site. The resulting loxR–URA3ex–loxP fragment encoding the orotidine 5′ phophate decarboxylase gene was rescued from JME803 by Isce I restriction and inserted into the corresponding site in JME1072. The final construct was named JME1077 (Fig. 2a).

Schematic diagrams of plasmids for deletion and MFE gene expression used in this study. a Schematic diagram of the MFE1 disruption vector. The promoter (P-Fragment) and terminator (T-fragment) regions (about 900 bp) were inserted into the pGMT-easy vector (Promega) as a P-IsceI-T fragment. The URA3ex excisable marker was introduced into the I-SceI site. The MFE1 disruption cassette, the PUT fragment, was amplified by PCR before transformation, as described in “Materials and methods”. b The various sequences encoding the multifunctional enzyme ABC, BC, and C domains (Fig. 2) were inserted into the JMP62 vector containing the URA3ex excisable marker and the promoter of the POX2 gene (pPOX2). The expression cassette was released by Not1 digestion
Deletion of the MFE1 gene, Cre recombinase expression and marker excision
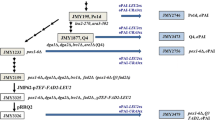
The MFE PUT deletion cassette was generated by PCR amplification, using MFE-P1/MFE-T2 as the primer pairs and plasmid JME1077 as the template. MFE deletion was performed in the JMY195 (Po1d) and JMY1877 (Q4, deficient in acyltransferases) strains. Yeast transformants were selected on YNB-casa. We checked that the MFE gene was correctly disrupted, by PCR, using MFE-Ver1/MFE-Ver2 and URA3 marker rescue was as previously described (Fickers et al. 2003), to generate the JMY1913 (Δmfe) and JMY1915 (Q4 Δmfe) strains (Fig. 3).

Schematic representation of strain construction. a The auxotrophic strain Po1d (JMY195, Leu− Ura−) was derived from WT strain W29. Strain JMY1888, containing a disrupted MFE gene (mfe::URA3), was obtained by transforming JMY195 with the PUTmfe cassette. The URA3 marker was rescued by transformation with the PUB-hygromycin plasmid, as previously described (Fickers et al. 2003). The expression cassette containing the P. aeruginosa PhaC gene encoding PHA synthase was introduced by transformation, with selection for Leu+ transformants. The expression cassette containing the Y. lipolytica MFE1 gene encoding the multifunctional enzyme was introduced by transformation, with selection for Ura+ transformants. The MFE1 cassettes contain the ABC domains (1), the BC domains (2), and the C domain only (3). b The Q4 strain was derived from Po1d by the successive deletion of the four acyltransferase genes (LRO1, DGA1, ARE1, and ARE2; Beopoulos and coworkers, unpublished data). The same transformation steps were used for the construction of strains JMY1955, JMY1957, and JMY1959, with deletions of the MFE gene (Δmfe), the PhaC expression cassette and the MFE expression cassettes MFE-ABC, MFE-BC, and MFE-C, respectively, in the Q4 genotype
Construction of MFE-ABC, MFE-BC, MFE-C, and MFE-A*BC expression vectors and transformation of yeast cells
The 2.7-kb fragment corresponding to the MFE1 ORF (ABC domain) was amplified by PCR with the MFE-Start/MFE-Cstop primers (Table 2) and W29 genomic DNA as the template. These primers contain BamHI and AvrII sites, for the insertion of these fragments into the JMP62-URA3ex vector. This vector contains a strong and inducible promoter, pPOX2. The digested PCR fragment was ligated into a vector digested with BamHI and AvrII. The resulting construct, JMP1076, was checked by sequencing with the 61-Start, 61-Stop, MFE-B interne, and MFE-C interne primers (Fig. 2b).
A Blast analysis comparing the human MFE-2 gene with Y. lipolytica MFE1 was used to define the presence of different domains. The ylMFE1 versions lacking the A or AB domains (encoding dehydrogenase activity) were obtained by PCR with the MFE-B Start/MFE-C Stop and MFE-C Start/MFE-C Stop pair primers, respectively. Strategies used to clone MFE-ABC were adopted for the cloning of both variants (MFE-BC and MFE-C) of the MFE1 gene, generating the plasmids JME1073 and JME1164, respectively.
For construction of the MFE1 gene with a non-functional A domain (MFE-A*BC), the amino acid Gly16 was replaced by a serine residue (Gly16Ser), by mutagenesis. Alignment of the amino-acid sequence of the nucleotide-binding site of the A domain of S. cerevisiae and Y. lipolytica with the Homo sapiens dehydrogenase domain revealed that Gly16 in Y. lipolytica MFE-2 corresponded to an identical residue, Gly16, in Hs MFE-2.
MFE-A*BC was amplified with the primer pair MFE-AG16S/MFE-C stop (Table 2), and the PCR fragment was inserted by the same strategy as for the MFE-ABC fragment into the JMP62-URA3ex vector, generating plasmid JME1248.
The JMP62-LEU2ex-PHA expression plasmids were constructed by inserting the PCR-amplified PhaC coding sequence from pART7-PHA into the JMP62-LEU2ex vectors, as previously described (Haddouche et al. 2010).
The strain with the quadruple acyltransferase deletion (JMY1877) and Po1d expressing the PHA synthase gene PhaC from P. aeruginosa were used as recipients for transformation with the MFE variant plasmids, as shown in Fig. 3.
The various expression cassettes harboring the different MFE1 genes and the PhaC gene under the control of the pPOX2 promoter were released by NotI digestion before the transformation of yeast cells by the lithium acetate transformation technique, generating the strains listed in Table 1, according to the procedure depicted in Fig. 3 (Barth and Gaillardin 1996).
PHA analysis
For PHA production, a stationary-phase culture was harvested by centrifugation; cells were washed once in water and resuspended at a dilution of 1:20 dilution in YNB-casa medium supplemented with 0.1% glucose, 2% Pluronic-127 (Sigma), and between 0.02% and 0.2% v/v fatty acids (Haddouche et al. 2010). Cells were cultured for 4 days before harvesting by centrifugation. PHA analysis was performed essentially as previously described (Poirier et al. 2001). Briefly, cells were harvested by centrifugation, washed twice in water, and freeze-dried. The dried material was then weighed (approximately 15–30 mg) and transferred to a glass tube. The material was extracted four or five times with warm (65°C) methanol to remove lipids, free fatty acids, and acyl-CoA, including 3-hydroxyacyl-CoA, whereas PHA, which is insoluble in methanol, remained associated with the cells. After centrifugation and removal of the residual methanol, the material was suspended in 0.5 ml of chloroform, to which 0.5 ml of 3% sulfuric acid in methanol was added. The mixture was heated at 95°C for 4 h and allowed to cool on ice. We added 1 ml of 0.1% NaCl to each tube, and the mixture was vortexed vigorously and centrifuged at 5,000×g for 5 min. The chloroform phase was harvested and dried over anhydrous MgCl2. The methyl esters of 3-hydroxy acids were identified and quantified by GC-MS with a Hewlett-Packard 5890 gas chromatograph (HP-5MS column) coupled to a Hewlett-Packard 5972 mass spectrometer. GC-MS analysis was carried out in the ion-selective mode (mass/charge ratio of 103).
Statistical tests
The PHA monomer composition of the Q4 strain harboring a native copy of the MFE1 gene (i.e., JMY1956) and the Q4 strains with a mutated dehydrogenase domain (i.e., JMY2025, JMY2026, and JMY2027) was evaluated by a combination of ANOVA and Tukey tests. ANOVA was used to determine whether there was a significant difference in monomer composition. Whenever a difference was observed, the Tukey test was carried out on all possible pairwise comparisons to identify the strains displaying significantly different compositions. Both ANOVA and Tukey tests were performed with R statistical software (Team 2010).
Results
Effect of modified MFE domains on PHA level and monomer composition in Yarrowia lipolytica
We evaluated the effects of various MFE-2 domains on PHA levels and monomer composition in Y. lipolytica by constructing three expression cassettes for expression of the ABC, BC, and C domains (Fig. 1c), as described in “Materials and methods”. For this purpose, the various expression cassettes were introduced into JMP62 vectors under the control of the POX2 promoter, a strong fatty acid-inducible promoter (Juretzek et al. 2000). In parallel, the disruption cassette for deletion of the MFE1 gene was constructed as previously described (Fickers et al. 2003). Strains were constructed as shown in Fig. 3. The JMY1963, JMY1965, and JMY2058 strains contain MFE-ABC, MFE-BC, and MFE-C, respectively.
We previously showed that PHA production levels in Y. lipolytica were highest when the cells were growing on tridecanoic acid (C13) (Haddouche et al. 2010). We therefore assessed the PHA production of the MFE variants on medium containing this fatty acid. No PHA was detected in the Δmfe strain (JMY1923), indicating that the PHA produced in this strain was generated solely by peroxisomal β-oxidation [Fig. 4a and Table 3, results represent the mean values and standard deviations (SD) from four independent experiments]. PHA levels equivalent to 1.3 % of cell dry weight (CDW) were restored in strain JMY1964 expressing the MFE-ABC domain (Table 3), and these production levels were similar to that observed for the wild-type strain JMY1645 expressing PhaC (1.4% CDW). Thus, production levels were similar whether the MFE gene was expressed under the control of either its own promoter or under the control of the POX2 promoter. By contrast, PHA levels were 2.5 and 1.8 times higher in the strains expressing the MFE-BC and MFE-C domains, respectively (JMY1966; 3.3% CDW, JMY2058; 2.3 % CDW).

PHA content of Y. lipolytica strains as a function of MFE1 and acyltransferases genotypes. a PHA content of PO1d derivatives expressing the Pseudomonas aeruginosa PhaC gene and the WT MFE1 (JMY1645), no MFE1 gene (JMY1923), and the MFE-ABC (JMY1664), MFE-BC (JMY1666), and MFE-C (JMY2056) domains, under the control of the POX2 promoter (pPOX2). b PHA content of Q4 derivatives expressing PhaC and MFE genes, as for PO1d; WT MFE1 (JMY1887), no MFE1 (JMY1925), MFE-ABC (JMY1956), MFE-BC (JMY1968), and MFE-C (JMY1960). The results shown are the means and SD from four independent experiments
We have previously demonstrated that Y. lipolytica expressing the P. aeruginosa PHA synthase gene produces PHA preferentially composed of the H9 monomer (3-hydroxy acid monomers are denoted by the prefix H, followed by the number of carbon atoms) (Haddouche et al. 2010). In this study work, the PHA profile of strain JMY1964 (MFE-ABC) also showed a predominance of the H9 monomer (Table 3). A wide distribution of PHA monomers was observed in the profile of this strain, with H9 accounting for 53.2%, H13, H11, and H7 being produced to a lesser extent (6.4%, 13.8%, and 24.1%, respectively) and the H5 monomer being detected in the smallest amounts (2.5%).
Expression of the BC or C domains in the WT background resulted in the production by the strains of homopolymer (≥ 99.8% H13). The absence of dehydrogenase activity may account for the redirection of all β-oxidation intermediates toward PHA, without shortening of the fatty acid used as the carbon source. However, the inability of the BC domain to mediate the complete β-oxidation of fatty acids, as deduced from the composition of the PHA polymer produced, may be accounted for by lack of 3-hydroxyacyl-CoA dehydrogenase activity of the B domain (inactive domain) or by the misconformation of this domain in the MFE-BC protein.
Modification of PHA monomer composition through expression of the MFE-A*BC variant
We investigated whether the Y. lipolytica B domain was inactive in the ABC or BC construct, by mutating the glycine residue in the conserved VVxxTGAGxGxGx10GAxVVVND nucleotide binding site motif of the A domain in MFE-ABC to generate the MFE-A*BC expression cassette, as described in the “Materials and methods”. Similar mutations have been shown to abolish dehydrogenase activity (van Grunsven et al. 1999; Qin et al. 1999).
The MFE-A*BC expression cassette was introduced into the JMY1923 strain (Δmfe, PhaC) by transformation. PHA production was analyzed for three independent transformants, JMY2025, JMY2026, and JMY2027 (Table 2). Mutation in the 3-hydroxyacyl-CoA dehydrogenase A domain of YlMFE1 had little effect on the amount of PHA produced. In JMY2025, JMY2026, and JMY2027, PHA levels reached 2.9%, 4.7%, 3.5% g/g CDW, respectively, values similar to those obtained for JMY1956 (about 3.4% g/g CDW) (Table 4).
Inactivation of the A domain of the MFE1 gene resulted in an enzyme displaying a significant preference for long 3-hydroxyacyl-CoA (Table 5). The H13 and H11 products accounted for 12–14% and 18–19%, respectively, of the monomers incorporated into the PHA polymers produced by strains harboring the MFE-A*BC variant, whereas the strain harboring the native copy of MFE1 (JMY 1956) incorporated about 9% and 15% H13 and H11, respectively. The rate of H7 monomer incorporation was significantly lower in the MFE-A*BC variant (about 19–22%) than in JMY1956 (MFE-ABC), in which the rate of H7 incorporation was about 27%. By contrast, the incorporation of H9 and H5 monomers was similar in both contexts.
PHA synthesis in a strain devoid of triacylglycerides
In Y. lipolytica, external fatty acids may enter the β-oxidation pathway or be diverted into the TAG synthesis pathway. We investigated possible competition between the TAG synthesis and PHA synthesis pathways by introducing various MFE expression cassettes into the Q4 strain, as described in the “Materials and methods” (Fig. 3b).
PHA production levels and monomer composition are summarized in Fig. 4b and Table 3. No PHA was detected in strain Q4 Δmfe (JMY1925), confirming that, as in the Po1d background, PHA was synthesized exclusively by peroxisomal β-oxidation. PHA production in the Q4 strain expressing the MFE-ABC (1.6% CDW) or MFE-BC (1.7% CDW) domain (JMY1956 and JMY1958, respectively) was similar to that observed for the Q4 strain expressing PhaC (JMY1887), which produced about 2.2% CDW. Thus, production levels were not markedly affected by expression of the MFE1 gene under the control of its own promoter or the POX2 promoter, even in the Q4 context. By contrast, PHA levels were 4.6 times higher in the strain expressing the MFE-C domain (JMY1960), which produced 7.3% CDW PHA (Fig. 4b and Table 3). Thus, inactivation of the TAG synthesis pathway can improve PHA synthesis.
PHA content and monomer composition depend on fatty acid chain length and MFE-2 variants
PHA content as a function of the length of the external fatty acid and MFE-ABC and MFE-C domains was analyzed in the Q4 context. We studied C13:0 (0.2%), C11 (0.05%), C9 (0.02%), and C7 (0.02%), and the results are summarized in Table 4 and Fig. 5. For the strain expressing the MFE-ABC cassette, PHA content depended on fatty acid chain length. The highest PHA content was observed for tridecanoic acid (C13) and corresponded to 1.3% of CDW, with the production of a broad range of monomers. H9 accounted for 50%, H7 and H11 for about 20% each, H13 for 10%, and H5 for only a small amount (2%). On undecanoic acid (C11), PHA levels were lower by a factor of 23 (0.055%), and the PHA produced consisted mostly of H9 and H7, which together accounted for almost 90% of the monomers (55% and 35%, respectively), whereas H5 and H11 accounted for the remaining 10%. PHA content was two orders of magnitude lower on nonanoic acid (C9) than on tridecanoic acid (0.009% of CDW), and the PHA produced in these conditions consisted mostly of H9 and H7 monomers. When the recombinant strain was grown on heptanoic acid (C7), PHA content was lower than that on C13, by a factor of 43, with 97% H7 homopolymer and with only trace amounts of H5 and H7.

PHA monomer composition as a function of MFE-2 domain and fatty-acid chain length. Percentage of PHA monomers at 72 h of culture, for Q4 strains expressing MFE-ABC (top panels) and MFE-C (bottom panels) on a medium containing a heptanoic acid, b nonanoic acid, c undecanoic acid, and d undodecanoic acid. The results shown are means and SD from four independent experiments
When the recombinant strains expressing MFE-C were grown in media containing the same fatty acids, PHA content was consistently significantly higher than in the MFE-ABC strains. The strain harboring MFE-C produced 5.6, 8.5, 188, and 11 times more PHA than the MFE-ABC strain, on C13, C11, C9, and C7, respectively. Indeed, on C11 and C9, PHA levels were lower than those on C13 by factors of only 16 and 4, respectively, whereas they were lower by factors of 23 and 145, respectively, for the MFE-ABC strain.
The abolition of dehydrogenase activity resulted in incomplete β-oxidation, generating a large pool of 3-hydroxyacyl-CoAs for polymerization by PHA synthase. This made it possible to produce much more PHA and also resulted in the production of homopolymers. Indeed, for each fatty acid added to the medium, the corresponding hydroxyacid accounted for more than 80% of the PHA.
On C13 and C7, only trace amounts of shorter monomers were observed. On C13, H11, and H9 accounted for <0.3% of the PHA generated, indicating that the corresponding shorter 3-hydroxyacyl-CoAs were produced only very inefficiently by β-oxidation. On C7, H5 again accounted for <2% of the monomers.
Surprisingly, we observed some shortening of the fatty acids when C11 and C9 were used as carbon sources, even in the strain expressing the MFE-C variant, which has no 3-hydroxyacyl-CoA dehydrogenase activity and should therefore produce only homopolymers. For example, the PHA produced by the Q4 MFE-C strain from C11 contained 12.3% H9 and 5.9% H7, whereas PHA produced from C9 contained 11.8% H7. As these shorter odd-chain fatty acids could not have resulted from de novo synthesis, we analyzed the level of purity of the external fatty acid. All fatty acids were found to be 99.99% pure (data no shown), excluding the possibility that shorter chain PHA monomers were derived from contaminating shorter-chain fatty acids. There may still be 3-hydroxyacyl-CoA dehydrogenase activity in the strain expressing the MFE-C variant. Blast analysis of the Y. lipolytica genome revealed the presence of two genes encoding potential mitochondrial proteins with significant sequence similarity to enoyl CoA-hydratase (YALI0B10406g and YALI0F22121g): 3-hydroxyacyl CoA dehydrogenase (YALI0C08811g) and 3-ketoacyl CoA thiolase (YALI0E11099g) (Shen and Burger 2009). Together with a putative mitochondrial acyl CoA dehydrogenase (YALI0D15708g), these genes identified by in silico analysis suggest that there may be a functional β-oxidation pathway in Y. lipolytica mitochondria that further degrades the medium- or short-chain intermediates of acyl-CoA generated in the peroxisome.
An additional source of fatty acids for PHA synthesis could be the elongation of the external fatty acid substrate via the endogenous fatty acid biosynthetic pathway. This process would account for the production of small amounts of H9 monomer in strains expressing MFE-ABC and MFE-C grown on heptanoic acid.
Discussion
We previously reported the production of PHA by the yeast Y. lipolytica following expression of the P. aeruginosa PHA synthase (PhaC) gene in peroxisomes. This yeast possesses six acyl-CoA oxidase (Aox) isoenzymes encoded by genes POX1-POX6. We have shown that the POX genotype significantly affects PHA levels, but not the monomer composition of PHA (Haddouche et al. 2010). We investigated possibilities for further manipulation of PHA production and monomer composition in this yeast by expressing MFE variants in the Δmfe strain expressing the PHA synthase gene. The absence of PHA production in the Δmfe strain demonstrates that a functional β-oxidation pathway is required for PHA production. Similar findings have been reported for the Δmfe strain of S. cerevisiae (Marchesini et al. 2003). PHA production in strains expressing MFE1 gene under the control of its own promoter was found to be similar to that in strains expressing the MFE1 gene under the control of the strong POX2 promoter, indicating that the MFE and POX2 promoters are of similar strength. In these strains, PHA monomer composition depends on the fatty acids added to the medium and reflects the specificity of the P. aeruginosa PHA synthase for R-3-hydroxy-nonanoyl-CoA (H9). Indeed, on media containing nonanoic, undecanoic, and undodecanoic acids, H9 is the most abundant monomer in the PHA produced, accounting for 45% to almost 50% of the monomers incorporated.
The amino-acid sequence of the yeast peroxisomal MFE-2 showed that the polypeptide contained two domains (domains A and B, Fig. 1b) coding for the 3-hydroxyacylCoA dehydrogenase activity and displaying 45% amino-acid sequence identity. The homopolymers produced in strains expressing the variants of the MFE1 gene with deletions of the A domain or both the A and B domains (Δmfe1 MFE-BC and Δmfe1 MFE-C) demonstrated an absence of 3-hydroxyacylCoA dehydrogenase activity. The broad monomer profiles observed in the MFE-A*BC mutant containing a point mutation inactivating the A domain of the dehydrogenase revealed that the B domain was not functional in the MFE-BC construct, but was active in the A*BC construct. This may be a consequence of misfolding of the B domain in the truncated protein.
Y. lipolytica is an oleaginous yeast capable of accumulating lipid as triglycerides (TAG) (Beopoulos et al. 2008). We therefore expected TAG synthesis to compete with PHA production, as previously observed in plants. The deletion of one diacyl glycerol acyltransferase (DGA2) increased the rate of fatty acid flow toward β-oxidation in developing seeds of Arabidopsis, resulting in a tenfold increase in PHA levels (Poirier et al. 1999). We checked for possible competition between TAG and PHA biosynthesis by expressing the wild-type MFE-2 and its variants in the Q4 strain. Sandager and coworkers have identified the four genes (LRO1, DGA1, ARE1, and ARE2) involved in TAG synthesis in S. cerevisiae. LRO1 encodes a phospholipid:diacylglycerol acyltransferase, DGA1 encodes an acyl-CoA:diacylglycerol acyltransferase of the DGAT2 family, and ARE1 and ARE2 each encode sterol:acyltransferases with little acyl-CoA:diacylglycerol acyltransferase activity (Sandager et al. 2002). Y. lipolytica has a single homolog for each of three of these acyltransferases namely LRO1, DGA1, and ARE1. It also has a DGA2 gene encoding an acyl-CoA:diacylglycerol acyltransferase of the DGAT1 family that is absent in S. cerevisiae but commonly found in mammals and plants (Yen et al. 2008). Y. lipolytica strain Q4, which lacks these four genes (LRO1, DGA1, DGA2, and ARE1), cannot accumulate TAG (Beopoulos et al., unpublished data). The expression of the MFE-2 variants and of PhaC in strains deficient in lipid storage (Q4) led to higher levels of PHA accumulation. This was particularly true for the JMY1960 strain (Q4-PhaC Δmfe MFE-C), which accumulated PHA to levels of up to 7.3% of dry weight. This metabolic route therefore competes strongly with the fatty acid degradation pathway, and PHA level may be increased by blocking both β-oxidation and TAG synthesis.
Surprisingly, in strain JMY1960, the Q4 strain expressing the MFE-C variant, we observed shortening of the fatty acid used as the carbon source, particularly for C11 and C9. The third step of peroxisomal β-oxidation is blocked in this strain, and we therefore did not expect to observe the incorporation of PHA monomers shorter than the fatty acids used for growth. Unexpectedly, substantially shorter PHA monomers were observed (Table 4). On medium containing C13, total shorter PHA monomers accounted for only 0.33% of the monomers incorporated (H5, H7, H9, and H11). However, on medium containing C11, these shorter PHA monomers accounted for almost 19% of the monomers incorporated (H5, H7, and H9), with H9 accounting for 12.75% by itself. On medium containing C9, the shorter PHA monomers accounted for 11.82% of the monomers incorporated (C5 and C7). On medium containing C7, H5 accounted for only 2% of the monomers incorporated. These results suggest that there may be an additional dehydrogenase activity complementary to the β-oxidation cycle. Indeed, the Y. lipolytica genome sequence was found to contain an additional NAD + dehydrogenase gene, a potential candidate for vestigial mitochondrial β-oxidation. This goes against the current view that yeasts, including the Ascomycetes, such as Saccharomyces cerevisiae, Y. lipolytica, and Candida tropicalis, have only peroxisomal β-oxidation capacity.
We suggest (Fig. 6), based on the results of this study, that 3-hydroxyfatty acid intermediates may enter the residual mitochondrial β-oxidation pathway, undergoing conversion by a mitochondrial dehydrogenase into the corresponding 3-ketoacyl-CoA, which is then either cleaved by the mitochondrial thiolase or returned to the peroxisome for cleavage by the peroxisomal thiolase. The shortened acetyl-CoA (n-2) may then enter the peroxisomal β-oxidation pathway, generating a shortened 3-hydroxyacyl-CoA, which could then be incorporated into PHA. This bypass pathway was observed principally for C9 and C7, but not for C11 and C5, demonstrating chain-length specificity for this residual mitochondrial β-oxidation pathway. These precursors did not result in PHA production in the Δmfe strain, indicating either the absence of a functional mitochondrial acyl-CoA dehydrogenase or a defect in the transport of these particular precursors into the mitochondria.

Schematic representation of peroxisomal and residual mitochondrial β-oxidation in Y. lipolytica. During growth on a fatty acid with a carbon chain of (n) carbons, in a Δmfe context (cross), no PHA was detected. When the hydratase (C-domain) was expressed, the 3-hydroxy-acyl-CoA (n) intermediate of n carbons could be incorporated into PHA, but could not be transformed into 3-β-ketoacyl-CoA, preventing the formation of (n-2) acyl-CoA. The presence of (n-2) 3-hydroxy-acyl-CoA indicates that the (n) 3-hydroxy-acyl-CoA could enter the mitochondria for conversion into a 3-β-ketoacyl-CoA (n) (1), which was either cleaved by the mitochondrial thiolase (2), or returned to the peroxisome for cleavage by the peroxisomal thiolase to generate Acyl-CoA (n-2), which was subsequently fed into the β-oxidation pathway to generate a 3-hydroxy-acyl-CoA (n-2), resulting in the presence of two and four carbons shorter even-numbered carbon chain fatty acid monomers in PHA
In summary, we have shown that, depending on the MFE-2 variants expressed, it is possible to obtain PHA with different monomer compositions, in the form of heteropolymers (in the presence of a functional MFE gene) or as homopolymers (expression of the MFE-C domain). We also demonstrate that, in Y. lipolytica, the TAG synthesis pathway competes with PHA synthesis and that the abolition of TAG synthesis results in the redirection of fatty acid intermediates towards PHA synthesis. Finally, the presence of shorter monomers in the strain expressing the MFE-C domain suggests that functional residual mitochondrial β-oxidation occurs, as demonstrated by genome exploration (Shen and Burger 2009). Nevertheless, further studies are required to identify the mitochondrial enzymes involved in this (partial) ß-oxidation cycle.
References
Barth G, Gaillardin C (1996) Yarrowia lipolytica. In: Wolf K (ed) Nonconventional yeasts in biotechology: a handbook. Springer, Berlin, pp 313–388
Beopoulos A, Mrozova Z, Thevenieau F, Le Dall M-T, Hapala I, Papanikolaou S, Chardot T, Nicaud J-M (2008) Control of lipid accumulation in the yeast Yarrowia lipolytica. Appl Environ Microbiol 74:7779–7789
Beopoulos A, Cescut J, Haddouche R, Uribelarrea JL, Molina-Jouve C, Nicaud JM (2009) Yarrowia lipolytica as a model for bio-oil production. Prog Lipid Res 48:375–387
Czabany T, Athenstaedt K, Daum G (2007) Synthesis, storage and degradation of neutral lipids in yeast. Biochim Biophys Acta 1771:299–309
Dear S, Staden R (1991) A sequence assembly and editing program for efficient management of large projects. Nucleic Acids Res 19:3907–3911
Fickers P, Le Dall MT, Gaillardin C, Thonart P, Nicaud JM (2003) New disruption cassettes for rapid gene disruption and marker rescue in the yeast Yarrowia lipolytica. J Microbiol Methods 55:727–737
Fickers P, Benetti PH, Wache Y, Marty A, Mauersberger S, Smit MS, Nicaud JM (2005) Hydrophobic substrate utilisation by the yeast Yarrowia lipolytica, and its potential applications. FEMS Yeast Res 5:527–543
Haddouche R, Delessert S, Sabirova J, Neuveglise C, Poirier Y, Nicaud JM (2010) Roles of multiple acyl-CoA oxidases in the routing of carbon flow towards beta-oxidation and polyhydroxyalkanoate biosynthesis in Yarrowia lipolytica. FEMS Yeast Res 10:917–927
Hiltunen JK, Qin Y (2000) Beta-oxidation - strategies for the metabolism of a wide variety of acyl-CoA esters. Biochim Biophys Acta 1484:117–128
Jung Y, Lee S, Tam T (2010) Towards systems metabolic engineering of PHA producers. In: Chen GG-Q (ed) Plastics from bacteria, vol 14, Microbiology monographs. Springer, Berlin, pp 63–84
Juretzek T, Wang H, Nicaud J-M, Mauersberger S, Barth G (2000) Comparison of promoters suitable for regulated overexpression of β-galactosidase in the alkane-utilizing yeast Yarrowia lipolytica. Biotechnol Bioprocess Eng 5:320–326
Kunau WH, Dommes V, Schulz H (1995) Beta-oxidation of fatty acids in mitochondria, peroxisomes, and bacteria: a century of continued progress. Prog Lipid Res 34:267–342
Le Dall MT, Nicaud JM, Gaillardin C (1994) Multiple-copy integration in the yeast Yarrowia lipolytica. Curr Genet 26:38–44
Leaf TA, Peterson MS, Stoup SK, Somers D, Srienc F (1996) Saccharomyces cerevisiae expressing bacterial polyhydroxybutyrate synthase produces poly-3-hydroxybutyrate. Microbiology 142:1169–1180
Luo YS, Wang HJ, Gopalan KV, Srivastava DK, Nicaud JM, Chardot T (2000) Purification and characterization of the recombinant form of acyl CoA oxidase 3 from the yeast Yarrowia lipolytica. Arch Biochem Biophys 384:1–8
Luo Y-S, Nicaud J-M, Van Veldhoven PP, Chardot T (2002) The acyl-CoA oxidases from the yeast Yarrowia lipolytica: characterization of Aox2p. Arch Biochem Biophys 407:32–38
Magliano P, Sanglard D, Poirier Y (2010) Repercussion of a deficiency in mitochondrial ss-oxidation on the carbon flux of short-chain fatty acids to the peroxisomal ss-oxidation cycle in Aspergillus nidulans. Biochim Biophys Acta 1801:1386–1392
Marchesini S, Erard N, Glumoff T, Hiltunen JK, Poirier Y (2003) Modification of the monomer composition of polyhydroxyalkanoate synthesized in Saccharomyces cerevisiae expressing variants of the beta-oxidation-associated multifunctional enzyme. Appl Environ Microbiol 69:6495–6499
Mlickova K, Roux E, Athenstaedt K, d'Andrea S, Daum G, Chardot T, Nicaud JM (2004) Lipid accumulation, lipid body formation, and acyl coenzyme A oxidases of the yeast Yarrowia lipolytica. Appl Environ Microbiol 70:3918–3924
Poirier Y, Brumbley S (2010) Metabolic engineering of plants for the synthesis of polyhydroxyalkanaotes. In: Chen G-Q (ed) Plastics from bacteria, vol 14, Microbiology monographs. Springer, Berlin, pp 187–211
Poirier Y, Ventre G, Caldelari D (1999) Increased flow of fatty acids toward beta-oxidation in developing seeds of Arabidopsis deficient in diacylglycerol acyltransferase activity or synthesizing medium-chain-length fatty acids. Plant Physiol 121:1359–1366
Poirier Y, Erard N, Petetot JM-C (2001) Synthesis of polyhydroxyalkanoate in the peroxisome of Saccharomyces cerevisiae by using intermediates of fatty acid beta-oxidation. Appl Environ Microbiol 67:5254–5260
Poirier Y, Erard N, MacDonald-Comber Petetot J (2002) Synthesis of polyhydroxyalkanoate in the peroxisome of Pichia pastoris. FEMS Microbiol Lett 207:97–102
Qin YM, Marttila MS, Haapalainen AM, Siivari KM, Glumoff T, Hiltunen JK (1999) Yeast peroxisomal multifunctional enzyme: (3R)-hydroxyacyl-CoA dehydrogenase domains A and B are required for optimal growth on oleic acid. J Biol Chem 274:28619–28625
Querol A, Barrio E, Huerta T, Ramon D (1992) Molecular monitoring of wine fermentations conducted by active dry yeast strains. Appl Environ Microbiol 58:2948–2953
Ratledge C (2005) Single cell oils for the 21st century. In: Ratledge C, Cohen Z (eds) Single cell oils. AOCS, Champaign
Sambrook J, Maniatis T, Fritsch EF (1989) Molecular cloning: a laboratory manual, 2nd edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor
Sandager L, Gustavsson MH, Stahl U, Dahlqvist A, Wiberg E, Banas A, Lenman M, Ronne H, Stymne S (2002) Storage lipid synthesis is non-essential in yeast. J Biol Chem 277:6478–6482
Shen Y-Q, Burger G (2009) Plasticity of a key metabolic pathway in fungi. Funct Integr Genomics 9:145–151
Smith JJ, Aitchison JD (2009) Regulation of peroxisome dynamics and number by fatty acid β-oxidation in the yeast Yarrowia lipolytica. Curr Opin Cell Biol 21:119–126
Steinbüchel A, Hein S (2001) Biochemical and molecular basis of microbial synthesis of polyhydroxyalkanoates in microorganisms. In: Babel W, Steinbüchel A (eds) Biopolyesters, vol 71, Advances in Biochemical Engineering/Biotechnology. Springer, Berlin, pp 81–123
Steinbüchel A, Lütke-Eversloh T (2003) Metabolic engineering and pathway construction for biotechnological production of relevant polyhydroxyalkanoates in microorganisms. Biochem Eng J 16:81–96
Steinbüchel A, Valentin HE (1995) Diversity of bacterial polyhydroxyalkanoic acids. FEMS Microbiol Lett 128:219–228
Sudesh K, Abe H, Doi Y (2000) Synthesis, structure and properties of polyhydroxyalkanoates: biological polyesters. Prog Polym Sci 25:1503–1555
Suriyamongkol P, Weselake R, Narine S, Moloney M, Shah S (2007) Biotechnological approaches for the production of polyhydroxyalkanoates in microorganisms and plants—a review. Biotechnol Adv 25:148–175
Team RDC (2010) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna
Terentiev Y, Breuer U, Babel W, Kunze G (2004) Non-conventional yeasts as producers of polyhydroxyalkanoates—genetic engineering of Arxula adeninivorans. Appl Microbiol Biotechnol 64:376–381
van Grunsven EG, van Berkel E, Mooijer PAW, Watkins PA, Moser HW, Suzuki Y, Jiang LL, Hashimoto T, Hoefler G, Adamski J, Wanders RJA (1999) Peroxisomal bifunctional protein deficiency revisited: resolution of its true enzymatic and molecular basis. Am J Hum Genet 64:99–107
Wang H, Le Clainche A, Le Dall MT, Wache Y, Pagot Y, Belin JM, Gaillardin C, Nicaud JM (1998) Cloning and characterization of the peroxisomal acyl CoA oxidase ACO3 gene from the alkane-utilizing yeast Yarrowia lipolytica. Yeast 14:1373–1386
Williams MD, Rahn JA, Sherman DH (1996) Production of a polyhydroxyalkanoate biopolymer in insect cells with a modified eucaryotic fatty acid synthase. Appl Environ Microbiol 62:2540–2546
Witholt B, Kessler B (1999) Perspectives of medium chain length poly(hydroxyalkanoates), a versatile set of bacterial bioplastics. Curr Opin Biotechnol 10:279–285
Yen C-LE, Stone SJ, Koliwad S, Harris C, Farese RV (2008) Thematic review series: glycerolipids. DGAT enzymes and triacylglycerol biosynthesis. J Lipid Res 49:2283–2301
Zhang B, Carlson R, Srienc F (2006) Engineering the monomer composition of polyhydroxyalkanoates synthesized in Saccharomyces cerevisiae. Appl Environ Microbiol 72:536–543
Acknowledgments
We acknowledge the research grants provided by the European Commission in the framework of the LipoYeasts project (contract number 213068). This work was also partly funded by a grant from the “Fond National Suisse de la Recherche Scientifique” (grant number 3100A0-122493) and the University of Lausanne to Y.P. We thank Julie Sappa of Alex Edelman and Associates for her excellent help in correcting the English version of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Haddouche, R., Poirier, Y., Delessert, S. et al. Engineering polyhydroxyalkanoate content and monomer composition in the oleaginous yeast Yarrowia lipolytica by modifying the ß-oxidation multifunctional protein. Appl Microbiol Biotechnol 91, 1327–1340 (2011). https://doi.org/10.1007/s00253-011-3331-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-011-3331-2