
Abstract
A laboratory experiment was conducted to identify key hydrocarbon degraders from a marine oil spill sample (Prestige fuel oil), to ascertain their role in the degradation of different hydrocarbons, and to assess their biodegradation potential for this complex heavy oil. After a 17-month enrichment in weathered fuel, the bacterial community, initially consisting mainly of Methylophaga species, underwent a major selective pressure in favor of obligate hydrocarbonoclastic microorganisms, such as Alcanivorax and Marinobacter spp. and other hydrocarbon-degrading taxa (Thalassospira and Alcaligenes), and showed strong biodegradation potential. This ranged from >99% for all low- and medium-molecular-weight alkanes (C15–C27) and polycyclic aromatic hydrocarbons (C0- to C2- naphthalene, anthracene, phenanthrene, dibenzothiophene, and carbazole), to 75–98% for higher molecular-weight alkanes (C28–C40) and to 55–80% for the C3 derivatives of tricyclic and tetracyclic polycyclic aromatic hydrocarbons (PAHs) (e.g., C3-chrysenes), in 60 days. The numbers of total heterotrophs and of n-alkane-, aliphatic-, and PAH degraders, as well as the structures of these populations, were monitored throughout the biodegradation process. The salinity of the counting medium affects the counts of PAH degraders, while the carbon source (n-hexadecane vs. a mixture of aliphatic hydrocarbons) is a key factor when counting aliphatic degraders. These limitations notwithstanding, some bacterial genera associated with hydrocarbon degradation (mainly belonging to α- and γ-Proteobacteria, including the hydrocarbonoclastic Alcanivorax and Marinobacter) were identified. We conclude that Thalassospira and Roseobacter contribute to the degradation of aliphatic hydrocarbons, whereas Mesorhizobium and Muricauda participate in the degradation of PAHs.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
In November 2002, the oil tanker Prestige sank about 240 km off Cape Finisterre (NW Spanish coast) to a depth of 3,500 m. Before it sank, it released around 60,000 tonnes of a Russian heavy fuel oil (type M-100), containing 22% of aliphatic hydrocarbons, 50% of aromatics, and 28% of resins and asphaltenes. Consequently, the microbial communities in Galician coastal waters were confronted with a highly recalcitrant product. As biodegradation plays a major role in oil depletion, a detailed understanding of the biodegradation processes and the bacterial communities involved is required in order to foresee the environmental damage and to develop effective remediation strategies.
Chronically hydrocarbon-polluted sites, such as the Spanish NW coasts, are likely to harbor autochthonous microbial communities that can biodegrade oil (Harayama et al. 1999). Hence, intrinsic biodegradation was detected after the Prestige oil spill (Medina-Bellver et al. 2005). These communities are necessarily highly heterogeneous as oils are complex mixtures containing hundreds, even thousands, of different compounds and most hydrocarbon-degrading bacteria usually degrade a limited range of hydrocarbons (McKew et al. 2007a).
In previous studies, we characterized microbial communities of oil-polluted sites and compared them to those from non-polluted sites (Alonso-Gutiérrez et al. 2008). In addition, we conducted a successful bioremediation experiment in a polluted beach (Jiménez et al. 2006, 2007), which showed the high biodegradation potential of autochthonous microbiota. However, the evolution of hydrocarbon-degrading populations and the role of specific taxa throughout the degradation of this fuel are yet to be clarified.
This paper evaluates a 60-day biodegradation process carried out by an autochthonous marine bacterial community obtained by enrichment in fuel oil. The inoculum was a sample of oil–water emulsion “chocolate mousse” recovered from the Galician coast a few days after the oil spill. Changes in the total eubacterial community and in the predominant hydrocarbon-degrading [aliphatic-, n-alkane, and polycyclic aromatic hydrocarbon (PAH) degrading] populations were tracked by denaturing gradient gel electrophoresis (DGGE) and by a most-probable number method. In addition, some hydrocarbon degradation-related organisms were identified.
Materials and methods
Inoculum and enrichment procedure
The emulsified fuel oil sample was collected from the Ría de Ortigueira (A Coruña, Spain) in May 2003. The microbial consortium was obtained from this sample by enrichment in autoclave-sterilized 250-mL Erlenmeyer flasks that contained 50 mL of artificial seawater, approximately 50 mg of the sample and inorganic nutrients (5 mM NH4NO3 and 0.35 mM K2HPO3; Díez et al. 2005). Flasks were shaken at 150 rpm and 25°C. The culture (500 μL) was transferred into fresh flasks every third week for 17 months.
Biodegradation assessment
The biodegradation experiment was conducted under the same conditions as the enrichment. In this case, as the 48 flasks contained 100 mg of original (not weathered) fuel oil, the volatile alkane and PAH contents were much higher. Microcosms (33) were inoculated with 500 μL of growing culture, and 15 more were kept as abiotic controls. Samples were taken at 0, 5, 10, 20, and 60 days for chemical analysis (three inoculated and three control each time) and at 0, 5, 10, 20, 40, and 60 days for microbial analysis (three flasks each time).
Chemical analysis
Aliquots of 5–10 mg of dehydrated fuel were dissolved in hexane and fractionated in a previously conditioned cartridge (6 mL hexane, Merck), contaning cyanopropyl-silica solid phase (SiO2/C3–CN, 1.0/0.5 g) (Interchim, Montluçon, France; Alzaga et al. 2004). The aliphatic and aromatic fractions were obtained by eluting with 4.0 mL of hexane (FI) and 5.0 mL of hexane-dichloromethane (1:1) (FII), respectively. Both fractions were analyzed by Gas chromatography–mass spectrometry (Jiménez et al. 2006). The weathering or extent of biodegradation of each compound was measured using 17α(H),21β-(H)-hopane (m/z 191) as an internal conservative molecular marker (Prince et al. 1994). The peak areas of the target analytes were measured in the reconstructed ion chromatograms at m/z 85 for aliphatics and at the corresponding molecular ion ratio for the aromatics.
Counting of total heterotrophic and hydrocarbon-degrading microbial populations
Bacterial counts of total heterotrophs n-hexadecane and PAH degraders were performed by a miniaturized most-probable number (MPN) method in 96-well microtiter plates with eight replicate wells per dilution, as described elsewhere (Alonso-Gutiérrez et al. 2009). The media were corrected to reach 3% NaCl. DNA was extracted from the highest dilute for further DGGE analysis, to provide an image of the predominant hydrocarbon-degrading taxa.
Isolation of culturable strains
Total culturable heterotrophs were isolated on artificial seawater agar (Scharlau Chemie SA, Barcelona), supplemented with tenfold diluted Luria–Bertani, and on tryptic soy agar corrected to reach 3% NaCl (Alonso-Gutiérrez et al. 2009). They were incubated at 20°C. n-Hexadecane degraders were isolated on mineral agar (BMTM agar, 3% NaCl), supplemented with n-hexadecane in the vapor phase (Sei et al. 2003). All isolated strains were stored at −80°C in 20% (v/v) glycerol for subsequent analysis.
Screening of the hydrocarbon-degrading capability of strains
To assess hydrocarbon-degrading capacity on solid media, we used artificial seawater agar supplemented with n-hexadecane and a mixture of PAHs containing phenanthrene, anthracene, fluorene, and dibenzothiophene (Wrenn and Venosa 1996). Microtiter plates with 200 μL of artificial seawater per well were used in liquid screenings. The carbon sources were n-tetradecane, n-hexadecane, n-heptadecane, n-octadecane, pristane and phenanthrene, dibenzothiophene, pyrene or fluorene, and yeast extract (Alonso-Gutiérrez et al. 2008).
DNA extraction
Total community DNA was extracted from the original marine sample, the microcosms with fuel oil and the MPN plates using a PowerSoil DNA extraction kit (MoBio Laboratories, Inc., Solano Beach, CA, USA), following the manufacturer’s instructions. Suitable yields of high-molecular weight DNA were obtained (5–20 μg g−1 fuel paste), and no further purification was required.
16S rRNA gene clone library
Two clone libraries were developed: one from the original sample (before enrichment) and the other from the consortium (after enrichment). Almost complete 16S rRNA genes were PCR-amplified from genomic DNA, using primers F27 and R1492 and Takara Ex Taq DNA polymerase (Takara Bio, Otsu, Shiga, Japan) in 50 μL of reaction medium. Each reaction mixture contained 2.5 U of polymerase, 25 mM TAPS (pH 9.3), 50 mM KCl, 2 mM MgCl2, 200 μM of each deoxynucleoside triphosphate, 0.5 μM of each primer, and 100 ng of template DNA quantified by a Low DNA Mass Ladder (Gibco BRL, Rockville, MD, USA). The mixtures were subjected to an initial denaturation and enzyme activation step (5 min at 95°C), 40 cycles of 30 s at 96°C, 30 s at 50°C and 2 min at 72°C, followed by an extension step of 10 min at 72°C. All PCR reactions were performed with a Mastercycler (Eppendorff, Hamburg, Germany).
PCR products were ligated in pGEM-T Easy Vector and transformed in competent Escherichia coli DH5α cells, following the manufacturer’s protocol. A restriction analysis of clones was performed to identify those that contained a suitable fragment. The PCR products were digested with 5 U of EcoRI (Roche) for 2.5 h at 37°C. Fifty recombinant plasmids were extracted with a Wizard Plus SV Minipreps DNA Purification System (Promega) and then reamplified with primers F27 and R1492.
Denaturing gradient gel electrophoresis
The V3–V5 variable regions of the 16S rRNA gene were amplified using F341-GC and R907 primers (Yu and Morrison 2004). The F341-GC primer included a GC clamp at the 5′ end (5′-CGCCCGCCGCGCCCCGCGCCCGTCCCGCCGCCCCCGCCCG-3′). Each reaction contained the mixture detailed above. After 9 min of initial denaturation and enzyme activation at 94°C, a touchdown thermal profile protocol was executed, and the annealing temperature was decreased by 1°C per cycle from 65°C to 55°C, followed by 20 additional cycles of 1 min denaturation at 94°C, 30 s of primer annealing at 55°C, 1 min of primer extension at 72°C, and a 10-min final primer extension.
Approximately 400 ng of purified PCR product was loaded onto a 6% (w/v) polyacrylamide gel (0.75 mm thick), with denaturing gradients ranging from 40% to 80% (100% denaturant contains 7 M urea and 40% formamide). DGGE was performed in 1× Tris–acetate–EDTA (TAE) buffer (40 mM Tris, 20 mM sodium acetate, 1 mM EDTA, pH 7.4), using a DGGE-2001 System (CBS Scientific, Del Mar, CA, USA) at 100 V and 60°C for 16 h. DGGE gels were stained for 45 min in 1× TAE buffer containing SybrGold (Molecular Probes, Eugene, OR, USA) and scanned by a Bio-Rad molecular imager FX Pro Plus multi-imaging system (Bio-Rad Laboratories, Hercules, CA, USA). DGGE images were processed using Quantity One version 4.5.1 image analysis software (Bio-Rad Laboratories), as reported elsewhere (Jiménez et al. 2007). Predominant DGGE bands were excised with a sterile razor blade, resuspended in 50 μL of sterilized Milli-Q water, stored at 4°C overnight, PCR-reamplified with primers F341 and R907, and cloned (when necessary) in pGEM-T.
Analysis of DGGE images
Bacterial diversity analysis and principal component analysis (PCA) of band types were performed, and the relative peak areas were calculated as previously described (Viñas et al. 2005) for the different DGGE profiles to consider possible shifts in the composition of the microbial populations.
Sequencing and phylogenetic analysis
Sequencing was accomplished with the ABI PRISM™ Big Dye Terminator Cycle Sequencing Ready Reaction Kit (version 3.1) and an ABI PRISM™ 3700 automated sequencer (Applied Biosystems, Foster City, CA, USA), following the manufacturer’s instructions. Sequences were edited and assembled with BioEdit 7.0.9 software (Hall 1999), inspected for the presence of ambiguous base assignments and subjected to the Check Chimera program of the Ribosomal Database Project (Maidak et al. 2000). Subsequently, they were examined and compared with the BLAST alignment tool comparison software (Altschul et al. 1990) and the classifier tool of RDP (Maidak et al. 2000). Sequences were aligned with reference ones obtained from GenBank using ClustalW (Thompson et al. 1994), prior to phylogenetic analysis by Mega4 software (Tamura et al. 2007).
Nucleotide sequence accession numbers
The nucleotide sequences identified in this study were deposited in the GenBank database under accession numbers HM171160 to HM171208 (clones from the original marine sample), HM171209 to HM171258 (clones from the consortium), HM171259 to HM171269 (isolates, M81.SA to M81.SK), and JF264441 to JF264462 (DGGE bands).
Results
Adaptation of the microbial community to the presence of a heavy fuel oil
In the original marine sample, the clones belonged mainly to γ-Proteobacteria genera, particularly Methylophaga (54%) (Fig. 1). Less abundant were the α-Proteobacteria and Bacteroidetes. However, after enrichment, the consortium was composed mainly of Alcanivorax spp. (66%), although Marinobacter and Methylophaga were also present (Fig. 1).

Distribution of the clones in the 16S rDNA libraries corresponding to the original sample (inoculum) and the M81 consortium. Percent id relates to the identity to the closest related sequence in the GenBank database determined via Blast analysis of ∼1,500 bp sequences (E. coli position F27-R1492). α, β, and γ represent alpha-, beta-, and gammaproteobacteria, respectively, and B Bacteroidetes
Biodegrading capabilities
The selected consortium caused a rapid and intense degradation of the resolved compounds and the unresolved complex mixture of both fractions. At day 60, ∼50% of aliphatics and 40% of aromatics were depleted, compared with the abiotic controls. The n-alkanes up to n-C27 were fully degraded (Table 1) and the higher molecular-weight alkanes (C28–C40) significantly depleted (75–98%), following well-established patterns for marine oil biodegradation. In the aromatic fraction, biodegradation mainly affected the lighter PAHs (including heteroatomic compounds, e.g., >99% of naphthalenes, phenanthrenes, dibenzothiophenes, and carbazoles), the extent and rates decreasing with alkylation (Table 1), although highly recalcitrant tetra- and penta-cyclic aromatic hydrocarbons (and even their alkyl-derivatives) were also significantly degraded. The degradation of C3-pyrenes and C3-chrysenes was 75% and ∼60%, respectively.
Growth of the total heterotrophic and hydrocarbon-degrading populations in a biodegradation process
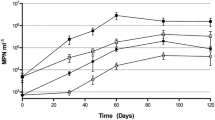
Total heterotrophic populations increased rapidly at the beginning of the incubation from 106 to 108 MPN per milliliter in 10 days and remained stable until the 40th day.
Aliphatic hydrocarbon degraders
Two distinct microbial populations were counted: n-alkane degraders (grown using n-hexadecane as a sole source of carbon and energy) and aliphatic degraders (grown with the aliphatic fraction of a crude oil). The growth of both populations is shown in Fig. 2. At the early stages of incubation, their counts were similar (105 to 108 MPN per milliliter in 10 days). However, at the end of the experiment, whereas the aliphatic-degrading population remained the same size as the total heterotrophs (107), the n-alkane degraders were a logarithmic unit lower.

Growth of the heterotrophic, n-alkane-, aliphatic-, and PAH-degrading populations and relative depletion of the aliphatic and aromatic fractions of oil in the inoculated microcosms with respect to the sterile controls over the 60 days of incubation. Error bars indicate 95% confidence interval and SD, respectively (n = 3)
PAH degraders
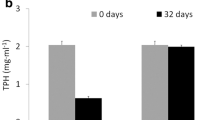
The aromatic-degrading population grew more slowly, and the corresponding counts were significantly lower (104 to 107 MPN per milliliter in about 15 days). To check the suitability of the culture medium used to count this population, the same medium with a 1% NaCl was assayed with several culture samples (Fig. 3).

Effect of salinity in the growth of total heterotrophic, n-alkane- and PAH-degrading bacteria. Error bars correspond to the 95% confidence interval (n = 3)
Changes in the composition of the bacterial community structure throughout the biodegradation process
In the total eubacterial community, 29 different bands were detected (21 sequenced; Fig. 4, Table 2). Profiles of triplicate microcosms were similar at the beginning, with Pearson product-moment correlation coefficients ranging from 0.95 ± 0.02 to 0.978 ± 0.009. At day 60, the variability was much higher (0.84 ± 0.09). α- and γ-Proteobacteria were the predominant taxonomic groups in all the sampling periods (Table 2). Whereas Thalassospira sp., Marinobacter hydrocarbonoclasticus, Roseobacter, Parvibaculum, and Alcaligenes were detected throughout the incubation, Alcanivorax was only present until day 20 and Lutibacterium and Muricauda only at the final stages. Microbial diversity (H′) and richness decreased significantly at the beginning of the incubation (in 5 days; Table 3) and then fluctuated slightly until the end of the experiment.

DGGE profiles of PCR-amplified 16s rRNA genes from microcosms samples showing the changes in the composition of the bacterial community at different sampling periods. Lanes marked with asterisks correspond to the DGGE markers. The codes (B1–B21) indicate predominant bands that were excised and sequenced (∼500 bp; see Table 2 for the description). Bacterial diversity was calculated (Table 3), and a PCA analysis was performed to assess possible shifts in its composition (Fig. 7)
Aliphatic degraders
The DGGE profiles of the main aliphatic hydrocarbon-degrading (FI) and n-alkane-degrading (C16) populations are shown in Fig. 5. In the FI profiles, 15 distinct bands were detected and sequenced, and matched one band or another of the total community profiles (Table 2). Most of them belonged to α-Proteobacteria, with Thalassospira sp. and Parvibaculum sp. dominating. Alcanivorax borkumensis was only detectable at day 10. By this day, there was a biodiversity peak in both indexes (Table 3), after which they gradually returned to the initial levels. The C16 profiles were much less diverse (seven bands). Biodiversity was extremely low at the beginning (only two different bands, Thalassospira sp. and Alcaligenes sp.), but it increased by day 20. Unexpectedly, Alcanivorax spp. was not detected in any of the profiles.

DGGE profiles of PCR-amplified 16s rRNA genes from microcosms samples showing the changes in the composition of the predominant n-alkane-degraders (C16) and aliphatic-degraders (FI) at different sampling periods. Lanes marked with asterisks correspond to the DGGE markers. The codes (A1–A13) indicate predominant bands that were excised and sequenced (∼500 bp; see Table 2 for the description). Bacterial diversity was calculated (Table 3), and a PCA analysis was performed to assess possible shifts in its composition (Fig. 7)
PAH degraders
Twenty-two different bands were detected (15 sequenced) in the PAH degraders profiles (Fig. 6), one of which was new (P16, related to Mesorhizobium sp.). Bands P8 (Thalassospira tepidiphila), P9 (Roseobacter sp.), and P11 (Muricauda aquimarina) were not present in any of the FI or C16 profiles. Richness in microbial diversity increased at the beginning of the experiment. The Shannon–Weaver index decreased, but by the 60th day, it was higher than at the beginning.

DGGE profiles of PCR-amplified 16s rRNA genes from microcosms samples showing the evolution of the structure of the predominant PAH-degraders at different sampling periods. Lanes marked with asterisks correspond to the DGGE markers. The codes (P1–P16) indicate predominant bands that were excised and sequenced (∼500 bp; see Table 2 for the description). Bacterial diversity was calculated (Table 3), and a PCA analysis was performed to assess possible shifts in its composition (Fig. 7)
Principal component analysis
Figure 7 shows the scatter plot for the first two components (PC1 and PC2, 47.5% of variability). The loadings for each band in PC1 and PC2 are shown in Table 2.

Principal component analysis scatter plot for the first two components (PC1, 32.8%; PC2, 14.7% of variability) of DGGE profiles of PCR-amplified 16S rDNA, representing the differences in the structures of the total bacterial community and the n-alkane- (C16), aliphatic- (FI), and PAH-degrading (PAHs) populations throughout the biodegradation process. The samples for total DNA at day 0 were significantly different (P < 0.05) from the rest
Isolation and identification of cultivable organisms
Eleven bacterial strains were isolated in different cultivation media; they were closely related to: Paracoccus yeei, Martelella mediterranea and Mesorhizobium sp., Alcanivorax spp., four strains; Marinobacter, two strains; Muricauda aquamarina, and Staphylococcus pasteuri. Almost all of them (Martelella mediterranea and Mesorhizobium were not tested) grew using n-alkanes as the sole carbon and energy sources, but only the Alcanivorax species grew on the isoprenoid pristane. Five strains, two of Alcanivorax and the ones affiliated to Paracoccus, Muricauda, and Staphylococcus, transformed the PAHs tested.
Discussion
Changes in the community structure within the enrichment process
The prevalence of genus Methylophaga in the original sample could be due to the presence in the ocean of C1 or C2 compounds, like methanol and methylamine (Gibb and Hatton 2004), which are carbon sources for this genus (Neufeld et al. 2007). Nevertheless, this microorganism has been reported in several hydrocarbon-polluted sites (Röling et al. 2002, Schwermer et al. 2008). Thus, its potential role in the degradation of the fuel has to be considered, particularly because this fuel has a high sulfur content (2.28%), and species like Methylophaga sulfidivorans use dimethyl sulfur as carbon source (de Zwart et al. 1996).
According to previous studies (Kasai et al. 2002; McKew et al. 2007b), during the enrichment process, the community evolved toward the predominance of two hydrocarbonoclastic genera (Alcanivorax and Marinobacter), considered to be early colonizers after an oil spill, as primary degraders of the most biodegradable fractions (Röling et al. 2002). They were thus found in the Japan Sea just after the Nakhodka spill (Kasai et al. 2002) and have been related to hydrocarbon biodegradation in other studies (Schleheck et al. 2004; Alonso-Gutiérrez et al. 2008). In addition, Alcanivorax predominates in oil-degrading communities when nutrients are abundant (Yakimov et al. 2007) and was one of the main microorganisms found just after the spill in the Ría de Vigo (Alonso-Gutiérrez et al. 2008), which has large concentrations of dissolved N and P (around 0.6 and 0.06 mg L−1, respectively; Nogueira et al. 1997).
Another interesting change was the detection (which may reflect an increase) of other genera related to hydrocarbon degradation (Thalassospira, Alcaligenes, Citreicella, or Roseobacter).
Biodegrading capabilities of the consortium
The Prestige’s fuel oil was a complex mixture. Thus, by enriching the original sample, we obtained a rich and highly efficient oil-degrading bacterial consortium, able to cause an exceptionally intense biodegradation in the aliphatic and aromatic fractions. Particularly remarkable was the degradation the alkylated PAHs, including C3-chrysenes (a recalcitrant group of compounds). In addition, the consortium degraded the carbazole compounds extensively, at similar rates than phenanthrenes and dibenzothiophenes. These heterocyclic hydrocarbons have carcinogenic and toxic properties (Tsuda et al. 1982), and their water solubility may make them bioavailable. However, their biodegradation has been only poorly assessed. Nevertheless, carbazole and alkylcarbazoles may supply nitrogen for the degrading microbiota (Huang et al. 2003).
Recently, only minor biodegradation was described for the strain AP1 of Mycobacterium sp., a well-known aromatic hydrocarbon degrader, growing on the same fuel in the same incubation time (Vila and Grifoll, 2009). Nevertheless, the potential capacity of a consortium is usually higher than that of a pure strain. In this respect, Gallego et al. (2007) combined bacteria with different metabolic capabilities in order to degrade a heavy oil refinery tank bottom sludge. They degraded 43% of tricyclic PAHs in 10 days, whereas our enrichment consortium degraded 70–75% of C3-phenanthrenes and C3-dibenzothiophenes, ∼60 C3-pyrenes and ∼40% C3-chrysenes in the same time (results not shown).
Enrichment procedures can provide a microbial community with potentially higher efficiency in degrading identified and non-identified components. In addition, degraders of dead-end products accumulated in co-metabolism processes (Casellas et al. 1998) are likely to be selected, too, which can help avoid inhibitory effects that might prevent further PAH biodegradation. In this respect, in the above-mentioned study with Mycobacterium sp., several intermediate metabolites were detected.
Microbial community dynamics
The predominance of α-Proteobacteria and γ-Proteobacteria groups through the biodegradation experiment is consistent with previous results from coasts affected by the Prestige oil spill (Jiménez et al. 2007; Alonso-Gutiérrez et al. 2009). Röling et al. (2002) reported that in artificial media the γ-Proteobacteria subclass is replaced by α-Proteobacteria. Nonetheless, the relative importance of α-Proteobacteria decreased at the end of the incubation period.
The PCA revealed changes in the microbial structure throughout the incubation, which reflect the changes in oil composition: from a labile mixture, rich in n-alkanes and low molecular weight PAHs, to a weathered product, enriched in recalcitrant compounds. In PC1, samples were ordered and clustered according to the incubation time (Fig. 7). The bands with the highest loadings corresponded to the Rhodobacteraceae group and Thalassospira (Table 2). These microorganisms may thus play a predominant role at the beginning of the degradation process. The most negative loadings were those of bands corresponding to Sphingopyxis, Parvibaculum, and Alcaligenes, which were more important in the final stages.
In addition, a microorganism related to Alcanivorax borkumensis was mainly detected in the first 20 days, in which the maximum depletion of the most labile components takes place. The ability to produce biosurfactants (Abraham et al. 1998) might help Alcanivorax being a first colonizer after a spill. However an enhancement of bioavailability (caused by oil emulsification) could promote the growth of other efficient degrading taxa (Gertler et al. 2009). This might explain the detection of these bacteria just after the Prestige oil spill took place (Alonso-Gutiérrez et al. 2009) and its absence in the later surveys (Jiménez et al. 2007).
Worthy of note is the absence of the Gram-positive Rhodococcus, present in various locations affected by the Prestige oil spill (Jiménez et al. 2007; Alonso-Gutiérrez et al. 2009). Nevertheless, it is not found among the prevailing bacteria in recent hydrocarbon-polluted environments (Margesin et al. 2003), and, in vitro, it is outcompeted by fast-growing taxa such as Alcanivorax or Thalassospira.
Microorganisms relevant to biodegradation
The community had a wide variety of hydrocarbon-degrading bacteria able to grow in aliphatic and aromatic hydrocarbons as the sole carbon and energy sources, most of which grew in all of the microtiter plates used for MPN counts: Marinobacter, Alcanivorax, Thalassospira, Lutibacterium, Parvibaculum, Roseobacter, and Alcaligenes (Gauthier et al. 1992; Chung and King 2001; Dutta and Harayama 2001; Schleheck et al. 2004; McKew et al. 2007a; Kodama et al. 2008).
In this respect, whereas salinity does not seem to affect the growth of total heterotrophs and aliphatic degraders (Fig. 3), it strongly affects the counting of PAH degraders, which was unexpected, since the consortium is a marine community. Nevertheless, several studies have reported a repression of PAH degradation with increasing salinity (Kästner et al. 1998, Tam et al. 2002).
In addition, two of the isolated Alcanivorax spp. not only grew in alkanes as the sole carbon and energy sources but also in PAHs. Hence, these strains are highly adapted to the degradation of oil or complex mixtures in which both aliphatic and aromatic hydrocarbons are abundant.
Genera Thalassospira and Roseobacter were detected in all the profiles, although they have been related to n-alkanes degradation (Hara et al. 2003; McKew et al. 2007a; Kodama et al. 2008). In this respect, PCA analysis associated band 20 (Thalassospira sp., with a low loading for PC2) to aliphatic degradation.
In relation to aliphatic hydrocarbon degradation, the FI profiles contained not only all the bands of C16 profiles but also some new bands. Thus, we highly recommend the replacement of n-hexadecane by a complex substrate such as the aliphatic fraction as sole source of carbon and energy for counting the alkane degraders. One of these bands corresponded to Alcanivorax borkumensis. Interestingly, although Alcanivorax species (including all of our isolates) degrade n-hexadecane, no bands corresponding to this genus were found in the C16 profiles. McKew et al. (2007a) suggested that in single-substrate n-alkane microcosms (with n-tetradecane or n-hexadecane), Alcanivorax could be out-competed by other n-alkane-degrading taxa. However, this genus is more efficient in branched alkanes (such as pristane or phytane) degradation (Hara et al. 2003, McKew et al. 2007a). In fact, among all the isolates obtained in the present study, only the ones belonging to Alcanivorax spp. grew in pristane. This could explain its major occurrence in oil-enriched communities.
Mesorhizobium and Muricauda aquimarina were only detected in the PAH’s MPN medium. However, both microorganisms were isolated, and Muricauda not only transformed all the PAHs tested but also grew with all of the short n-alkanes (up to n-C18). The relationship of Muricauda aquimarina to hydrocarbon biodegradation had not been proven before, unlike Mesorhizobium sp., which was recently related to the degradation of phenanthrene and fluorene (Wang et al. 2008). In this respect, according to their loadings in PC2, Mesorhizobium, Lutibacterium anuloederans, and a Sphingobacteriales might be related to PAH degradation.
Our results suggest that α and γ-Proteobacteria are the predominant oil-degrading taxonomic groups. In addition, key microorganisms involved in the degradation of a heavy fuel oil were identified and the hypothesis that Alcanivorax plays an important role in branched alkane degradation was reinforced. The Thalassospira and Roseobacter genera may be important to aliphatic hydrocarbon degradation, whereas Mesorhizobium and Muricauda are related to PAH degradation. In conclusion, we strongly recommend the review of established methods for the counting of aliphatic and aromatic hydrocarbon-degrading bacteria, as the carbon source is a key factor when counting aliphatic hydrocarbon degraders and PAH-degrader counts are significantly affected by salinity.
References
Abraham WR, Meyer H, Yakimov M (1998) Novel glycine containing glucolipids from the alkane using bacterium Alcanivorax borkumensis. Biochim Biophys Acta 1393:57–62
Alonso-Gutiérrez J, Costa MM, Figueras A, Albaigés J, Viñas M, Solanas AM, Novoa B (2008) Alcanivorax strain detected among the cultured bacterial community from sediments affected by the 'Prestige' oil spill. Mar Ecol Prog Ser 362:25–36
Alonso-Gutiérrez J, Figueras A, Albaigés J, Jiménez N, Viñas M, Solanas AM, Novoa B (2009) Bacterial communities from shoreline environments (Costa da Morte, Northwestern Spain) affected by the Prestige oil spill. Appl Environ Microbiol 75(11):3407–3418
Altschul S, Gish W, Miller W, Myers E, Lipman J (1990) Basic local alignment search tool. J Mol Biol 215(3):403–410
Alzaga R, Montuori P, Ortiz L, Bayona JM, Albaigés J (2004) (2003) Fast solid-phase extraction–gas chromatography–mass spectrometry procedure for oil fingerprinting application to the Prestige oil spill. J Chromatogr A 1025:133–138
Casellas M, Grifoll M, Sabaté J, Solanas AM (1998) Isolation and characterization of a 9-fluorenone-degrading bacterial strain and its role in synergistic degradation of fluorene by a consortium. Can J Microbiol 44(8):734–742
Chung W, King G (2001) Isolation, characterization, and polyaromatic hydrocarbon degradation potential of aerobic bacteria from marine macrofaunal burrow sediments and description of Lutibacterium anuloederans gen. nov., sp. nov., and Cycloclasticus spirillensus sp. nov. Appl Environ Microbiol 67(12):5585–5592
de Zwart J, Nelisse P, Kuenen J (1996) Isolation and characterization of Methylophaga sulfidovorans sp. nov.: an obligately methylotrophic, aerobic, dimethylsulfide oxidizing bacterium from a microbial mat. FEMS Microbiol Ecol 20(4):261–270
Díez S, Sabaté J, Viñas M, Bayona JM, Solanas AM, Albaigés J (2005) The Prestige oil spill I. Biodegradation of a heavy fuel oil under simulated conditions. Environ Toxicol Chem 24(9):2203–2217
Dutta T, Harayama S (2001) Biodegradation of n-alkylcycloalkanes and n-alkylbenzenes via new pathways in Alcanivorax sp. strain MBIC 4326. Appl Environ Microbiol 67(4):1970–1974
Gallego JLR, García-Martínez MJ, Llamas JF, Belloch C, Peláez AI, Sánchez J (2007) Biodegradation of oil tank bottom sludge using microbial consortia. Biodegradation 18:269–281
Gauthier MJ, Lafay B, Christen R, Fernandez L, Acquaviva M, Bonin P, Bertrand JC (1992) Marinobacter hydrocarbonoclasticus gen. nov., sp. nov., a new, extremely halotolerant, hydrocarbon-degrading marine bacterium. Int J Syst Evol Microbiol 42:568–576
Gertler C, Gerdts G, Timmis KN, Yakimov MM, Golyshin PN (2009) Populations of heavy fuel oil-degrading marine microbial community in presence of oil sorbent materials. J Appl Microbiol 107(2):590–605
Gibb S, Hatton A (2004) The occurrence and distribution of trimethylamine-N-oxide in Antarctic coastal waters. Mar Chem 91(1–4):65–75
Hall T (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser 41:95–98
Hara A, Syutsubo K, Harayama S (2003) Alcanivorax which prevails in oil-contaminated seawater exhibits broad substrate specificity for alkane degradation. Environ Microbiol 5(9):746–753
Harayama S, Kishira H, Kasai Y, Shutsubo K (1999) Petroleum biodegradation in marine environments. J Molec Microbiol Biotechnol 1(1):63–70
Huang H, Bowler BFJ, Zhang Z, Oldenburg TBP, Larter SR (2003) Influence of biodegradation on carbazole and benzocarbazole distributions in oil columns from the Liaohe basin, NE China. Org Geochem 34:951–969
Jiménez N, Viñas M, Bayona JM, Albaigés J, Solanas AM (2007) The Prestige oil spill: bacterial community dynamics during a field biostimulation assay. Appl Microbiol Biotechnol 77:935–945
Jiménez N, Viñas M, Sabaté J, Díez S, Bayona JM, Solanas AM, Albaigés J (2006) The Prestige oil spill. 2. Enhanced biodegradation of a heavy fuel oil under field conditions by the use of an oleophilic fertilizer. Environ Sci Technol 40:2578–2585
Kasai Y, Kishira H, Sasaki T, Syutsubo K, Watanabe K, Harayama S (2002) Predominant growth of Alcanivorax strains in oil-contaminated and nutrient-supplemented sea water. Environ Microbiol 4(3):141–147
Kästner M, Breuer-Jammali M, Mahro B (1998) Impact of inoculation protocols, salinity, and pH on the degradation of polycyclic aromatic hydrocarbons (PAHs) and survival of PAH-degrading bacteria introduced into soil. Appl Environ Microbiol 64(1):359
Kodama Y, Sutiknowati L, Ueki A, Watanabe K (2008) Thalassospira tepidiphila sp. nov., a polycyclic aromatic hydrocarbon-degrading bacterium isolated from seawater. Int J Syst Evol Microbiol 58:711–715
Maidak BL, Cole JR, Lilburn TG, Parker CT, Saxman PR, Stredwick JM, Garrity GM, Li B, Olsen GJ, Pramanik S, Schmidt TM, Tiedje JM (2000) The RDP (ribosomal database project) continues. Nucleic Acids Res 28(1):173–174
Margesin R, Labbé D, Schinner F, Greer C, Whyte L (2003) Characterization of hydrocarbon-degrading microbial populations in contaminated and pristine alpine soils. Appl Environ Microbiol 69(6):3085–3092
McKew BA, Coulon F, Osborn A, Timmis KN, McGenity TJ (2007a) Determining the identity and roles of oil-metabolizing marine bacteria from the Thames estuary, UK. Environ Microbiol 9(1):165–176
McKew BA, Coulon F, Yakimov MM, Denaro R, Genovese M, Smith CJ, Osborn AM, Timmis KN, McGenity TJ (2007b) Efficacy of intervention strategies for bioremediation of crude oil in marine systems and effects on indigenous hydrocarbonoclastic bacteria. Environ Microbiol 9(6):1562–1571
Medina-Bellver JI, Marin P, Delgado A, Rodríguez-Sanchez A, Reyes E, Ramos JL, Marqués S (2005) Evidence for in situ crude oil biodegradation after tha Prestige oil spill Environ Microbiol 7(6):773–779
Neufeld J, Schäfer H, Cox M, Boden R, McDonald I, Murrell J (2007) Stable-isotope probing implicates Methylophaga spp and novel Gammaproteobacteria in marine methanol and methylamine metabolism. ISME J 1:480–491
Nogueira E, Pérez F, Ríos A (1997) Seasonal patterns and long-term trends in an estuarine upwelling ecosystem (Ría de Vigo, NW Spain). Estuar Coast Shelf Sci 44(3):285–300
Prince RC, Elmendorf DL, Lute JR, Hsu CS, Haith CE, Senius JD, Dechert GJ, Douglas GS, Butler EL (1994) 17α(H)-21β(H)-hopane as a conserved internal marker for estimating the biodegradation of crude oil. Environ Sci Technol 28(1):142–145
Röling WFM, Milner MG, Jones DM, Lee K, Daniel F, Swannell RPJ, Head IM (2002) Robust hydrocarbon degradation and dynamics of bacterial communities uring nutrient-enhanced oil spill bioremediation. Appl Environ Microbiol 68(11):5537–5548
Schleheck D, Tindall B, Rosselló-Mora R, Cook A (2004) Parvibaculum lavamentivorans gen. nov., sp. nov., a novel heterotroph that initiates catabolism of linear alkylbenzenesulfonate. Int J Syst Evol Microbiol 54:1489–1497
Schwermer CU, Lavik G, Abed RMM, Dunsmore B, Ferdelman TG, Stoodley P, Gieseke A, de Beer D (2008) Impact of nitrate on the structure and function of bacterial biofilm communities in pipelines used for injection of seawater into oil fields. Appl Environ Microbiol 74(9):2841–2851
Sei K, Sugimoto Y, Mori K, Maki H, Kohno T (2003) Monitoring of alkane-degrading bacteria in a sea-water microcosm during crude oil degradation by polymerase chain reaction based on alkane-catabolic genes. Environ Microbiol 5(6):517–522
Tam N, Guo C, Yau W, Wong Y (2002) Preliminary study on biodegradation of phenanthrene by bacteria isolated from mangrove sediments in Hong Kong. Mar Pollut Bull 45(1–12):316–324
Tamura K, Dudley J, Nei M, Kumar S (2007) MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol Biol Evol 24(8):1596–1599
Thompson J, Higgins D, Gibson T (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix. Nucleic Acids Res 22:4673–4680
Tsuda H, Hagiwara A, Shibata M, Ito N (1982) Carcinogenic effect of carbazole in the liver of a (C57BL/6NxC3H/HeN)F1 mice. J Natl Cancer Inst 69:1389–1393
Vila J, Grifoll M (2009) Actions of Mycobacterium sp. strain AP1 on the saturated-and aromatic-hydrocarbon fractions of fuel oil in a marine medium. Appl Environ Microbiol 75(19):6232–6239
Viñas M, Sabaté J, Espuny M, Solanas AM (2005) Bacterial community dynamics and polycyclic aromatic hydrocarbon degradation during bioremediation of heavily creosote-contaminated soil. Appl Environ Microbiol 71(11):7008–7018
Wang B, Lai Q, Cui Z, Tan T, Shao Z (2008) A pyrene-degrading consortium from deep-sea sediment of the west pacific and its key member Cycloclasticus sp. P1. Environ Microbiol 10(8):1948–1963
Wrenn B, Venosa A (1996) Selective enumeration of aromatic and aliphatic hydrocarbon degrading bacteria by a most-probable-number procedure. Can J Microbiol 42:252–258
Yakimov MM, Timmis KN, Golyshin P (2007) Obligate oil-degrading marine bacteria. Curr Opin Biotechnol 18(3):257–266
Yu Z, Morrison M (2004) Comparisons of different hypervariable regions of rrs genes for use in fingerprinting of microbial communities by PCR-denaturing gradient gel electrophoresis. Appl Environ Microbiol 70(8):4800–4806
Acknowledgment
This research was supported by the Spanish Ministry of Education and Science (VEM2003-20068-C05 and CTM2007-61097/TECHNO). N.J. is grateful for a PhD fellowship from the Spanish Ministry of Education and Science.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Jiménez, N., Viñas, M., Guiu-Aragonés, C. et al. Polyphasic approach for assessing changes in an autochthonous marine bacterial community in the presence of Prestige fuel oil and its biodegradation potential. Appl Microbiol Biotechnol 91, 823–834 (2011). https://doi.org/10.1007/s00253-011-3321-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-011-3321-4