
Abstract
Clostridium thermocellum, an anaerobic, thermophilic, and ethanogenic bacterium produces a large cellulase complex termed the cellulosome and many free glycosyl hydrolases. Most cellulase genes scatter around the genome. We mapped the transcripts of the six-gene cluster celC–glyR3–licA–orf4–manB–celT and determined their transcription initiation sites by primer extension. Northern blot showed that celC–glyR3–licA were co-transcribed into a polycistronic messenger with the transcription initiation site at −20 bp. Furthermore, RT-PCR mapping showed that manB and celT, two cellulosomal genes immediately downstream, were co-transcribed into a bicistronic messenger with the initiation site at −233 bp. In contrast, rf4 was transcribed alone with the two initiation sites at −130 and −138 bp, respectively. Finally, quantitative RT-PCR analysis showed that celC, glyR3, and licA were coordinately induced by growing on laminarin, a β-1,3 glucan. Gene expression peaked at the late exponential phase. Taking together with our previous report that GlyR3 binds to the celC promoter in the absence of laminaribiose, a β-1,3 glucose dimer, these results indicate that celC, glyR3, and licA form an operon repressible by GlyR3 and inducible by laminaribiose, signaling the availability of β-1,3 glucan. The celC operon is the first glycosyl hydrolase operon reported in this bacterium.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Clostridium thermocellum is an anaerobic, thermophilic, cellulolytic, and ethanogenic bacterium. It produces a cellulase system highly active on crystalline cellulose (Johnson et al. 1982). The extracellular cellulase components form an ordered protein complex termed the cellulosome (Lamed et al. 1983). In addition, many free glycosyl hydrolases are produced. The core of the cellulosome is a 250-kDa non-catalytic, scaffold protein, CipA (Gerngross et al. 1993; Kruus et al. 1995; Lamed et al. 1983; Romaniec et al. 1991). CipA contains domains for binding to cellulose, to the surface of the cell and to the catalytic components, respectively (Leibovitz and Beguin 1996; Leibovitz et al. 1997). Searching the genome sequence of C. thermocellum revealed more than 70 genes encoding dockerin-containing proteins, presumed to be the cellulosome components (Demain et al. 2005; Zverlov et al. 2005). Thus, including the genes encoding the cellulosome components, the scaffold and anchorage proteins, and the free enzymes but without counting the regulatory and sugar-transport genes, there are likely more than 100 genes involving in biomass degradation in this bacterium. How the organism regulates the expression of such a large number of genes and proteins involved in biomass degradation is an intriguing, yet largely unresolved, question. Previous studies have shown that the cellulase expression can be dependent on growth rate (Dror et al. 2003), yet no mechanisms have been reported. In addition, only one cellulase transcription factor has been identified in this bacterium (Newcomb et al. 2007). Recently, a set of putative sigma and anti-sigma factors that include extracellular polysaccharide sensing domains have been proposed to regulate cellulosomal genes via an extracellular carbohydrate-sensing mechanism (Bahari et al. 2010; Kahel-Raifer et al. 2010; Nataf et al. 2010). In Clostridium celluylolyticum, it has been shown that different protein levels can be a result of different mRNA half-lives (Maamar et al. 2006).
Co-transcription of genes is a method of coordinated gene regulation in prokaryotes. In mesophilic Clostridia, clusters of cellulase genes are prevalent (Schwarz 2001). In Clostridium cellulovorans, a nine-gene cluster encoding cellulase components has been found (Tamaru and Doi 2000; Tamaru et al. 2000). Northern blot analysis revealed two polycistronic transcripts corresponding to the first two and four genes, respectively (Han et al. 2003a). Cellulase gene clusters have also been reported in other Clostridia including C. cellulolyticum (Bagnara-Tardif et al. 1992), Clostridium acetobutylicum (Nolling et al. 2001; Sabathe et al. 2002), and Clostridium josui (Kakiuchi et al. 1998).
In contrast, most cellulase and hemicellulase genes of C. thermocellum are scattered around the genome (Guglielmi and Beguin 1998). Nonetheless, several clusters have been found. A gene for a non-cellulosomal endoglucanase, CelI, is clustered with the cellulosomal cellulase CelN gene and a possible cellulosomal structural component CseP (cellulosomal element protein) gene (Zverlov et al. 2003). The second gene cluster consists of celA, chiA, and orfZ (Zverlov et al. 2002). Recently, we reported a previously unidentified gene cluster consisting of five genes, all of which encode uncharacterized dockerin-containing proteins (Demain et al. 2005). The fourth cluster (Fuchs et al. 2003) encodes the non-cellulosomal endoglucanase CelC (Petre et al. 1986; Schwarz et al. 1988), a Lac-I like protein GlyR3 (Newcomb et al. 2007), and LicA (Fuchs et al. 2003).
Although, two pairs of co-transcribed genes exist in the cipA gene cluster cipA–olpB–orf2p–olpA, i.e., cipA–olpB and orfp2–olpA (Fujino et al. 1993), no co-transcription of glycosyl hydrolase genes in C. thermocellum has been reported. In this work, we mapped transcripts of the celC gene cluster celC–glyR3–licA–orf4–manB–celT using Northern blot and determined the celC transcription initiation site by primer extension. In addition, we quantified the expression of all six genes in the cluster using quantitative reverse transcriptase (RT)-PCR on three different carbon sources. The results showed that celC–glyR3–licA are co-transcribed with the transcription initiation site 8 bp upstream from the putative ribosome binding site. Transcript mapping further revealed that manB (Kurokawa et al. 2001) and celT (Kurokawa et al. 2002), two cellulosomal genes immediately downstream from the celC–glyR3–licA cluster, are co-transcribed into a bicistronic messenger. Primer extension showed that rf4 has two transcription initiation sites, 130 and 138 bp upstream of the start codon, respectively, while manB–celT has one initiation site, 233 bp upstream of its start codon. Finally, the results of quantitative RT-PCR analysis indicate that when the cells are grown on laminarin, celC, glyR3, and licA show an expression peak at the late exponential phase of growth that is at least 2.5-fold greater than when the cells are grown on cellobiose or cellulose.
Materials and methods
Bacterial strain
Clostridium thermocellum ATCC 27405 was used throughout the study and as the source of all genomic DNA and RNA.
Culture conditions
Hungate tubes were used to culture C. thermocellum in chemically defined MJ medium (Johnson et al. 1981). The medium contained 0.5% of carbon source [cellobiose (Sigma, St. Louis, MO), cotton, or laminarin (Sigma)]. Seed cultures were grown on cellobiose for 12 h. All cultures were incubated at 60°C.
Measuring cellular protein
Due to the solid content of some media, cell growth kinetics was monitored by measuring cellular protein. The cells were harvested by centrifugation at 12,000×g for 15 min. Cotton cultures were filtered through layers of cheesecloth, the remaining cotton fibers in the culture washed with 0.85% NaCl solution to remove the remaining adherent cells before the cell suspension were centrifuged. The cell pellet was washed twice with 0.85% NaCl solution. Before the last centrifugation step, 1 ml of the resuspended pellet was taken for cellular protein analysis, the rest of the resuspended pellet was spun down for RNA isolation. The removed 1 ml aliquot was centrifuged and the final pellet was resuspended in 1 ml of 0.2 N NaOH and heated in boiling water for 15 min to extract cellular protein. After boiling, the sample was centrifuged again to remove the debris. Finally, 1 ml of 0.2 N HCl was added to neutralize the supernatant. Protein in the supernatant was measured using the Bradford (Bradford 1976) reagent (Bio-Rad, Hercules, CA) and bovine serum albumin (Sigma) as a standard.
RNA isolation
Clostridium thermocellum cell pellets were obtained as outlined above. Total RNA was isolated by using the Trizol (Invitrogen, Carlsbad, CA) method. The RNA sample was digested with DNase I (ABgene, Cambridge, UK) and quantified using a spectrophotometer at 260/280 nm.
Reverse transcription
Reverse transcription was carried out using a standard protocol and MMLV Reverse Transcriptase (Invitrogen).
Preparation of Northern blot probes
Northern blot probes were made using PCR, the gene-specific primers listed in Table 1, and Thermostart Taq Polymerase (ABgene). The annealing temperature was 58°C and an extension time of 20 s were used for each probe. The PCR product was purified using the Wizard SV Gel and PCR Clean-Up System (Promega, Madison, WI) prior to use.
Northern blot
Clostridium thermocellum was grown on cotton for 60 h or laminarin to the late exponential phase. Total RNA was isolated as described above and Northern blot carried out using the NorthernMax-Gly kit (Ambion, Foster City, CA) following the manufacturer’s protocol. Briefly, 20 μg of RNA or RNA molecular weight markers (Novagen, Gibbstown, NJ) were loaded to a 0.8% agarose gel. After the electrophoresis, RNA in the gel was transferred to a Biodyne B (Pall, Port Washington, NY) positively charged membrane using downward capillary blot. The RNA markers were stained using 0.2% methylene blue to verify the success of the blotting. The blot was mixed with ULTRAhyb buffer from the NorthernMax kit and allowed to prehybridize for 45 min at 42°C. Probes were prepared by using a PCR reaction with primers from Table 1 as described above. Deoxynucleotide triphosphates were used in the reaction that had the deoxyadenosine triphosphate labeled with biotin in a 5.25:1 ratio (New England Biolabs, Ipswitch, MA). Probes were heated at 95°C for 10 min then added to the prehybridization solution to a final concentration of 2 ng/ml. The hybridization was carried out for 24 h. The membrane was then washed with low stringency buffer for 10 min at room temperature and washed twice with high stringency buffer at 42°C for 15 min each. The blot was developed following the manufacturer’s protocol for the Chemi-luminescent Nucleic Acid Detection Module (Pierce, Rockford, IL).
Reverse transcription mapping
To determine if orf4, manB, and celT of the gene cluster are co-transcribed, we mapped the mRNA species transcribed from the celC gene cluster using RT-PCR as previously described (Han et al. 2003a). In this analysis, the PCR primers each represents a sequence from one of the two adjacent genes. Successful amplification would indicate the co-transcription of the two genes.
Primer extension
The gene-specific primer (Table 1) was end-labeled with fluoroscein (Invitrogen). A standard reverse transcription reaction was carried out using MMLV Reverse Transcriptase (Invitrogen). The size of the resulting cDNA was determined by capillary electrophoresis using a Model 3100 Genetic Analyzer (Applied Biosystems, Carlsbad, CA).
Quantitative RT-PCR
Quantitative gene expression analysis was carried out using the iQ Syber Green Supermix (Bio-Rad) reagent on a iCycler (Bio-Rad) with gene-specific primers. All primer sets were tested to make sure data fall within the linear range of quantification, and that amplification efficiency was greater than 90%. The two-step PCR protocol consisted of activation of the polymerase at 95°C for 5 min, followed by 40 cycles of 95°C for 15 s and 60°C for 30 s. All sample/primer combinations were analyzed in triplicate. The 16S ribosomal RNA was used to normalize the experiment for sample-to-sample variation; expression data for genes of interest was reported relative to 16S expression.
Results
The celC gene cluster consists of six genes
We searched the genome sequence of C. thermocellum for potential genes encoding glycosyl hydrolases and found that the celC cluster is larger than reported (Fuchs et al. 2003). The cluster celC–glyR3–licA was adjacent to orf4–manB–celT (Fig. 1). The six-member gene cluster encodes both the cellulosomal (manB and celT) and non-cellulosomal (celC and licA) components of the cellulase system as well as glyR3, a gene homologous to lacI, and orf4 encoding a putative transmembrane protein.

Schematic drawing of the celC–celT gene cluster of C. thermocellum. The cluster consists of six genes, including four encoding previously identified glycosyl hydrolases, celC (Cthe_2807), licA (Cthe_2809), manB (Cthe_2811), and celT (Cthe_2812). The function of Orf4 (Cthe_2810) is unknown. GlyR3 (Cthe_2808) is a regulatory protein that binds to the celC promoter region (Newcomb et al. 2007)
Northern blot analysis of the celC–licA gene cluster
To determine if the members of the celC gene cluster are co-transcribed, we mapped the mRNA species transcribed from the celC gene cluster using Northern blot analysis. Probes for celC, glyR3, and licA all hybridized to a RNA species with a size just over 6 kb (Fig. 2). The expected size of celC, glyR3, and licA, if they were all co-transcribed, would be 6,261 bp. Thus, the result indicates that celC, glyR3, and licA are all transcribed as one transcription unit.

Northern blot analysis of celC, glyR3, and licA. All of the three gene probes hybridize to the same transcript of ∼6.2 kbp, consistent with the size of 6,261 bp as expected if celC, glyR3, and licA are co-transcribed. Cells were grown on laminarin to the late exponential phase
RT-PCR mapping of the licA–celT gene cluster
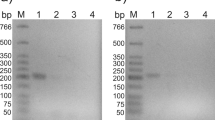
Northern blot was attempted using probes for orf4, manB, and celT, but was unsuccessful due to low expression levels of the transcript(s) (data not shown). As an alternative, we mapped the mRNA species transcribed from the celC gene cluster using RT-PCR. In this analysis, the PCR primers, each representing a sequence from one of the two adjacent genes, were used as previously described (Han et al. 2003a). In principle, to determine if two adjacent genes are co-transcribed, the primers were designed to amplify the 3′ end of the upstream gene, the intergenic sequence, and 5′ end of the downstream gene. If a pair of genes is co-transcribed, the PCR product with the expected size would be obtained (Fig. 3a).

Co-transcriptions of manB–celT as evidenced by RT-PCR mapping. a A schematic drawing of the licA–celT gene cluster, indicating the results of RT-PCR using primers (Table 1), each corresponding to a sequence of one of the two adjacent genes. A horizontal bar (—) between two adjacent genes indicates that the respective RT-PCR product of the expected size was observed; a big letter X (X) indicates the respective RT-PCR product was not obtained after repeated attempts. b Agarose gel electrophoresis of the PCR-amplified cDNA. Lane 1, molecular weight marker; lanes 2, 5, and 8, positive controls of PCR using genomic DNA as the template; lanes 4, 7, and 10, negative controls omitting the reverse transcriptase in the RT reaction; lanes 3 and 6, the negative results, indicating the lack of co-transcription of licA–orf4 and orf4–manB, respectively; lane 9, the 450 bp RT-PCR product indicating co-transcription of manB–celT. Cells were grown on cotton for 60 h
Positive controls using C. thermocellum genomic DNA as the template were included to verify that the primers would correctly amplify the DNA segment (Fig. 3b; lanes 2, 5, and 8). Negative controls using the total RNA as the template but omitting RT in the RT reaction were also included to detect any genomic DNA background or contamination of the RNA sample (Fig. 3b; lanes 4, 7, and 10). orf4 appears to be transcribed alone, as it was not possible to amplify the upstream bridging fragment (290 bp; Fig. 3b, lane 3) and downstream bridging fragment (445 bp; Fig. 3b, lane 6) above background levels (lanes 4 and 7, respectively) despite repeated attempts. For positive controls, PCR products of correct sizes were obtained (lanes 2 and 5, respectively). Finally, it was possible to amplify the bridging region between manB and celT (450 bp; Fig. 3b, lane 9), indicating that these two genes are co-transcribed. Therefore, under the growth conditions, the celC gene cluster, with its six members, is transcribed into a celC–glyR3–licA polycistronic mRNA, an orf4 monocistronic mRNA, and a manB–celT bicistronic mRNA.
Primer extension analysis of the celC, orf4, and manB transcripts
We used a non-radioisotope technique for primer extension analysis to determine the transcription initiation sites for the three transcription units of the celC–glyR3–licA–orf4–manB–celT gene cluster. In this method, total RNA was isolated at the exponential growth phase as mentioned above. A reverse primer corresponding to the nucleotide positions 14–33 of celC was end-labeled with fluoroscein (Table 1) and used for primer extension. The resulting cDNA was analyzed by capillary electrophoresis to determine its size as described in “Materials and methods.” As shown in Fig. 4a, a 53 bp extended cDNA fragment was obtained, corresponding to the transcription initiation site located at the −20 nucleotide position of celC (Fig. 4b). The transcription initiation site is 8 bp upstream of the putative Shine–Dalgarno sequence.

Transcription initiation site as revealed by primer extension using a primer corresponding to the celC sequence. a Capillary electrophoresis of the extended cDNA, showing a 53 bp fragment, matching up to the transcription initiation site; b DNA sequence of the celC promoter region. The transcription initiation site matching the 53 bp extended cDNA fragment is indicated with an arrow and the putative Shine–Dalgarno sequence is underlined. The palindromic GlyR3 binding site (Newcomb et al. 2007) is bolded. The −10 and −35 sigma factor binding sites predicted by BProm (http://linux1.softberry.com/berry.phtml, accessed 3 January 2011) are noted by a solid and a dashed box, respectively
The technique was applied to identify the transcription initiation sites for orf4 and manB (Fig. 5). orf4 was shown to have two starts sites that were 130 and 138 bp upstream from the start codon, respectively (Fig. 5a). The single transcription initiation start site for manB was found to be 233 bp upstream of the start codon (Fig. 5b).

Transcription initiation site as revealed by primer extension using primers corresponding to the orf4 and manB sequences, respectively. Arrows indicate the transcription initiation sites located in the promoter regions of the respective genes. a Capillary electrophoresis of the extended cDNA, showing a 170 and 178 bp fragments, matching up to the transcription initiation sites (−130 and −138 bp, respectively) for orf4; b the extended 278 bp cDNA fragment, matching up to the transcription initiation site (−233 bp) for manB
The results of the primer extension analysis are consistent with the observation that the six genes are organized into three separate transcription units.
Expression profiles of the gene cluster on different carbon sources
To determine if celC, glyR3, and licA are indeed co-regulated, we compared the expression profiles of these three genes on three different carbon sources, cellobiose, cotton, and laminarin, a β-1,3-linked glucan. The growth kinetics was monitored for each culture condition based on the cellular protein level (Fig. 6). At different growth stage, the RNA was harvested from the cells and subjected to quantitative RT-PCR analysis to determine the gene expression kinetics using gene-specific primer sets. A comparison was made between the expression of all three genes at each growth phase on all three carbon sources (Fig. 7).

Growth kinetics of C. thermocellum on cellobiose, cellulose, or laminarin as the sole carbon source as measured by cellular protein. Errors bars represent the standard deviations of triplicate runs

Expression profiles of celC, glyR3, and licA of C. thermocellum on cellobiose, cellulose, or laminarin as the sole carbon source as determined by quantitative RT-PCR. Transcript levels are shown relative to 16S expression and error bars represent standard deviations of triplicate runs
The most striking result is that, at late exponential growth, celC, glyR3, and licA all had a spike in expression when the cultures were grown on laminarin (Fig. 7). The spike was not seen when the cells were grown on cellobiose or cellulose, indicating that these three genes are induced by growing on laminarin. Once the cells enter early stationary phase, the expression levels of all three genes sharply diminished, indicating that the genes were highly repressed at this growth phase. In addition, the expression profiles of these three genes were parallel to one another, as expected if they are transcribed into a polycistronic messenger. As will be discussed later, these results corroborate well with the mechanism we have proposed for GlyR3 regulation in the celC gene cluster (Newcomb et al. 2007).
In contrast, the expression of orf4, manB, and celT did not show a spike when grown on any of the substrates (Fig. 8). It is possible that different carbon sources are involved in inducing orf4, manB, and celT. Nonetheless, the expression patterns of manB and celT on the three carbon sources are similar, indicating co-regulation of the two genes and consistent with the observation that the two genes are co-transcribed into a bicistronic messenger.

Expression profiles of orf4, manB, and celT of C. thermocellum on cellobiose, cellulose, or laminarin as the sole carbon source as determined by quantitative RT-PCR. Transcript levels are shown relative to 16S expression and error bars represent standard deviations of triplicate runs
Discussion
Clostridium thermocellum produces a highly active cellulase system that has over 100 different components. At this time, only one transcription factor has been reported for the regulation of the cellulase system components (Newcomb et al. 2007). GlyR3 has been shown to bind to the celC promoter region, inhibiting transcription of celC until the inducer, laminaribiose, a β-1,3 glucose dimer, is present. One important goal of this study was to determine whether the six genes found in this cluster were co-regulated by GlyR3 and laminaribiose.
Co-transcription of celC–glyR3–licA and of manA–celT, respectively, as demonstrated in this work, represents the first two cases of co-transcription of glycosyl hydrolase genes in C. thermocellum. Co-transcription of genes is a method of coordinated gene regulation in prokaryotes. In this regard, we have previously found that GlyR3 serves as a negative regulator that binds to a palindromic sequence in the celC promoter region (Newcomb et al. 2007). The binding is inhibited by laminaribiose, leading to the induction of the celC gene. The results of this work further indicate that the induction is for celC–glyR3–licA, but not for orf4 or manB–celT. Since CelC (Petre et al. 1986; Schwarz et al. 1988) and LicA (Fuchs et al. 2003) are both active on β-1,3 glucans, the results indicate that C. thermocellum coordinates the expression of glycosyl hydrolases in response to the availability of a particular biomass substrate and, despite the insolubility of biomass substrate, this regulation can be mediated by a soluble sugar released from the biomass substrate.
The first three genes of the cluster celC, glyR3, and licA are all co-transcribed into a polycistronic messenger (Fig. 2), leading to their co-regulation. A basal level of expression is present for these three genes, as shown by the low level transcript present at the exponential stage of growth on all three substrates (Fig. 7). When the bacterium encounters a substrate that contains β-1,3 linkages, some degree of degradation occurs by the action of the low level expression of celC and licA. The degradation product laminaribiose is presumed to enter the cell (Nataf et al. 2009) and inactivate GlyR3’s binding to the celC promoter region (Newcomb et al. 2007). Full level transcription of celC–glyR3–licA is now allowed to proceed, as evidenced by the significant increase in the expression level detected at late exponential growth for all three genes on laminarin (Fig. 7). In addition, by having GlyR3 regulate its own expression, a feedback loop is created. Once the level of the inducer laminaribiose dwindles, GlyR3 is freed up to bind the promoter region and quickly downregulates celC–glyR3–licA, as demonstrated by the dramatic drop in the expression of these three genes at early stationary phase (Fig. 7). The sharp spike of celC–glyR3–licA expression occurs only when the cell is grown with a substrate containing β-1,3 linkages (laminarin). The same expression pattern was observed when lichenan (β-1,3 and β-1,4 linkages) or laminaribiose was used as the sole carbon source (data not shown). The parallel expression profiles of these three genes as determined by quantitative RT-PCR are consistent with the result of Northern blot, which indicates that celC, glyR3, and licA are co-transcribed into a polycistronic messenger. It is of note that, although the three protein products are to be translated from this messenger, other factors such as the efficiency of ribosome binding site may still lead to differential expression of the members of the celC operon.
Concurrent with the spike in celC operon expression is the transition from exponential phase to stationary phase (Fig. 7), which may be indicative of a growth phase- or growth rate-dependent expression (Dror et al. 2003). Studies in C. cellulovorans show a sharp increase in the expression of genes encoding cellulolytic proteins as cultures reach late log phase (Han et al. 2003b). The mechanism of this growth phase- or growth rate-dependence remains to be investigated.
It is noteworthy that in the CelC cluster in C. thermocellum, the non-cellulosomal glycosyl hydrolase genes (celC and licA) and the cellulosomal glycosyl hydrolase genes (manB and celT) belong to two different transcription units. The disparate expression kinetics (Figs. 7 and 8) clearly indicates that the two sets of genes are regulated by different mechanisms under the culture conditions. Whether the cellulosomal and non-cellulosomal glycosyl hydrolase genes are always regulated by different mechanisms remains to be further investigated. It is would be of interest to determine how the manB–celT gene cluster is regulated. Our results also demonstrated that orf4 encoding a hitherto unknown transmembrane protein is transcribed alone and has a relatively low level of expression on all of three substrates studied. It would be interesting to determine if orf4 is related to biomass degradation, either as a part of the transport system or in some other capacity.
Our results demonstrated that a β-1,3-linked glucan is able to upregulate celC, glyR3, and licA. These three genes are co-transcribed and therefore form an operon. The celC operon is the first operon with a known molecular mechanism reported in C. thermocellum. The bacterium clearly tailors the expression of the celC operon in response to the substrate present in the environment. As the study of the C. thermocellum cellulase system continues, other such regulatory units will be discovered along with novel molecular mechanisms.
References
Bagnara-Tardif C, Gaudin C, Belaich A, Hoest P, Citard T, Belaich JP (1992) Sequence analysis of a gene cluster encoding cellulases from Clostridium cellulolyticum. Gene 119(1):17–28
Bahari L, Gilad Y, Borovok I, Kahel-Raifer H, Dassa B, Nataf Y, Shoham Y, Lamed R, Bayer EA (2010) Glycoside hydrolases as components of putative carbohydrate biosensor proteins in Clostridium thermocellum. J Ind Microbiol Biotechnol. doi:10.1007/s10295-010-0848-9
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Demain AL, Newcomb M, Wu JH (2005) Cellulase, clostridia, and ethanol. Microbiol Mol Biol Rev 69(1):124–154
Dror TW, Morag E, Rolider A, Bayer EA, Lamed R, Shoham Y (2003) Regulation of the cellulosomal celS (cel48A) gene of Clostridium thermocellum is growth rate dependent. J Bacteriol 185(10):3042–3048
Fuchs KP, Zverlov VV, Velikodvorskaya GA, Lottspeich F, Schwarz WH (2003) Lic16A of Clostridium thermocellum, a non-cellulosomal, highly complex endo-β-1,3-glucanase bound to the outer cell surface. Microbiology 149(Pt 4):1021–1031
Fujino T, Beguin P, Aubert JP (1993) Organization of a Clostridium thermocellum gene cluster encoding the cellulosomal scaffolding protein CipA and a protein possibly involved in attachment of the cellulosome to the cell surface. J Bacteriol 175(7):1891–1899
Gerngross UT, Romaniec MP, Kobayashi T, Huskisson NS, Demain AL (1993) Sequencing of a Clostridium thermocellum gene (cipA) encoding the cellulosomal SL-protein reveals an unusual degree of internal homology. Mol Microbiol 8(2):325–334
Guglielmi G, Beguin P (1998) Cellulase and hemicellulase genes of Clostridium thermocellum from five independent collections contain few overlaps and are widely scattered across the chromosome. FEMS Microbiol Lett 161(1):209–215
Han SO, Yukawa H, Inui M, Doi RH (2003a) Transcription of Clostridium cellulovorans cellulosomal cellulase and hemicellulase genes. J Bacteriol 185(8):2520–2527
Han SO, Yukawa H, Inui M, Doi RH (2003b) Regulation of expression of cellulosomal cellulase and hemicellulase genes in Clostridium cellulovorans. J Bacteriol 185(20):6067–6075
Johnson EA, Madia A, Demain AL (1981) Chemically defined minimal medium for growth of the anaerobic cellulolytic thermophile Clostridium thermocellum. Appl Environ Microbiol 41(4):1060–1062
Johnson EA, Sakajoh M, Halliwell G, Madia A, Demain AL (1982) Saccharification of complex cellulosic substrates by the cellulase system from Clostridium thermocellum. Appl Environ Microbiol 43(5):1125–1132
Kahel-Raifer H, Jindou S, Bahari L, Nataf Y, Shoham Y, Bayer EA, Borovok I, Lamed R (2010) The unique set of putative membrane-associated anti-sigma factors in Clostridium thermocellum suggests a novel extracellular carbohydrate-sensing mechanism involved in gene regulation. FEMS Microbiol Lett 308(1):84–93
Kakiuchi M, Isui A, Suzuki K, Fujino T, Fujino E, Kimura T, Karita S, Sakka K, Ohmiya K (1998) Cloning and DNA sequencing of the genes encoding Clostridium josui scaffolding protein CipA and cellulase CelD and identification of their gene products as major components of the cellulosome. J Bacteriol 180(16):4303–4308
Kruus K, Lua AC, Demain AL, Wu JH (1995) The anchorage function of CipA (CelL), a scaffolding protein of the Clostridium thermocellum cellulosome. Proc Natl Acad Sci USA 92(20):9254–9258
Kurokawa J, Hemjinda E, Arai T, Karita S, Kimura T, Sakka K, Ohmiya K (2001) Sequence of the Clostridium thermocellum mannanase gene man26B and characterization of the translated product. Biosci Biotechnol Biochem 65(3):548–554
Kurokawa J, Hemjinda E, Arai T, Kimura T, Sakka K, Ohmiya K (2002) Clostridium thermocellum cellulase CelT, a family 9 endoglucanase without an Ig-like domain or family 3c carbohydrate-binding module. Appl Microbiol Biotechnol 59(4–5):455–461
Lamed R, Setter E, Bayer EA (1983) Characterization of a cellulose-binding, cellulase-containing complex in Clostridium thermocellum. J Bacteriol 156(2):828–836
Leibovitz E, Beguin P (1996) A new type of cohesin domain that specifically binds the dockerin domain of the Clostridium thermocellum cellulosome-integrating protein CipA. J Bacteriol 178(11):3077–3084
Leibovitz E, Ohayon H, Gounon P, Beguin P (1997) Characterization and subcellular localization of the Clostridium thermocellum scaffoldin dockerin binding protein SdbA. J Bacteriol 179(8):2519–2523
Maamar H, Abdou L, Boileau C, Valette O, Tardif C (2006) Transcriptional analysis of the cip-cel gene cluster from Clostridium cellulolyticum. J Bacteriol 188(7):2614–2624
Nataf Y, Yaron S, Stahl F, Lamed R, Bayer EA, Scheper TH, Sonenshein AL, Shoham Y (2009) Cellodextrin and laminaribiose ABC transporters in Clostridium thermocellum. J Bacteriol 191(1):203–209
Nataf Y, Bahari L, Kahel-Raifer H, Borovok I, Lamed R, Bayer EA, Sonenshein AL, Shoham Y (2010) Clostridium thermocellum cellulosomal genes are regulated by extracytoplasmic polysaccharides via alternative sigma factors. Proc Natl Acad Sci USA 107(43):18646–18651
Newcomb M, Chen CY, Wu JH (2007) Induction of the celC operon of Clostridium thermocellum by laminaribiose. Proc Natl Acad Sci USA 104(10):3747–3752
Nolling J, Breton G, Omelchenko MV, Makarova KS, Zeng Q, Gibson R, Lee HM, Dubois J, Qiu D, Hitti J, Wolf YI, Tatusov RL, Sabathe F, Doucette-Stamm L, Soucaille P, Daly MJ, Bennett GN, Koonin EV, Smith DR (2001) Genome sequence and comparative analysis of the solvent-producing bacterium Clostridium acetobutylicum. J Bacteriol 183(16):4823–4838
Petre D, Millet J, Longin R, Beguin P, Girard H, Aubert JP (1986) Purification and properties of the endoglucanase C of Clostridium thermocellum produced in Escherichia coli. Biochimie 68(5):687–695
Romaniec MP, Kobayashi T, Fauth U, Gerngross UT, Demain AL (1991) Cloning and expression of a Clostridium thermocellum DNA fragment that encodes a protein related to cellulosome component SL. Appl Biochem Biotechnol 31(2):119–134
Sabathe F, Belaich A, Soucaille P (2002) Characterization of the cellulolytic complex (cellulosome) of Clostridium acetobutylicum. FEMS Microbiol Lett 217(1):15–22
Schwarz WH (2001) The cellulosome and cellulose degradation by anaerobic bacteria. Appl Microbiol Biotechnol 56(5–6):634–649
Schwarz WH, Schimming S, Rucknagel KP, Burgschwaiger S, Kreil G, Staudenbauer WL (1988) Nucleotide sequence of the celC gene encoding endoglucanase C of Clostridium thermocellum. Gene 63(1):23–30
Tamaru Y, Doi RH (2000) The engL gene cluster of Clostridium cellulovorans contains a gene for cellulosomal manA. J Bacteriol 182(1):244–247
Tamaru Y, Karita S, Ibrahim A, Chan H, Doi RH (2000) A large gene cluster for the Clostridium cellulovorans cellulosome. J Bacteriol 182(20):5906–5910
Zverlov VV, Fuchs KP, Schwarz WH (2002) Chi18A, the endochitinase in the cellulosome of the thermophilic, cellulolytic bacterium Clostridium thermocellum. Appl Environ Microbiol 68(6):3176–3179
Zverlov VV, Velikodvorskaya GA, Schwarz WH (2003) Two new cellulosome components encoded downstream of celI in the genome of Clostridium thermocellum: the non-processive endoglucanase CelN and the possibly structural protein CseP. Microbiology 149(Pt 2):515–524
Zverlov VV, Kellermann J, Schwarz WH (2005) Functional subgenomics of Clostridium thermocellum cellulosomal genes: identification of the major catalytic components in the extracellular complex and detection of three new enzymes. Proteomics 5(14):3646–3653
Acknowledgments
The research was supported by a grant (DE-FG02-94ER20155) from the Office of Science (BES) and in part by a grant (DE-FG02-08ER64692) from the Office of Science (BER), US Department of Energy to JHDW. We thank David Russell and Robert Zagursky for assistance in the analysis of the genomic sequence.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Newcomb, M., Millen, J., Chen, CY. et al. Co-transcription of the celC gene cluster in Clostridium thermocellum . Appl Microbiol Biotechnol 90, 625–634 (2011). https://doi.org/10.1007/s00253-011-3121-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-011-3121-x