
Abstract
The activity of a dye-linked l-proline dehydrogenase (dye-l-proDH) was found in the crude extract of an aerobic hyperthermophilic archaeon, Pyrobaculum calidifontis JCM 11548, and was purified 163-fold through four sequential chromatography steps. The enzyme has a molecular mass of about 108 kDa and is a homodimer with a subunit molecular mass of about 46 kDa. The enzyme retained more than 90% of its activity after incubation at 100 °C for 120 min (pH 7.5) or after incubation at pHs 4.5–9.0 for 30 min at 50 °C. The enzyme catalyzed l-proline dehydrogenation to Δ1-pyroline-5-carboxylate using 2,6-dichloroindophenol (DCIP) as the electron acceptor and the Michaelis constants for l-proline and DCIP were 1.67 and 0.026 mM, respectively. The prosthetic group on the enzyme was identified as flavin adenine dinucleotide by high-performance liquid chromatography. The subunit N-terminal amino acid sequence was MYDYVVVGAG. Using that sequence and previously reported genome information, the gene encoding the enzyme (Pcal_1655) was identified. The gene was then cloned and expressed in Escherichia coli and found to encode a polypeptide of 415 amino acids with a calculated molecular weight of 46,259. The dye-l-proDH gene cluster in P. calidifontis inherently differs from those in the other hyperthermophiles reported so far.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Dye-linked dehydrogenases (dye-DHs) belong to a group of oxidoreductases that catalyze the oxidation of various organic acids, amino acids, and alcohols in the presence of an artificial acceptor, like 2,6-dichloroindophenol (DCIP) or potassium ferricyanide (Ohshima and Tanaka 1993; Sakuraba et al. 2001). Many kinds of dye-DHs have been found in mesophilic microorganisms, and their use as specific elements in biosensors has been anticipated (Frew and Hill 1987). So far, however, their instability has limited the practical application of mesophilic dye-DHs. On the other hand, enzymes from hyperthermophiles are known to be much more stable than those from mesophiles and moderate thermophiles. This increases our ability to obtain useful information about their structure and function and makes them more amenable to practical application (Sakuraba et al. 2001; Kawakami et al. 2005; Satomura et al. 2008).
We have been extensively screening dye-DHs from hyperthermophiles for several years. During that period, we have identified two kinds of dye-linked l-proline dehydrogenase (dye-l-proDH) from several anaerobic hyperthermophiles, including Thermococcus profundus (Sakuraba et al. 2001) and Pyrococcus horikoshii OT-3 (Kawakami et al. 2005), a dye-linked d-proline dehydrogenase from the anaerobe Pyrobaculum islandicum (Satomura et al. 2002) and a dye-linked d-lactate dehydrogenase from the aerobe Sulfolobus tokodaii (Satomura et al. 2008). We found that these enzymes remain stable for longer times under a variety of conditions and are applicable to a novel type of electrochemical detector for l-proline and d-proline (Kawakami et al. 2004; Tani et al. 2008). In addition, we just recently identified a dye-l-proDH from an aerobic hyperthermophilic archaeon P. calidifontis, isolated from a hot spring in the Philippines (Amo et al. 2002), and found that some enzymological properties of this enzyme are completely different from those of the Thermococcus and Pyrococcus strains. In this paper, we describe the purification and characterization of the P. calidifontis enzyme and the cloning of its gene and propose that this enzyme is a novel type of dye-l-proDH.
Materials and methods
Microorganism strains, plasmids, and chemicals
P. calidifontis JCM 11548 was obtained from the Japan Collection of Microorganisms (Wako, Japan). Escherichia coli strains JM109 and BL21(DE3) codon plus RIL were from Stratagene (La Jolla, CA, USA). The plasmid pET22b was from Novagen (Madison, WI, USA). DCIP and l-proline were purchased from Nacalai Tesque (Kyoto, Japan). Restriction endonucleases were from New England Biolabs (Beverly, MA, USA). All other chemicals were of reagent grade.
Conditions of cell growth
P. calidifontis was cultured in 2-L Erlenmeyer flasks in medium (600 mL, pH 7.0, adjusted with NaOH) containing 10 g L−1 of tryptone, 1 g L−1 yeast extract, and 3 g L−1 Na2S2O3·5H2O at 90 °C under aerobic conditions. After 24 h in culture, the cells (1.2 L medium) were harvested by centrifugation at 10,000×g for 10 min, washed first with 0.85% NaCl, and then with 10 mM potassium phosphate buffer (pH 7.2), and stored at −20 °C until use.
Enzyme assay and protein determination
Dye-l-proDH activity was assayed by measuring the rate of DCIP reduction at 50 °C. The standard reaction mixture contained 100 mM l-proline, 0.1 mM DCIP, 200 mM Tris–HCl buffer (pH 8.0) and the enzyme in a total volume of 1.0 mL. The mixture without the substrate (l-proline) was incubated at 50 °C for about 3 min in a cuvette with a 0.4-cm light path length, after which the reaction was started by the addition of l-proline. The initial decrease in the absorbance at 600 nm was measured using a Shimadzu UV-160A recording spectrophotometer (Kyoto, Japan). One unit was defined as the amount of the enzyme catalyzing the reduction of 1 μmol of DCIP/min at 50 °C. An absorption coefficient (ε) of 21.5 mM−1 cm−1 at 600 nm was used for DCIP (Ohshima and Tanaka 1993). The reductions in ferricyanide, p-iodonitrotetrazolium violet (INT), and horse liver cytochrome c were monitored at 405 nm (ε = 1.04 mM−1 cm−1), 490 nm (ε = 15.0 mM−1 cm−1), and 553 nm (ε = 15.3 mM−1 cm−1), respectively. For reduction in INT, phenazine methosulfate (PMS) was used as an electron transfer intermediate. Protein concentration was determined by the method of Bradford, using bovine serum albumin as the standard (Bradford 1976).
Purification of l-Pro DH from P. calidifontis cells
All steps in the purification procedure were carried out at a room temperature, using 10 mM potassium phosphate buffer (pH 7.2) as the standard buffer. P. calidifontis cells (about 2 g wet weight) suspended in standard buffer were disrupted by ultrasonication. After centrifugation at 10,000×g for 20 min to remove any remaining intact cells and the cell debris, the supernatant was used as the crude extract. The enzyme solution was applied to a DEAE-Toyopearl 650M column (2.8 × 17.0 cm; Tosoh, Tokyo) previously equilibrated with the standard buffer. The enzyme was eluted with a linear gradient of 0–0.5 M NaCl in standard buffer, after which the active fractions were pooled and dialyzed against the standard buffer. Thereafter, preparative slab polyacrylamide gel electrophoresis (PAGE) was carried out according to the method of Ohshima and Ishida (1992), and the enzyme was extracted from the gel pieces using the standard buffer. The resultant enzyme solution was applied to a Superdex 200 pg column (2.6 × 60 cm; GE Healthcare UK Ltd, UK) previously equilibrated with the standard buffer and then eluted with the same buffer. The active fractions were pooled, and the solution was used for experimentation.
Polyacrylamide gel electrophoresis and molecular mass determination
Native PAGE using 7.5% polyacrylamide gel was performed according to the method of Davis (1964). Activity staining was performed at 50 °C in a mixture containing 0.3 M Tris–HCl buffer (pH 8.0), 100 mM l-proline, 0.04 mM PMS, and 0.1 mM INT until a red band of sufficient intensity was visible. Protein was stained using 0.025% Coomassie brilliant blue G-250 in 50% methanol and 10% acetic acid.
Sodium dodecyl sulfate (SDS)-PAGE was carried out using 15% polyacrylamide gel containing 0.1% SDS according to the method of Leammli (1970). Precision Plus protein standards (BIO-RAD, Tokyo, Japan) were used as the molecular mass standards. The protein sample was boiled for 5 min in 10 mM Tris–HCl buffer (pH 7.0) containing 1% SDS and 1% 2-mercaptoethanol. Protein bands were visualized by staining with 0.025% Coomassie brilliant blue R-250.
The molecular masses of the native enzymes were determined by gel filtration column chromatography using a Superdex 200 pg column (2.6 × 60 cm) with thyroglobulin (669 kDa), ferritin (440 kDa), catalase (232 kDa), aldolase (158 kDa), albumin (67 kDa), and ribonuclease A (13.7 kDa) serving as molecular standards (GE Healthcare UK Ltd).
Extraction and determination of flavin
The flavin compound from the enzyme was extracted using 1% perchloric acid (PCA) as described elsewhere (Sakuraba et al. 2001; Kawakami et al. 2005). After centrifugation to remove the precipitate formed, the flavin compound in the supernatant was identified using high-pressure liquid chromatography with a TSKgel ODS-80Ts column (4.6 × 150 mm; Tosoh, Tokyo). A linear gradient between 10 mM potassium phosphate buffer (pH 6.0) and methanol (100% v/v) was used for the elution. The flow rate was 1.0 mL/min, and the total elution time was 45 min. FAD and FMN were monitored by determining the absorbance at 260 nm. FAD content was determined spectrophotometrically as previously described (Kawakami et al. 2004). After solutions of the enzyme was denatured by 1% (v/v) PCA and the precipitants were removed by centrifugation, the resultant supernatants were used for determination of FAD content. FAD concentration was estimated using an extinction coefficient of 11.3 mM−1 cm−1 at 450 nm.
Identification of the reaction product
The reaction product after oxidation of l-proline was identified using thin layer chromatography as previously described (Satomura et al. 2002). The postulated reaction products are Δ1-pyrroline-5-carboxylate or Δ1-pyrroline-2-carboxylate, which are converted into glutamate or 4-aminobutyrate, respectively, through oxidation by H2O2 (Meister 1954).
N-terminal amino acid analysis
The N-terminal amino acid sequence of the enzyme was determined using an Edman degradation protein sequencer. The phenylthiohydantatoin derivatives (Phh-Xaa) were separated and identified using a PPSQ-10 protein sequencer (Shimadzu).
Cloning of the dye-l-proDH gene
Sequencing of the genome of P. calidifontis was completed in 2007 (http://www.genome.jp/kegg-bin/show_organism?org=pcl). On the basis of the N-terminal amino acid sequence of the original dye-l-proDH, the open reading frame of an FAD-dependent oxidoreductase homologue (ORF ID, Pcal_1655) was identified in the P. calidifontis genome using BLAST (Altschul et al. 1997). To collect the genomic DNA containing Pcal_1655, P. calidifontis cells were harvested by centrifugation (10,000×g, 10 min) and washed first with 0.85% NaCl and then with 10 mM potassium phosphate buffer (pH 7.2), after which the genomic DNA was prepared as described previously (Murray and Thompson 1980).
Overexpression and purification of the recombinant protein
To construct an expression plasmid, a 1.3-kbp gene fragment comprised of the gene encoding dye-l-proDH and NdeI and EcoRI linkers was amplified by PCR using the primers 5′-TGTCTCCATATGTATGACTATG-3′, which was designed to contain the N-terminal region of the dye-L-proDH gene and an NdeI digestion sequence, and 5′-GCCGTAGGAATTCCTCAATCATATCACAA-3′, which was designed to contain the C-terminal region and an EcoRI digestion sequence. The respective restriction sequences are underlined. The genomic DNA was used as the template. The amplified 1.3-kbp fragment was digested with NdeI and EcoRI and then ligated into the expression vector pET22b, linearized using NdeI and EcoRI, to generate pLPDH. Thereafter, E. coli strain BL21(DE3) codon plus RIL cells were transformed with pLPDH, and the transformants were cultivated at 37 °C in 1 L of Luria–Bertani medium containing 50 μg mL−1 ampicillin until the optical density at 600 nm reached 0.6. Expression was then induced by adding 0.1 mM isopropyl-β-D-thiogalacto-pyranoside to the medium, and cultivation was continued for an additional 4 h. The cells were then harvested by centrifugation, suspended in 10 mM potassium phosphate buffer (pH 7.2), which was used as the standard buffer, and disrupted by ultrasonication. The crude extract was heated at 70 °C for 30 min, and the denatured protein was removed by centrifugation (10,000×g for 10 min). The supernatant was purified by preparative slab PAGE, and the enzyme was extracted from the gel pieces using the standard buffer. The resultant enzyme solution was applied on a Superdex 200 pg column (2.6 × 60 cm) previously equilibrated with the standard buffer and eluted with the same buffer. The active fractions were pooled, and the solution was used for experimentation.
Kinetic parameters
The Michaelis constant (K m) was determined from double-reciprocal plots of the initial rate data using l-proline or l-hydoxyproline as the electron donor and DCIP as the electron acceptor at 50 °C.
Results
Purification of dye-l-proDH from P. calidifontis
Many dye-DHs are membrane-bound enzymes. With that in mind, we initially disrupted P. calidifontis cells by sonication and fractionated them into particulate and supernatant fractions by ultracentrifugation (Beckman Ultracentrifuge at 140,000×g for 90 min). Dye-l-proDH activity was then measured separately in the two fractions. More than 90% of the activity was found in the supernatant fraction, which indicates that the enzyme is either not bound to the cytoplasmic membrane or is easily solubilized from the membrane. We therefore omitted the step for solubilization and fractionation of the enzyme from the purification procedure. The purification of dye-l-proDH from P. calidifontis is summarized in Table 1. After the final step, the enzyme had been purified about 163-fold, with an overall yield of about 18%.
Molecular mass and subunit structure
Using native-gradient PAGE, the molecular mass of the P. calidifontis dye-l-proDH was determined to be about 108 kDa (data not shown); moreover, SDS-PAGE showed one major band (Fig. 1), indicating the enzyme is composed of identical subunits. The molecular mass of the subunit was estimated to be about 46 kDa, indicating that the enzyme is a homodimer.

SDS-PAGE of the purified enzyme. Left column molecular maker proteins, right column the enzyme
pH and temperature optima and thermostability
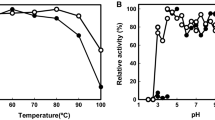
To determine the pH optimum for this dye-l-proDH, its activity was measured at various pHs at 50 °C. Acetate (pH 4.5–5.5), MES-NaOH (pH 5.5–6.5), potassium phosphate (pH 6.0–7.5), MOPS-NaOH (pH 6.5–7.5), and Tris-HCl (pH 7.5–9.0) buffers were used, and the pH of the reaction mixture was measured at 25 °C after each assay. The optimum pH was about 7.0, and half of the activity was retained at pH 6.0 and 9.0.
We determined the optimum temperature for the enzyme by assaying activity at temperatures ranging from 50 to 90 °C. Because DCIP is unstable above about 70 °C, we used ferricyanide in 200 mM Tris-HCl buffer (pH 7.5) as the electron acceptor instead of DCIP. The assay was started by adding the enzyme after preincubation of the reaction mixture for 5 min at the temperature of interest. We found that the optimum temperature for dye-l-proDH activity was above 80 °C, but more precise determination was not possible due to heat-induced destruction of the reaction system.
When we assessed the thermostability of this dye-l-proDH, we found that it retained about 90% of its activity after incubation at 100 °C for 120 min, and the half-life at 105 °C was about 90 min (Fig. 2). In addition, when the enzyme was incubated at pHs between 4.5 and 9.0 for 30 min at 50 °C, it retained more than 90% of the highest activity seen at pH 7. Thus, dye-l-proDH from P. calidifontis appears to be very stable under a variety of conditions.

The effect of temperature on dye-l-proDH. The enzyme was incubated at 100 °C (filled diamond) and 105 °C (filled square), and the residual activity was measured at 50 °C
Substrate and electron acceptor specificity
We found that this dye-l-proDH catalyzed the dehydrogenation of l-proline as the most preferred substrate, though it also showed activity with l-hydroxyproline (relative activity, 72% as compared to l-proline). On the other hand, sarcosine, d-proline, l-ornithine, l-glutamate, l-arginine, l-serine, l-leucine, l-valine, and l-alanine were all inert as substrates. When we examined the electron acceptor specificity of the enzyme, we found that DCIP was the most preferred electron acceptor, though ferricyanide and PMS-INT also exhibited electron acceptor activity (relative activities, 93% and 9%, respectively, as compared to DCIP). NAD, NADP, and bovine heart cytochrome c were inert as electron acceptors.
Steady-state kinetics
We next carried out steady-state kinetic analysis of l-proline dehydrogenation with DCIP as the electron acceptor. Initial velocity experiments were done by varying the concentration of one substrate while keeping the concentration of the other substrates constant, as previously described (Ohshima and Tanaka 1993). Double reciprocal plots of the initial velocity and the substrate concentrations (l-proline and DCIP) showed a series of parallel lines. The K m values were calculated from the secondary plot of the intercepts vs. the reciprocal of the substrate concentration. From the secondary plots of the intercepts vs. reciprocals of l-proline and DCIP concentrations, the K m value for l-proline and DCIP was determined to be 1.67 and 0.026 mM, respectively. The K m value for l-hydroxyproline and DCIP was similarly calculated to be 6.05 and 0.035 mM, respectively. In addition, the K cat/K m value for l-proline (DCIP) and l-hydroxyproline (DCIP) were 1.25 (79.5) and 0.125 (21.9) s−1 mM−1, respectively.
N-terminal amino acid sequences
Using an automated Edman degradation protein sequencer, the sequence of the ten N-terminal amino acid residues of the subunit was determined to be MYDYVVVGAG.
Amino acid sequence alignment of P. calidifontis dye-l-proDH
The similarity between the amino acid sequence of P. calidifontis dye-l-proDH and other enzymes was searched using the BLAST server in the Kyoto Encyclopedia of Genes and Genomes. We found that P. calidifontis dye-l-proDH exhibited a small degree of similarity to the T. profundus dye-proDH β subunit (PdhB, 16%; originally assigned as a putative sarcosine oxidase β subunit), and to the subunits of two different Pc. horikoshii ProDHs, PH1364 and PH1751 (26% and 25% identity, respectively; Fig. 3).

Alignment of amino acid sequence of dye-l-proDH from four strains of hyperhtermophilic arachaea. Highly conserved residues are depicted in black boxes. Amino acid sequence identity with Pcal1655 is given in parentheses. The asterisks are the FAD-binding motif. Proteins shown on figure: PdhB dye-L-proDH β-subunit from T. profundus (NCBI accession number BAD13509.1); PH1364 PDH1 β-subunit from Pc. horikoshii (NCBI accession number BAA30470.1); PH1751 PDH2 β-subunit from Pc.horikoshii (NCBI accession number BAA30865.1); Pcal1655 dye-l-proDH from P. calidofontis (NCBI accession number YP_001056538.1)
Expression dye-l-proDH gene in E. coli and purification of its product
E. coli BL21(DE3) codon plus RIL cells transformed with the expression vector pLPDH exhibited dye-l-proDH activity (total protein = 251 mg, specific activity = 0.048 U/mg) that was not lost after incubation at 70 °C for 30 min. We were then able to easily obtain the purified enzyme (1.8 mg) in three steps: heating of the cell extract at 70 °C for 30 min followed by successive preparative slab PAGE and Superdex 200 pg chromatography. The yield was about 19%.
Properties of recombinant dye-l-proDH
The pH and temperature optima, thermostability, and substrate specificity of the recombinant enzyme were nearly identical to those of the native enzyme (data not shown).
The spectrum of the recombinant dye-l-proDH showed the characteristics of typical flavoprotein (Fig. 4). The flavin prosthetic group was extracted from the enzyme using 1% PCA, and analysis by high-performance liquid chromatography revealed it to be FAD; no FMN was detected. The content of FAD was determined to be 0.74 mol of FAD per mole of enzyme. In addition, when the enzyme reaction product after oxidation by H2O2 was analyzed by thin layer chromatography, the enzyme reaction produced a ninhydrin spot corresponding to l-glutamate, but not 4-aminobutyrate (data not shown). This means that the reaction product was Δ1-pyrroline-5-carboxylate (P5C), not Δ1-pyrroline-2-carboxylate.

Absorption spectra of recombinant dye-l-pro DH in 10 mM potassium phosphate (pH 7.2)
Discussion
In this study, we identified dye-l-proDH activity in an aerobic hyperthermophilic archaeon, P. calidifontis, and were able to purify and characterize the enzyme. Following cell disruption by sonication, the enzyme appeared in the soluble fraction without the need for a special solubilization procedure, suggesting that perhaps the enzyme is distributed on the surface of the cytoplasmic membrane and is easily released, which has been seen previously with dye-l-proline dehydrogenases from T. profundus and Pc. horikoshii (Sakuraba et al. 2001, Kawakami et al. 2005) but not with a dye-d-proline dehydrogenase from P. islandicum (Satomura et al. 2002).
Two different types of dye-l-proDH, PDH1 and PDH2, have been identified in the anaerobic hyperthermophile P. horikoshii OT-3, belonging to the phylum Euryarchaeota (Kawakami et al. 2005). PDH1 is a heterooctameric complex (α4β4; molecular mass, 440 kDa) containing FAD, FMN, Fe, and ATP (Kawakami et al. 2005), while PDH2 is a tetrameric complex (αβγδ; molecular mass, 120 kDa). Structural analysis of the PDH1 complex showed the enzyme to be a unique diflavin dehydrogenase containing a novel electron transfer system that is totally different from that of the PDH2 complex, which contains four components: proDH, NADH dehydrogenase, a ferredoxin-like protein, and a functionally unknown protein (Tuge et al. 2005). The structure of β-subunit, which is a PDH1 catalytic component containing FAD as a cofactor, was similar to that of monomeric sarcosine oxidase (MSOX). Based on the structural comparison with MSOX, the residues forming a substrate-binding pocket could be predicted (Trickey et al. 1999). As shown in Fig. 3, several sequence motifs in β-subunit of the Pc. horikoshii PDH1 (PH1364) are well conserved in all dye-l-pro DHs. Among these motifs, 12GXGXXG17 (residue numbers of PH1364 are given) is typical of FAD-binding domain. The residues in the motifs of 86GYLF89, 304WAGXY308, and 332GXSGXGXM339 are involved in the construction of the binding sites for the substrate and isoalloxazine ring of FAD in β-subunit of the Pc. horikoshii PDH1. These motifs are conserved in all the dye-l-pro DHs. Thus, these motifs might be important for the catalytic role of dye-l-pro DHs.
Here, we showed that, like PDH1 and PDH2, a dye-l-proDH from the aerobic hyperthermophilic archaeon P. calidifontis, belonging to the phylum Crenarchaeota, catalyzes the conversion of l-proline to P5C in the presence of DCIP, but it differs from those other enzymes in that it has a homodimeric structure (2 × 46 kDa). Thus, the molecular structure of the P. calidifontis enzyme is totally unlike those of PDH1 and PDH2 (Kawakami et al. 2005). In addition, we found that the sequence of the P. calidifontis subunit shows little similarity to those of the PDH1 and PDH2 β-subunits (26% and 25% identity, respectively), which are responsible for l-proline dehydrogenation—i.e., P. calidifontis dye-l-proDH is inherently different from the two dye-l-proDHs from Pc. horikoshii OT-3, with respect to sequence. We have thus identified a novel type of dye-l-proDH expressed by the aerobic hyperethermophilic archaeon P. calidifontis.
In addition to l-proline, dye-l-proDH from P. calidifontis catalyzes the dehydrogenation of l-hydroxyproline. By contrast, neither PDH1 nor PDH2 are able to use l-hydroxyproline as a substrate. Thus, the substrate specificity of the P. calidifontis enzyme appears to be somewhat broader than those of the Pc. horikoshii and T. profundus enzymes. In addition, the P. calidifontis enzyme is still active after incubation at 100 °C for 120 min, which makes it more thermostable than the enzymes from Pc. horikoshii and T. profundus. We have developed an application for T. profundus enzymes in an electrochemical sensor used to assay l-proline (Zheng et al. 2006). In addition, the dye-d-proDH from P. islandicum has proven useful in a novel biosensor for determining levels d-amino acids, such as d-proline and d-valine, in biological samples. In that sensor, the electrode is prepared by immobilizing the thermostable dye-d-ProDH within an agar gel membrane. At temperatures above 80 °C, liquid agar and the enzyme solution can be mixed and simply co-immobilized on a glassy carbon electrode using the spin-coating method (Tani et al. 2008). Moreover, carbon nanotube gel, which is composed of a mixture of single-wall carbon nanotubes, an ionic liquid, and an immobilized thermostable dye-D-proDH, can serve as the basis for electrodes that enable determination of d-amino acid levels in food samples with greater sensitivity than is obtained with the glassy agar-based electrode described above (Tani et al. 2009). In these cases, the high thermostability of dye-d-proDH supports the development of novel types of electrodes. That said, P. calidifontis dye-l-proDH loses no activity with incubation at 100 °C for 120 min and is much more thermostable than the l-proline sensor enzyme, T. profundus l-proDH, which loses activity at temperatures above 80 °C. In addition, the simpler subunit structure (homodimer) of the P. calidifontis enzyme may facilitate its immobilization during preparation of enzyme electrodes, as compared to the Pc. horikoshii heterotetrameric and heterooctermeric enzymes.
References
Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402
Amo T, Paje ML, Inagaki A, Ezaki S, Atomi H, Imanaka T (2002) Pyrobaculum calidifontis sp. nov., a novel hyperthermophilic archaeon that grows in atmospheric air. Archaea 1:113–121
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Davis BJ (1964) Disc electrophoresis—2. Method and application to human serum proteins. Ann NY Acad Sci 121:404–427
Frew JE, Hill HA (1987) Electrochemical biosensors. Anal Chem 59:933A–944A
Kawakami R, Sakuraba H, Ohshima T (2004) Gene and primary structures of dye-linked L-proline dehydrogenase from a hyperthermophilic archaeon Thermococcus profundus show the presence of a novel heterotetrameric amino acid dehydrogenase complex. Extremophiles 8:99–108
Kawakami R, Sakuraba H, Ohshima T (2005) A second novel dye-linked L-proline dehydrogenase complex is present in the hyperthermophilic archaeon Pyrococcus horikoshii OT-3. FEBS J 272:4044–4054
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the bacteriophase T4. Nature 227:680–685
Meister A (1954) The alpha-keto analogues of arginine, ornithine, and lysine. J Biol Chem 206:577–585
Murray MG, Thompson WF (1980) Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res 8:4321–4325
Ohshima T, Ishida M (1992) A large-scale preparative electrophoretic method for the purification of pyridine nucleotide-linked dehydrogenases. Protein Expr Purif 3:121–125
Ohshima T, Tanaka S (1993) Dye-linked L-malate dehydrogenase from thermophilic Bacillus species DSM 465, purification and characterization. Eur J Biochem 214:37–42
Sakuraba H, Takamatsu Y, Satomura T, Kawakami R, Ohshima T (2001) Purification, characterization, and application of a novel dye-linked L-proline dehydrogenase from a hyperthermophilic archaeon, Thermococcus profundus. Appl Environ Microbiol 67:1470–1475
Satomura T, Kawakami R, Sakuraba H, Ohshima T (2002) Dye-linked D-proline dehydrogenase from hyperthermophilic archaeon Pyrobaculum islandicum is a novel FAD-dependent amino acid dehydrogenase. J Biol Chem 277:12861–12867
Satomura T, Kawakami R, Sakuraba H, Ohshima T (2008) A novel flavin adenine dinucleotide (FAD) containing D-lactate dehydrogenase from the thermoacidophilic crenarchaeota Sulfolobus tokodaii strain 7: purification, characterization and expression in Escherichia coli. J Biosci Bioeng 106:16–21
Tani Y, Tanaka K, Yabutani T, Mishima Y, Sakuraba H, Ohshima T, Motonaka J (2008) Development of a D-amino acids electrochemical sensor based on immobilization of thermostable D-proline dehydrogenase within agar gel membrane. Anal Chim Acta 619:215–220
Tani Y, Itoyama Y, Nishi K, Wada C, Shoda Y, Satomura T, Sakuraba H, Ohshima T, Hayashi Y, Yabutani T, Motonaka J (2009) An amperometric D-amino acid biosensor prepared with the a thermostable D-proline dehydrogenase and carbon nanotube-ionic liquid gel. Anal Sci 25:919–923
Trickey P, Wagner MA, Jorns MS, Mathews FS (1999) Monomeric sarcosine oxidase: structure of a covalently flavinylated amine oxidizing enzyme. Structure 7:331–345
Tuge H, Kawakami R, Sakuraba H, Ago H, Miyano M, Aki K, Katunuma N, Ohshima T (2005) Crystal structure of a novel FAD-, FMN-, and ATP-containing L-proline dehydrogenase complex from Pyrococcus horikoshii. J Biol Chem 280:31045–31049
Zheng H, Hirose Y, Kimura T, Hori T, Suye S, Katayama H, Arai H, Kawakami R, Ohshima T (2006) L-Proline sensor based on layer-by-layer immobilization of thermostable dye-linked L-proline dehydrogenase and polymerized mediator. Sci Technol Adv Mater 7:243–248
Acknowledgments
This work was supported in part by a Grant-in-Aid for Scientific Research (no. 18380060) from the Japan Society for the Promotion of Science and the Japan Foundation of Applied Enzymology and by a grant from the Bio-oriented Technology Research Advancement Institution.
Author information
Authors and Affiliations
Corresponding author
Additional information
T. Satomura and X.-D. Zhang contributed equally to this work.
Rights and permissions
About this article
Cite this article
Satomura, T., Zhang, XD., Hara, Y. et al. Characterization of a novel dye-linked l-proline dehydrogenase from an aerobic hyperthermophilic archaeon, Pyrobaculum calidifontis . Appl Microbiol Biotechnol 89, 1075–1082 (2011). https://doi.org/10.1007/s00253-010-2914-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-010-2914-7