
Abstract
We have identified a carboxylesterase produced in liquid cultures of the thermophilic actinomycete Thermobifida fusca KW3 that were supplemented with poly(ethylene terephthalate) fibers. The enzyme hydrolyzed highly hydrophobic, synthetic cyclic poly(ethylene terephthalate) trimers with an optimal activity at 60°C and a pH of 6. V max and K m values for the hydrolysis were 9.3 µmol−1 min−1 mg−1 and 0.5 mM, respectively. The esterase showed high specificity towards short and middle chain-length fatty acyl esters of p-nitrophenol. The enzyme retained 37% of its activity after 96 h of incubation at 50°C and a pH of 8. Enzyme inhibition studies and analysis of substitution mutants of the carboxylesterase revealed the typical catalytic mechanism of a serine hydrolase with a catalytic triad composed of serine, glutamic acid, and histidine.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Carboxylesterases (EC 3.1.1.1) are ubiquitous enzymes that are found in both prokaryotes and eukaryotes. They differ from lipases (EC 3.1.1.3) in their substrate specificity. Carboxylesterases typically cleave water-soluble, short-chain acylglycerols (<10 carbon atoms) and p-nitrophenyl esters with short and medium acyl chain lengths while lipases preferentially catalyze the hydrolysis of water-insoluble long-chain triglycerides (>10 carbon atoms) (Arpigny and Jäger 1999; Chahinian et al. 2005).
The active site of esterases and lipases typically contains a catalytic triad composed of serine and glutamate or aspartate and histidine (Bornscheuer 2002). While many lipases have an amphiphilic peptide segment that regulates access to the active site of the enzyme, this “lid” structure is not found in esterases (Bornscheuer 2002; Jäger et al. 1994). Therefore, in contrast to many lipases, carboxylesterases do not show any interfacial activation behavior requiring a lipid/water interface for their activity (Bornscheuer 2002; Carvalho et al. 1998; Jäger et al. 1999).
Thermobifida fusca is a thermophilic actinomycete that plays an important role in the degradation of plant residues in composts and soils (Zhang et al. 1998). The complete genomic DNA sequence of T. fusca YX has been determined and a number of putative esterases have been identified (Lykidis et al. 2007). An acetylxylan esterase produced by T. fusca NTU22 that is involved in the degradation of hemicellulose and a polyhydroxyalkanoate depolymerase from the Thermobifida sp. BCC23166 that hydrolyzes poly-[(R)-3-hydroxybutyrate] have been reported (Yang and Liu 2008; Phithakrotchanakoon et al. 2009). T. fusca also produces serine hydrolases that have been described as cutinases due to their ability to hydrolyze the aliphatic plant polyester cutin (Fett et al. 1999). Similar to esterases, cutinases do not exhibit a lid structure (Carvalho et al. 1998). Two cutinases from a T. fusca strain with a 93% similarity in DNA sequence have been identified and correspond to the putative esterases Tfu_0882 and Tfu_0883 produced by T. fusca YX (Kleeberg et al. 2005; Chen et al. 2008).
Synthetic aromatic polymers such as poly(ethylene terephthalate) (PET) have previously been thought to be highly resistant to enzymatic hydrolysis (Müller et al. 2001). A cutinase from T. fusca DSM 43793 with a high sequence similarity to a triacylglycerol lipase from Streptomyces albus G has been reported to be able to degrade aliphatic–aromatic co-polyesters and PET sheets (Müller et al. 2005). Cutinases with the ability to hydrolyze PET fibers and oligomers have also been found in a number of other bacteria and fungi (Riegels et al. 2001; Yoon et al. 2002; Hooker et al. 2003; Figueroa et al. 2006; Liebminger et al. 2007; Nimchua et al. 2007; Brückner et al. 2008; Ronkvist et al. 2009). The structure of a fungal cutinase from Fusarium solani pisi has been solved. This enzyme belongs to the α/β-hydrolase fold superfamily and contains a Ser-His-Asp catalytic triad (Longhi et al. 1997). Cutinases can be used to modify the surface of PET fibers by increasing the hydrophilicity of the polyester (Yoon et al. 2002; Alisch-Mark et al. 2006; Heumann et al. 2006). T. fusca KW3 (DSM 6013) has previously been reported to produce extracellular esterases that are able to modify the surface of PET fibers when cultivated with suberin, a plant polyester containing aromatic moieties (Alisch et al. 2004; Feuerhack et al. 2008).
Recently, we reported the recombinant production of the carboxylesterase TfCa (T. fusca carboxylesterase) from T. fusca KW3 following optimization for codon usage in E. coli (Oeser et al. 2010). In this study, we have further characterized this unusual enzyme with PET-hydrolyzing activity that is distinct from cutinases. The thermostable TfCa may be useful in applications that require hydrolysis or modification of synthetic aromatic polymers and oligomers.
Materials and methods
Chemicals and PET substrates
Diethyl terephthalate (DET; Acros Organics, Geel, Belgium), terephthalic acid (Sigma-Aldrich Chemie GmbH, Steinheim, Germany), and bis(2-hydroxyethyl) terephthalate (BHET; Sigma-Aldrich Chemie GmbH, Steinheim, Germany) were purchased from commercial vendors. PET yarn was a kind gift from Dr. Th. Böhme KG (Geretsried, Germany) and poly(butylene terephthalate) (PBT) yarn was a kind gift from AMTEX Ldt. (Barcelona, Spain). The yarns were cut into pieces of approximately 10 mm in length. The recycled PET granulate (TEXPET A P800, particle size 4–8 mm) was a kind gift from Texplast GmbH (Wolfen, Germany). A cyclic PET trimer preparation was obtained by extracting PET fabric, which was a kind gift from Dr. Th. Böhme KG (Geretsried, Germany), with 1,4-dioxane in a soxhlet apparatus.
Microorganisms, culture conditions, and preparation of cell-free extracts
T. fusca KW3 (DSM 6013) was from the laboratory culture collection. T. fusca DSM 43792 and T. fusca DSM 43793 were obtained from the German Collection of Microorganisms and Cell Cultures (DSMZ, Braunschweig, Germany).
Preculture
The preculture medium for T. fusca DSM 6013 and DSM 43792 was composed of the following dissolved in demineralized water: 30 g/L saccharose, 3.0 g/L NaNO3, 1.0 g/L K2HPO4, 0.5 g/L MgSO4 · 7 H2O, 0.5 g/L KCl, 0.001 g/L FeSO4 · 7 H2O, 2.0 g/L yeast extract, and 5.0 g/L peptone. The pH was adjusted to 7.3. A modified Czapek medium containing 6.1 g/L of casein hydrolysate (Merck 2239) instead of 5 g/L of peptone at pH 7.2 ± 0.1 was used for T. fusca DSM 43793.
Flasks (1 L) with 250 ml preculture media were inoculated with a suspension of spores and hyphae of the T. fusca species at 50°C for 48 h on a shaker at 100 rpm. The culture was centrifuged at 4°C for 20 min at 4,000 rpm. The pellet was resuspended in the same volume of 50 mM phosphate buffer (pH 8.0) and used for inoculation of the culture medium.
Esterase production
The culture medium was composed of 50 mM phosphate buffer (pH 8.0) containing 5.0 g/L peptone, 2.0 g/L polyester monomers or polymers (PET fibers, PBT fibers, PET granulate, DET, BHET, or terephthalic acid), 12.5 ml/L mineral salt solution, 1.0 ml/L trace element solution, and 1.0 ml/L vitamin solution.
The mineral salt solution contained the following dissolved in demineralized water: 3.0 g/L (NH4)2SO4, 1.0 g/L NaCl, 10.0 g/L NH4Cl, 1.0 g/L MgSO4 · 7 H2O, 0.4 g/L CaCO3, and 6.0 g/L KH2PO4. The trace element solution contained the following dissolved in demineralized water: 0.04 g/L ZnCl2, 0.2 g/L FeCl3 · 6 H2O, 0.01 g/L CuCl2 · 2 H2O, 0.01 g/L MnCl2 · 4 H2O, 0.01 g/L Na2B4O7 · 10 H2O, and 0.01 g/L (NH4)6Mo7O24 · 4 H2O. The vitamin solution contained 6 mg/L biotine, 20 mg/L niacinamide, 20 mg/L p-aminobenzoate, 10 mg/L d-pantothenic acid, 100 mg/L pyridoxal hydrochloride, 50 mg/L cyanocobalamin, 20 mg/L folic acid, 50 mg/L riboflavin, 50 mg/L d,l-6,8-dithiooctanic acid, and 10 mg/L thiamin hydrochloride all dissolved in demineralized water and sterile filtered (pore size 0.45 µm).
Following inoculation, flasks (2 L) containing 500 ml medium were incubated at 50°C for 10 days on a shaker at 150 rpm. Aliquots of the centrifuged supernatants (4,000 rpm, 20 min) from the cultures were removed at time intervals and assayed for esterase activity. After cultivation, the supernatants of the cultures were centrifuged (4,000 rpm, 20 min) and lyophilized.
Comparison of esterases
Samples of supernatants from cultures grown with PET fibers and DET were loaded onto an SDS gel (Lämmli 1970). After separation, the gel was stained with Coomassie Brilliant Blue to visualize protein bands and with Fast Red to detect esterase activity (Traub 2000).
Determination of esterase activity
The esterase activity of TfCa was determined spectrophotometrically using para-nitrophenyl butyrate (p-NPB) as the substrate. Phosphate buffer (100 mM, pH 7.5, 940 μL), p-NPB (10 mM dissolved in ethanol, 10 μL), and enzyme solution (50 μL) were added to a cuvette and mixed. The reaction was monitored at 410 nm for 1 min using a Cary 50 Bio spectrophotometer (Varian Inc., Palo Alto, USA). One unit of esterase activity was defined as the amount of enzyme required to convert 1 µmol of p-NPB per minute (Alisch et al. 2004).
Enzyme purification
The lyophilized crude enzyme preparation was dissolved in demineralized water and dialyzed (Sectra/Por membrane, MWCO 6-8000, Spectrum Laboratories Inc., Raucho Dominguez, USA) against demineralized water for 5–6 h. After dialysis, the enzyme solution was centrifuged for 15 min at 11,000×g.
For chromatographic purification, an Äkta Basic 10 high-performance liquid chromatography (HPLC)-system (GE Healthcare, Uppsala, Sweden) was used. The enzyme solution was loaded onto a Phenylsepharose 6 Fast Flow (high sub) XK 16/20 column (GE Healthcare, Uppsala, Sweden) that was preequilibrated with 20 mM phosphate buffer (pH 8.0). Elution was carried out with a gradient from 0% to 100% of 20 mM phosphate buffer (pH 8.0) containing 30% isopropanol. Fractions containing esterase activity were pooled and the isopropanol was removed under reduced pressure. The pooled fractions were loaded onto a Toyopearl DEAE-650 M XK 16/40 column (Tosoh Bioscience GmbH, Stuttgart, Germany) that was preequilibrated with 20 mM phosphate buffer (pH 8.0) and eluted with a gradient from 0% to 100% of 1 M NaCl in 20 mM phosphate buffer (pH 8.0). Fractions containing esterase activity were pooled and dialyzed against 20 mM phosphate buffer (pH 8.0). The enzyme solution was loaded onto a HiTrap Octyl FF column (GE Healthcare, Uppsala, Sweden) and eluted with a gradient from 0% to 100% of 20 mM phosphate buffer (pH 8.0) containing 30% isopropanol. Fractions containing esterase activity were pooled and the isopropanol was removed under reduced pressure. The pooled fractions were loaded onto a Superdex 200 prep grade XK 16/70 column (GE Healthcare, Uppsala, Sweden) and eluted with 0.15 M NaCl in 20 mM phosphate buffer (pH 8.0).
The protein content was determined by the Bradford method (Bradford 1976). Bovine serum albumin was used as the standard.
SDS-PAGE
Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) was carried out according to the Lämmli protocol (Lämmli 1970). The protein concentration was 3 μg for protein samples from culture supernatants that were analyzed (Fig. 1). For analysis of the proteins from the purification steps, 3.3 μg of protein was loaded in lanes 1 and 2 and 0.3 μg was loaded in lanes 3, 4, and 5 (Fig. 2).

SDS-PAGE of extracellular proteins in the culture fluids of T. fusca DSM 43792, T. fusca DSM 43793, and T. fusca KW3 supplemented with PET fibers and DET. The gels were stained with Coomassie Brilliant Blue R-250 (blue bands) and Fast Red to visualize protein bands and esterase activity (red bands). St molecular mass standard; lane 1, T. fusca DSM 43792 supplemented with PET fibers; lane 2, T. fusca DSM 43792 supplemented with DET; lane 3, T. fusca DSM 43793 supplemented with PET fibers; lane 4, T. fusca DSM 43793 supplemented with DET; lane 5, T. fusca KW3 supplemented with PET fibers; and lane 6, T. fusca KW3 supplemented with DET. A Indicates an esterase with a size of approximately 50 kDa and B indicates an esterase with a size of approximately 30 kDa

SDS-PAGE of TfCa following different purification steps. St molecular mass standard; lane 1, dialyzed extracellular fluid; lane 2, Phenylsepharose 6 Fast Flow; lane 3, Toyopearl DEAE-650 M; lane 4, HiTrap Octyl Fast Flow; and lane 5, Superdex 200 prep grade
Proteins were visualized by Coomassie Brilliant Blue staining. The molecular mass of the enzymes was estimated by comparing their relative mobility to those of standard proteins (Roti®-Mark, Carl Roth GmbH, Karlsruhe, Germany). Esterase activity in the gel was detected by staining with 2-naphthyl acetate and Fast Red (Traub 2000).
Determination of the isoelectric point of TfCa
Isoelectric focusing was performed with a PhastSystem (Amersham Bioscience, Uppsala, Sweden) and PhastGel IEF pH 3.0–9.0 (Amersham Bioscience, Uppsala, Sweden). A 2 μl sample containing approximately 0.03 μg of protein was loaded into each well. The standard proteins used to calculate the pI of the enzyme were amyloglucosidase (pI 3.5), trypsin inhibitor (pI 4.55), β-lactoglobulin A (pI 5.20), bovine carbonic anhydrase B (pI 5.85), human carbonic anhydrase B (pI 6.55), myoglobin-acid band (pI 6.85), myoglobin-base band (pI 7.35), lentil lectin acidic band (pI 8.15), lentil lectin middle band (pI 8.45), lentil lectin basic band (pI 8.65), and trypsinogen (pI 9.3).
Analysis of enzymatic hydrolysis products of cyclic PET trimers
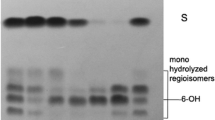
A stock solution of the cyclic PET trimer preparation in dioxane was diluted with 25 mM sodium phosphate buffer (pH 8.0) to a final concentration between 0.025 and 1 mM. Enzyme (0.15 μg) was added to 1 ml of reaction mixture. The incubation was performed at 50°C for 60 min while shaking on an orbital shaker (150 rpm). Aliquots (100 μl) of the reaction mixture were removed at intervals, adjusted to pH 6.0 with 1 M HCl, and analyzed by reversed phase HPLC (Vertommen et al. 2005). A Chromsep Microsorb 300-5 C18 column (250 × 4.6 mm, Varian Inc., Palo Alto, USA) was used with an Äkta Basic 10 chromatography system (GE Healthcare, Uppsala, Sweden). Hydrolysis products were detected at 241 nm. The mobile phase consisted of 0.1% trifluoroacetic acid (Acros Organics, Geel, Belgium, 99% purity) in water and acetonitrile (Roth GmbH, Karlsruhe, Germany, HPLC grade). The determinations were performed in duplicate.
The identity of the detected hydrolysis products was confirmed by reversed phase HPLC/mass spectrometry. An HP 1100 series pump (Agilent Technologies, Santa Clara, USA) and a Gemini column (5 μ C18 110 A, 150 × 2.00 mm, Phenomenex, Aschaffenburg, Germany) were used. Mass spectra were acquired using an ion trap-type instrument (Esquire 3000 plus, Bruker Daltonik GmbH, Bremen, Germany) equipped with an electrospray ion source. The detected mass per charge ratios obtained for each compound matched the following ions: terephthalic acid with m/z = 165 (M−−H)−, 353 (2 M−−2H +Na), 121 (M−−COOH)−; mono(2-hydroxyethyl) terephthalate (MHET) with m/z = 209 (M−−H)−, 441 (2 M−−2H +Na)−, 121 (M−−COO(CH2)2OH)−; BHET with m/z = 253 (M−−H)−, 121 (M−−COO(CH2)2OH −(CH2)2OH)−, and 1,2-ethylene-mono-terephthalate-mono(2-hydroxyethyl terephthalate (EMT) with m/z = 401 (M−−H)−.
Optimal temperature, pH, and stability analysis of TfCa
The optimal temperature for hydrolysis of cyclic PET trimers was determined by incubating TfCa (0.15 μg) with 500 μM of cyclic PET trimers in 25 mM sodium phosphate buffer (pH 8.0) in a total volume of 1 ml reaction mixture. The reaction was performed at 30°C, 40°C, 50°C, 55°C, 60°C, and 70°C for 60 min while shaking on an orbital shaker (150 rpm). The optimal pH for hydrolysis of the cyclic PET trimer preparation was determined by incubating the enzyme (0.15 µg) with 500 µM of cyclic PET trimers in 100 mM buffer (citrate-phosphate-buffer, pH 4.0 to 7.0; and phosphate buffer, pH 8.0) at 60°C. Reactions were stopped after 60 min of incubation and the hydrolysis products were analyzed by reversed phase HPLC as described above. For the determination of the temperature and pH stability of TfCa, the enzyme was incubated at 50°C, 55°C, and 60°C in Davies buffer (Davies 1959) at pH of 7.0, 8.0, and 9.0, respectively. Aliquots were removed at intervals and the residual esterase activity was determined using p-NPB as the substrate. All determinations were performed in triplicate for each temperature and pH value.
Determination of the catalytic properties of TfCa
The Michaelis–Menten constant (K m) and the maximal velocity (V max) for the hydrolysis of the acetate, butyrate, caprylate, and palmitate esters of p-nitrophenol by TfCa were determined. Stock solutions of the esters dissolved in ethanol were diluted with 100 mM sodium phosphate buffer (pH 7.5) to a final concentration between 0.05 and 1 mM. Each 1 ml of reaction mixture contained 0.03 μg of enzyme. The esterase activity with the different p-nitrophenyl esters was determined as described above. The kinetic constants were calculated from Lineweaver–Burk plots. All determinations were performed in triplicate. The K m and V max values for the hydrolysis of cyclic PET trimers by TfCa were determined by quantifying the hydrolysis products separated by reversed phase HPLC. The determinations were performed in duplicate.
Effect of inhibitors and reducing agents on the activity of TfCa
The enzyme was incubated with 1 and 10 mM ethylenediaminetetraacetic acid (EDTA) (Sigma-Aldrich Chemie GmbH, Steinheim, Germany), 0.1, 1, and 10 mM of phenylmethylsulfonyl fluoride (PMSF) dissolved in isopropanol (Acros Acros Organics, Geel, Belgium), and 0.1, 1, and 10 mM of tosyl-l-phenylalanine-chloromethyl ketone (TPCK) dissolved in methanol (Sigma-Aldrich Chemie GmbH, Steinheim, Germany) at 30°C for 15 min in a 25 mM sodium phosphate buffer (pH 8.0). In addition, TfCa was incubated with 0.1 mM, 1 mM, and 10 mM of dithiothreitol (DTT; Sigma-Aldrich Chemie GmbH, Steinheim, Germany) at 30°C for 15 min in a 25 mM sodium phosphate buffer (pH 8.0). The residual esterase activity was determined using p-NPB as the substrate as described above. The determinations were performed in duplicate.
Analysis of enzymatic hydrolysis products of apple cutin
Cutin was isolated from an apple peel according to Fernando et al. (1984). TfCa (0, 1, 2, and 4 U) was added to the cutin preparation (15 mg) suspended in 500 μl of 50 mM phosphate buffer (pH 8.0). The mixture was incubated at 50°C for 64 h on an orbital shaker (150 rpm). The reaction mixture was acidified with glacial acetic acid (500 µl) and liberated cutin monomers were extracted with 3 ml chloroform. The organic phase was removed under a stream of nitrogen. The residue was dissolved in 1 ml of chloroform/methanol (85:15), transferred into glass vials, and dried. Derivatization was performed with bis-(trimethylsilyl) trifluoroacetamide. The methyl esters were dissolved in hexane and analyzed by gas chromatography/mass spectrometry (GC/MS) (Chen et al. 2008). A Finnigan Trace GC ultra GC/MS system, Finnigan MAT 95XP (Thermo Electron Corporation, Bremen, Germany), and a Thermo TR-5MS column (30 × 0.25 mm ID 0.25 µm, C12) (Thermo Electron Corporation, Bremen, Germany) were used.
Amino acid sequencing of TfCa
For N-terminal amino acid sequencing, a sample of purified TfCa was subjected to electrophoresis on a 12.5% SDS-PAGE, transferred to a polyvinylidene difluoride membrane, dried, and stained with Coomassie Brilliant Blue. The protein band was excised and used for direct sequencing by the automated Edman degradation method with a Procise cLC Sequencing System (Model 492cLC, Applied Biosystems, Foster City, USA). The N-terminal amino acid sequence of TfCa (MEIVIRTGSG) was analyzed using the Basic Linear Alignment Tool (BLAST, (http://blast.ncbi.nlm.nih.gov/Blast.cgi) against the protein database of T. fusca YX (Lykidis et al. 2007).
For the determination of the complete amino acid sequence, aliquots of a TfCa preparation were separated by SDS-PAGE, stained with Coomassie Brilliant Blue, and protein bands excised. The enzyme was digested with trypsin and chymotrypsin and the peptides were separated by reversed phase chromatography. The fragments were analyzed by matrix-assisted laser desorption ionization mass spectrometry using a matrix-assisted laser desorption ionization–time-of-flight/time-of-flight (MALDI-TOF/TOF) Ultraflex III system (Bruker Daltonics, Bremen, Germany).
The sequences of TfCa and the putative carboxylesterase Tfu_2427 from T. fusca YX were compared using BLAST. The alignment with sequences from esterases Arth_3983 from Arthrobacter FB24 (accession number A0K238, UniProt knowledgebase), SCO6127 from Streptomyces coelicolor (accession number Q9Z545, UniProt knowledgebase), FRAAL0556 from Frankia alni (accession number Q0RT70, UniProt knowledgebase), and SACE_2933 from Saccharopolyspora erythraea (accession number A4FDT6, UniProt knowledgebase) was performed with BLAST against the Kyoto Encyclopaedia of Genes and Genomes (KEGG) protein database (http://www.genome.jp/kegg/).
DNA sequencing of TfCa
The TfCa gene was sequenced using the Big Dye® Terminator v3.1 Cycle Sequencing Kit and the Abi Prism® 3100 Genetic Analyzer (Applied Biosystems Inc., Foster City, USA). The complete sequencing procedure was repeated twice in both forward and reverse directions using different aliquots of template DNA.
Homology modeling of TfCa and the cutinases from T. fusca YX
Modeling of TfCa from T. fusca KW3 was performed with PHYRE (Bennett-Lovsey et al. 2008) using the thermostable carboxylesterase from G. stearothermophilus (PDB code 2OGS resp. 2OGT) as the template (Liu et al. 2007). A validity check of the final model using VERIFY3D (Eisenberg et al. 1997) resulted in 83% of the amino acids measuring above 0.2 confirming the consistency of the model. The modeled structure of the Tfu_0882 cutinase from T. fusca YX was based on the Streptomyces exfoliates lipase (SeL) and constructed by the SWISS-Model homology modeling web server (Chen et al. 2008; Schwede et al. 2003). Modeling of the Tfu_0883 cutinase from T. fusca YX was performed with PHYRE (Bennett-Lovsey et al. 2008) using the lipase from S. exfoliates (SeL) as the template (Chen et al. 2008; Liu et al. 2007). The sequences of the cutinase from F. solani sp. pisi (Longhi et al. 1997) and the cutinase from P. mendocina (Bott et al. 2003) were obtained from the NCBI protein database (http://www.ncbi.nlm.nih.gov). Modeling of the cutinase from H. insolens was performed using the WURST protein threading server (Sandal et al. 1996; Torda et al. 2004). Images of the modeled structures were constructed with PyMOL (DeLano 2002).
Substitutions of variable amino acids of the active site of TfCa
Site-directed mutagenesis of the active site of TfCa was performed using the QuikChange II Site-Directed Mutagenesis Kit from Stratagene (La Jolla, USA). The following primers were used (amino acid substitutions are underlined): Glu319Asp, forward primer: GCACCACCACCGATGATTATCGTCTGTTTCTGGC; Glu319Asp, reverse primer: GCCAGAAACAGACGATAATCATCGGTGGTGGTGC; Glu184His and Ala186Met, forward primer: GTTACCGTTTTTGGTCATAGCATGGGTGCAATGAGCG; Glu184His and Ala186Met, reverse primer: CGCTCATTGCACCCATGCTATGACCAAAAACGGTAAC; Glu184Gln, forward primer: GTTACCGTTTTTGGTCAGAGCGCAGGTGCAATGAGCG; Glu184Gln, reverse primer: CGCTCATTGCACCTGCGCTCTGACCAAAAACGGTAAC.
The activities of the mutated enzymes were determined spectrophotometrically using p-NPB as the substrate as described above.
Results
Production of esterase activity by T. fusca KW3
T. fusca KW3 was cultured in the presence of PET and PBT fibers, PET granulate, DET, or BHET. The p-NPB-hydrolyzing activity was then measured in the culture supernatant (Table 1). While the highest activity was found in cultures containing DET and PET fibers, no esterase activity was detected following cultivation with terephthalic acid. These results are comparable with those previously obtained with the PET-degrading strain T. fusca DSM 43793 (Kleeberg et al. 2005).
The activities of extracellular esterases from liquid cultures that were produced by T. fusca KW3 (DSM 6013), T. fusca DSM 43793, and T. fusca DSM 43792 were compared after SDS-PAGE protein separation (Fig. 1). An esterase with a molecular mass of approximately 30 kDa was detected in the culture fluids of all three strains supplemented with DET. In contrast, the T. fusca strains produced a second esterase with a molecular mass of approximately 50 kDa in cultures supplemented with PET fibers.
Purification of TfCa
The 50 kDa esterase from T. fusca KW3 was purified to homogeneity from the supernatants of cultures supplemented with PET fibers using ion exchange, hydrophobic interaction, and size exclusion chromatography. After purification, 7% of the enzymatic activity was recovered and determined to be 3,050 U/mg of protein, using p-NPB as the substrate (Table 2). Loss of activity during purification has been reported previously for the PET-degrading cutinase produced by DSM 43793 due to the hydrophobic nature of the enzyme (Kleeberg et al. 2005). When the purified TfCa was subjected to gel electrophoresis under denaturing conditions, a single protein band with a molecular mass of 52.4 kDa was obtained (calculated molecular mass: 52.94 kDa) (Fig. 2). The TfCa exhibited an acidic isoelectric point at 4.8 that was in agreement with the theoretical value of 4.79.
Optimum temperature, pH, and stability of TfCa
The optimal temperature for the enzymatic activity of TfCa was determined to be 60°C using cyclic PET trimers as substrates (Fig. 3a). The optimal pH for cyclic PET trimer hydrolysis at 60°C was found to be pH 6 (Fig. 3b). The half-life of TfCa at 50°C was determined using p-NPB as a substrate and found to be 57 h at pH 8.0, 40 h at pH 7.0, and 19 h at pH 9.0. The residual enzymatic activity was less than 10% when the enzyme was incubated with substrate between pH 7.0 and 9.0 at 60°C for 1 h (Fig. 4).

Optimal temperature (A) and pH (B) of the TfCa using cyclic PET trimers as substrates

Stability of TfCa at different temperatures and pH values determined using p-NPB as the substrate ( pH 7 and 50°C,
pH 7 and 50°C,  pH 7 and 55°C,
pH 7 and 55°C,  pH 7 and 60°C,
pH 7 and 60°C,  pH 8 and 50°C,
pH 8 and 50°C,  pH 8 and 55°C,
pH 8 and 55°C,  pH 8 and 60°C,
pH 8 and 60°C,  pH 9 and 50°C,
pH 9 and 50°C,  pH 9 and 55°C, and
pH 9 and 55°C, and  pH 9 and 60°C)
pH 9 and 60°C)
Enzymatic properties of TfCa
TfCa was assayed for its ability to hydrolyze PET by incubation with cyclic PET trimers. The enzyme hydrolyzed the PET trimers with a Michaelis–Menten constant (K m) of 0.5 mM and a maximal velocity (V max) of 9.3 µmol/min/mg. The main hydrolysis product formed (95% of total products) was EMT. Other products detected by reversed phase HPLC were MHET (3%) and BHET (2%). From a 1 mM solution of cyclic PET trimers, 45 µM of hydrolyzed products were released within 1 h of incubation with 0.53 U/ml TfCa.
Analysis of the substrate specificity of TfCa with p-nitrophenyl esters of different acyl chain lengths showed that the enzyme cleaved p-nitrophenyl esters with short and medium acyl chain lengths (C2 to C8) but did not cleave long chain acyl esters (C16) (Table 3).
Incubation of TfCa with PMSF at concentrations of 1 mM and 10 mM resulted in a 31% and 65% loss of activity, respectively (Table 4). Incubation of the enzyme with TPCK at concentrations of 1 and 10 mM also resulted in an 81% and 100% loss of activity, respectively. Treatment with the disulphide bond-reducing compound dithiothreitol at concentrations of 1 and 10 mM did not result in inactivation of the enzyme. Similarly, TfCa was not inhibited in the presence of EDTA at concentrations of 1 and 10 mM, indicating that the enzyme does not require metal ions for its activity.
The ability of TfCa to hydrolyze the plant polyester cutin was determined using apple cutin as the substrate. However, none of the characteristic cutin monomers (10,16-dihydroxyhexadecanoic acid, 18-hydroxyoctadeca-9-enoic acid, 18-hydroxyoctadeca-9,12-dienoic acid, or 9,10,18-trihydroxyoctadecanoic acid) could be detected by gas chromatographic/mass spectrometric analysis of the reaction products (Chen et al. 2008; Fett et al. 1992). The properties of TfCa compared with T. fusca cutinases (Chen et al. 2008; Kleeberg et al. 2005) are summarized in Table 5.
Amino acid composition and sequence comparison
The amino acid compositions of TfCa and of the two cutinases from T. fusca YX (Lykidis et al. 2007) were compared (Electronic supplementary material, Online resource 1). Ala, Gly, Pro, and Val were the most abundant amino acids found in TfCa, while Ala, Gly, and Val were the predominant amino acids in the T. fusca YX cutinases. TfCa was composed of 11.9% acidic and 9.3% basic amino acid residues compared with 8.4–10.3% acidic and 10–12.3% basic amino acid residues in the T. fusca YX cutinases, respectively. N-terminal sequencing analysis determined that the first 10 amino acids of TfCa were 100% identical to those of the putative carboxylesterase Tfu_2427 from T. fusca YX (Lykidis et al. 2007).
The complete amino acid sequence of TfCa was determined by MALDI-TOF/TOF analysis and verified by DNA sequencing. A comparison with the amino acid sequence of Tfu_2427 indicated 93.5% identity between the two enzymes. However, when the DNA sequences of TfCa and Tfu_2427 were compared, only two amino acid differences were detected, resulting in 99.6% identity (Electronic supplementary material, Online resource 2). Post-translational modifications of side chains or signal sequences were not detected by MALDI-TOF/TOF analysis and DNA sequencing. An alignment with the amino acid sequences of other carboxylesterases showed 42% identity with the carboxylesterase Arth_3983 from Arthrobacter FB24, 42% similarity with the carboxylesterase SCO6127 from S. coelicolor, 43% similarity with the putative carboxylesterase FRAAL0556 from F. alni, and 42% similarity with a putative p-nitrobenzyl esterase SACE_2933 from S. erythraea (Electronic supplementary material, Online resource 3).
Homology modeling of TfCa
To further compare TfCa with other hydrolases that have a reported ability to degrade PET, a homology model was constructed (Fig. 5) (Bennett-Lovsey et al. 2008). The thermostable carboxylesterase from Geobacillus stearothermophilus that has a 40% sequence similarity to TfCa was used as a template (Liu et al. 2007). The consistency of the chosen model was confirmed by a validity check (Eisenberg et al. 1997).

Homology modeling of TfCa from T. fusca KW3 compared with PET-hydrolyzing cutinases. Ribbon diagram of the modeled structure of a TfCa, b F. solani sp. pisi (Longhi et al. 1997), c H. insolens (Sandal et al. 1996), d cutinases Tfu_0882 from T. fusca YX (Lykidis et al. 2007; Chen et al. 2008), e Tfu_0883 from T. fusca YX (Lykidis et al. 2007; Chen et al. 2008), and f P. mendocina (Bott et al. 2003)
Substitution of variable amino acids in the active site of TfCa
The active sites and variable amino acids of the G-X-S-X-G motif of six carboxylesterases and cutinases are shown in Fig. 6. A comparison of the specific activities of the wild-type enzyme and enzyme variants of TfCa where the Glu of the active site and Glu and Ala of the G-E-S-A-G motif were substituted with Asp, His, and Met is shown in Fig. 7. Substitution of Glu184Gln, one variable amino acid of the serine motif, resulted in an 84% decrease in the specific activity compared with the wild-type enzyme. Substitution of Glu319Asp in the acid position of the active site resulted in a 75% decrease in the specific activity. Combination of both substitutions (E184Q and E319D) resulted in an 80% decrease of its specific activity compared with wild-type TfCa. A mutant TfCa carrying a double mutation (E184H/A186M) retained only 5% specific activity compared with wild-type TfCa. Introduction of a third substitution at the acid position (Glu to Asp) of the catalytic triad created a mutant (E184H/A186M/E319D). The specific activity of this mutant decreased by 88% compared with the wild-type TfCa.

Active site and variable amino acids of the G-X-S-X-G motif of carboxylesterases and cutinases

Specific activities of the wild-type enzyme and substitution mutants of TfCa
Discussion
The thermophilic actinomycete T. fusca produces a carboxylesterase (TfCa) with a molecular mass of 50 kDa when cultivated in minimal media supplemented with PET fibers. Previously reported enzymes able to degrade synthetic aromatic polyesters such as PET were cutinases or lipases isolated from various fungi and bacteria including T. fusca (Gübitz and Cavaco-Paulo 2003; Vertommen et al. 2005; Müller et al. 2005; Chen et al. 2008; Ronkvist et al. 2009). Cultivation of T. fusca in minimal media supplemented with DET indeed showed the production of an esterase with a molecular mass of 30 kDa (Fig. 1). This enzyme corresponded to the previously described cutinase TfH (Tfu_0883) produced by T. fusca (Chen et al. 2008; Kleeberg et al. 2005).
TfCa hydrolyzed cyclic PET trimers composed of three molecules of terephthalic acid and ethylene glycol linked by ester bonds. The degradation of cyclic PET and poly(trimethylene terephthalate) oligomers by cutinases has been previously reported (Hooker et al. 2003; Figueroa et al. 2006; Riegels et al. 2001; Eberl et al. 2008). A cutinase from Humicola insolens was shown to hydrolyze cyclic PET trimers to terephthalic acid, MHET, and BHET (Riegels et al. 2001). In addition, an unidentified cutinase completely hydrolyzed cyclic PET trimers within 24 h and predominantly formed terephthalic acid and MHET (Hooker et al. 2003). The cutinase from T. fusca DSM 43793 hydrolyzed cyclic poly(trimethylene terephthalate) dimers and degraded bis(3-hydroxypropyl) terephthalate first to mono(3-hydroxypropyl) terephthalate and subsequently to terephthalic acid (Eberl et al. 2008).
TfCa showed hydrolytic activity against p-nitrophenyl esters with short and medium acyl chain lengths (C2 to C8) but did not cleave long chain acyl esters (C16) (Table 3). Thereby, TfCa exhibited the typical substrate specificity of a carboxylesterase. In contrast, the cutinase from T. fusca DSM 43793 showed the highest enzymatic activities with C2 to C6 acyl chain length p-nitrophenyl esters and C8, C10, C12, and C18 acyl chain esters. Thus, cutinases have properties of both esterases and lipases (Kleeberg et al. 2005). While cutinases readily hydrolyze the plant polyester cutin, we showed that TfCa did not exhibit any cutinolytic activity. These results indicate that TfCa is clearly distinct from the previously described fungal and bacterial hydrolases that were able to degrade PET (Chen et al. 2008; Kleeberg et al. 2005; Purdy and Kolattukudy 1975).
TfCa was inactivated by PMSF, an inhibitor that irreversibly binds to serine at the active site of serine esterases and by TPCK, an inhibitor that specifically binds to histidine at the active site of serine proteases. These results indicate an essential role of these amino acids in the catalytic mechanism of TfCa.
Substitution mutagenesis experiments at the active site of TfCa showed that a substitution of Glu184Gln resulted in an strong decrease in the specific activity. TfCa of T. fusca and the putative carboxylesterase FRAAL0556 both display Glu at this position while the carboxylesterases SCO6127, SACE_2933 and Arth_3983 contain Gln (Electronic supplementary material, Online resource 2, Fig. 6). The decrease in activity as a result of the substitution indicates an important role of the motif amino acids on the enzyme activity, though they are not directly involved in the binding of the substrate. Substitution of Glu319Asp in the acid position of the active site resulted in a 75% decrease in the specific activity and the combination of both substitutions (E184Q and E319D) resulted in an 80% decrease of its specific activity. The mutant E184Q/E319D had the same active site and serine motif as the carboxylesterase Arth_3983. A TfCa mutant with another double mutation (E184H/A186M), comparable to the cutinases from T. fusca, retained only 5% specific activity compared with wild-type TfCa. This severe loss of activity further confirmed the important role of the variable amino acids of the serine motif in maintaining the catalytic activity of TfCa. A third substitution (E184H/A186M/E319D) yielding the same catalytic triad and serine motif as the cutinases from T. fusca also showed a strongly decreased specific activity. However, the additional substitution resulted in an increase of the activity compared with the E184H/A186M mutant, suggesting a stabilizing effect of the E319D substitution.
The catalytic center of serine hydrolases typically contains a highly conserved G-X-S-X-G sequence around the serine that was also present in the G-E-S-A-G sequence of TfCa (Bornscheuer 2002). The putative carboxylesterase FRAAL0556 from F. alni showed the same consensus sequence while the other esterases contained the G-Q-S-A-G consensus sequence. These results confirmed the differences found between the TfCa and the T. fusca cutinases and showed high similarity between TfCa and the carboxylesterase Tfu_2427 from T. fusca YX. The enzyme from Arthrobacter FB24 differed from the other aligned esterases with the presence of Asp instead of Glu in the catalytic triad, indicating that both amino acids can occur in the catalytic triad of these carboxylesterases.
The modeled TfCa protein structure showed similar features when compared with other PET-hydrolyzing bacterial and fungal cutinases (Fig. 5) (Chen et al. 2008; Longhi et al. 1997; Sandal et al. 1996; Bott et al. 2003). The cutinases display their active site on the surface of the enzyme without the mobile lid structure that is often found in lipases (Arpigny and Jäger 1999; Chahinian et al. 2005; Bornscheuer 2002; Jäger et al. 1994). Likewise, the TfCa did not show a lid structure. The results further indicate that the catalytic triad of TfCa is composed of Ser185, Glu319, and His415 also reported in other lipases. However, the typical catalytic triad of carboxylesterases is S-D-H with the consensus sequence G-X-S-X-G (Bornscheuer 2002). TfCa exhibited the motif G-E-S-A-G with an acidic and aliphatic amino acid residue, whereas both cutinases from T. fusca DSM 43793 (Kleeberg et al. 2005) and T. fusca YX (Chen et al. 2008) showed the G-H-S-M-G motif containing a basic and an aliphatic amino acid residue. As a consequence, the area around the nucleophilic serine of TfCa appeared to be more acidic compared with the cutinases. Instead of Glu in the catalytic triad of TfCa, the cutinases contain Asp. The catalytic triad of the T. fusca cutinases is composed of Ser132, Asp176, and His210 while the fungal cutinases from F. solani f. sp. pisi and H. insolens show an aromatic neutral G-Y-S-Q-G motif with Ser120, Asp175, His188 and Ser140, Asp195, His208, respectively (Carvalho et al. 1998; Sandal et al. 1996). The cutinase from P. mendocina shows a G-H-S-Q-G motif and has Ser126, Asp176, and His206 in the catalytic triad (Bott et al. 2003).
TfCa from T. fusca KW3 shares similar properties with cutinase enzymes with respect to the lack of a lid structure and their ability to hydrolyze PET substrates. The active site of TfCa appears to be located in the middle part of the enzyme and forms a pocket-like structure. In spite of these similarities with cutinases, TfCa was not able to degrade the biopolymer cutin, though it showed strong activity with hydrophobic, insoluble, synthetic aromatic polyester oligomers.
References
Alisch M, Feuerhack A, Müller H, Mensak B, Andreaus J, Zimmermann W (2004) Biocatalytic modification of polyethylene terephthalate fibres esterases from actinomycete isolates. Biocatal Biotransform 22:347–351
Alisch-Mark M, Herrmann A, Zimmermann W (2006) Increase of the hydrophilicity of polyethylene terephthalate fibres by hydrolases from Thermomonospora fusca and Fusarium solani f. sp pisi. Biotechnol Lett 28:681–685
Arpigny JL, Jäger K-E (1999) Bacterial lipolytic enzymes: classification and properties. Biochem J 343:177–183
Bennett-Lovsey RM, Herbert AD, Sternberg MJ, Kelley LA (2008) Exploring the extremes of sequence/structure space with ensemble fold recognition in the program Phyre. Proteins 70:611–625
Bornscheuer UT (2002) Microbial carboxyl esterases: classification, properties and application in biocatalysis. FEMS Microbiol Rev 26:73–81
Bott R, Kellis JT, Morrison TB (2003) High throughput mutagenesis screening method. WO Patent 03/076580 A2
Bradford MM (1976) A rapid and sensitive method for the quantization of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Brückner T, Eberl A, Heumann S, Rabe M, Gübitz GM (2008) Enzymatic and chemical hydrolysis of poly(ethylene terephthalate) fabrics. J Polym Sci, Part A, Polym Chem 46:6435–6443
Carvalho CM, Aires-Barros MR, Cabral JMS (1998) Cutinase structure, function and biocatalytic applications. Electron J Biotechnol 1:160–173
Chahinian H, Ali YB, Abousalham A, Petry S, Mandrich L, Manco G, Canaan S, Sarda L (2005) Substrate specificity and kinetic properties of enzymes belonging to the hormone-sensitive lipase family: comparison with non-lipolytic and lipolytic carboxylesterases. Biochim Biophys Acta 1738:29–36
Chen S, Tong X, Woodard RW, Du G, Wu J, Chen J (2008) Identification and characterization of bacterial cutinase. J Biol Chem 283:25854–25862
Davies MT (1959) A universal buffer solution for use in ultra-violet spectrophotometry. The Analyst 84:248–251
DeLano WL (2002) The PyMOL molecular graphics system. DeLano Scientific, Palo Alto
Eberl A, Heumann S, Kotek R, Kaufmann F, Mitsche S, Cavaco-Paulo A, Gübitz GM (2008) Enzymatic hydrolysis of PTT polymers and oligomers. J Biotechnol 135:45–51
Eisenberg D, Lüthy R, Bowie JU (1997) VERIFY3D: assessment of protein models with three-dimensional profiles. Meth Enzymol 277:396–404
Fernando G, Zimmermann W, Kolattukudy PE (1984) Suberin-grown Fusarium solani f. sp. pisi generates a cutinase-like esterase which depolymerizes the aliphatic components of suberin. Physiol Plant Pathol 24:143–155
Fett WF, Gerard RA, Osman SF (1992) Screening of nonfilamentous bacteria for production of cutin-degrading enzymes. Appl Environ Microbiol 58:2123–2130
Fett WF, Wijey C, Moreau RA, Osman SF (1999) Production of cutinase by Thermomonospora fusca ATCC 27730. J Appl Microbiol 86:561–568
Feuerhack A, Alisch-Mark M, Kisner A, Pezzin SH, Zimmermann W, Andreaus J (2008) Biocatalytic surface modification of knitted fabrics made of poly (ethylene terephthalate) with hydrolytic enzymes from Thermobifida fusca KW3b. Biocatal Biotransform 26:357–364
Figueroa Y, Hinks D, Montero GA (2006) A heterogeneous kinetic model for the cutinase-catalyzed hydrolysis of cyclo-tris-ethylene terephthalate. Biotechnol Prog 22:1209–1214
Gübitz GM, Cavaco-Paulo A (2003) New substrates for reliable enzymes: enzymatic modification of polymers. Curr Opin Biotechnol 14:577–582
Heumann S, Eberl A, Pobeheim H, Liebminger S, Fischer-Colbrie G, Almansa E, Cavaco-Paulo A, Gübitz GM (2006) New model substrates for enzymes hydrolyzing polyethyleneterephthalate and polyamide fibres. J Biochem Biophys Meth 39:89–99
Hooker J, Hinks D, Montero GA, Icherenska M (2003) Enzyme catalyzed hydrolysis of poly(ethylene terephthalate) cyclic trimer. J Appl Polym Sci 89:2545–2552
Jäger K-E, Ransac S, Dijkstra BW, Colson C, van Heuvel M, Misset O (1994) Bacterial lipases. FEMS Microbiol Rev 15:29–63
Jäger K-E, Dijkstra BW, Reetz MT (1999) Bacterial biocatalysts: molecular biology, three-dimensional structures, and biotechnological applications of lipases. Annu Rev Microbiol 53:315–351
Kleeberg I, Welzel K, VandenHeuvel J, Müller R-J, Deckwer W-D (2005) Characterization of a new extracellular hydrolase from Thermobifida fusca degrading aliphatic-aromatic copolyester. Biomacromolecules 6:262–270
Lämmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nat 227:680–685
Liebminger S, Eberl A, Fischer-Colbrie G, Heumann S, Gübitz GM (2007) Hydrolysis of PET and bis (benzoyloxyethyl) terephthalate with a new polyesterase from Penicillium citrinum. Biocat Biotrans 25:171–177
Liu P, Ewis HE, Tai PC, Lu CD, Weber IT (2007) Crystal structure of the Geobacillus stearothermophilus carboxylesterase Est55 and its activation of prodrug CPT-11. J Mol Biol 367:212–223
Longhi S, Mannesse M, Verheij HM, Haas GH, Egmond M, Knoops-Mouthuy E, Cambillau C (1997) Crystal structure of cutinase covalently inhibited by a triglyceride analogue. Protein Sci 6:275–286
Lykidis A, Mavromatis K, Ivanova I, Anderson I, Land M, DiBartolo G, Martinez M, Lapidus A, Lucas S, Copeland A, Richardson P, Wilson DB, Kyrpides N (2007) Genome sequence and analysis of the soil cellulolytic actinomycete Thermobifida fusca YX. J Bacteriol 189:2477–2486
Müller R-J, Kleeberg I, Deckwer W-D (2001) Biodegradation of polyesters containing aromatic constituents. J Biotechnol 86:87–95
Müller R-J, Schrader H, Profe J, Dresler K, Deckwer W-D (2005) Enzymatic degradation of poly(ethylene terephthalate): rapid hydrolyse using a hydrolase from T. fusca. Macromol Rapid Commun 26:1400–1405
Nimchua T, Punnapayak H, Zimmermann W (2007) Comparison of the hydrolysis of polyethylene terephthalate fibers by a hydrolase from Fusarium oxysporum LCH I and Fusarium solani f. sp. pisi. Biotechnol J 2:361–364
Oeser T, Wei R, Baumgarten T, Billig S, Föllner C, Zimmermann W (2010) High level expression of a hydrophobic poly(ethylene terephthalate)-hydrolyzing carboxylesterase from Thermobifida fusca KW3 in Escherichia coli BL21(DE3). J Biotech 146:100–104
Phithakrotchanakoon C, Daduang R, Thamchaipenet A, Wangkam T, Srikhirin T, Eurwilaichitr L, Champreda V (2009) Heterologous expression of polyhydroxyalkanoatedepolymerase from Thermobifida sp. in Pichia pastoris and catalytic analysis by surface plasmon resonance. Appl Microbiol Biotechnol 82:131–140
Purdy RE, Kolattukudy PE (1975) Hydrolysis of plant cutin by plant pathogens. Purification, amino acids composition, and molecular weight of two isoenzymes of cutinase and a nonspecific esterase from Fusarium solani f. pisi. Biochemistry 14:2824–2831
Riegels M, Kock R, Pedersen LS, Lund H (2001) Enzymatic hydrolysis of cyclic oligomers. US Patent 6(184):010
Ronkvist ÅM, Xie W, Lu W, Gross RA (2009) Cutinase-catalyzed hydrolysis of poly(ethylene terephthalate). Macromol 42:5128–5138
Sandal T, Kauppinen S, Kofod LV (1996) An enzyme with lipolytic activity. WO Patent 96/13580
Schwede T, Kopp J, Guex N, Peitsch MC (2003) SWISS-MODEL: an automated protein homology-modeling server. Nucleic Acids Res 31:3381–3385
Torda AE, Procter JB, Huber T (2004) Wurst: a protein threading server with a structural scoring function, sequence profiles and optimized substitution matrices. Nucleic Acids Res 32:W532–W535
Traub PC (2000) Gensynthese, Expression und Refolding der Lipasen aus Pseudomonas species KWI 56 und Chromobacterium viscosum. Dissertation, Universität Stuttgart, Germany
Vertommen MAME, Nierstrasz VA, van der Veer M, Warmoeskerken MMCG (2005) Enzymatic surface modification of poly(ethylene terephthalate). J Biotechnol 120:376–386
Yang C-H, Liu W-H (2008) Purification and properties of an acetylxylan esterase from Thermobifida fusca. Enzyme Microb Technol 42:181–186
Yoon MY, Kellis JT, Poulouse AJ (2002) Enzymatic modification of polyester. AATCC Rev 2:33–36
Zhang Z, Wang Y, Ruan J (1998) Reclassification of Thermomonospora and Microtetraspora. Int J Syst Bacteriol 48:411–422
Acknowledgement
S. Billig was supported by grant no. 20004/730, Deutsche Bundesstiftung Umwelt and T. Oeser by grant no. 13-8811.61/215-1, Sächsisches Staatsministerium für Umwelt und Landwirtschaft. C. Roth from the University of Leipzig is acknowledged for his assistance in the pI determination. I. Neundorf and J. Stichel from the University of Leipzig are acknowledged for the amino acid sequence determination and MALDI-TOF/TOF analysis.
Author information
Authors and Affiliations
Corresponding author
Additional information
Nucleotide sequence data reported in this paper will appear in the EMBL database under the accession number FN401519 and the protein sequence data in the UniProt knowledgebase under the accession number P86325.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Online resource 1
Amino acid composition of TfCa and the cutinases Tfu_0882 and Tfu_0883 from T. fusca YX (PDF 6 kb)
Online resource 2
Comparison of the amino acid sequence of TfCa from T. fusca KW3 with the putative carboxylesterase Tfu_2427 from T. fusca YX. The proteins differ in two amino acids (marked in grey and bold). Tfu_2427 has a glycine residue instead of a serine at position 335 and a threonine residue instead of an alanine at position 340. The identity of the two proteins was 99.6%. Amino acid residues of the catalytic triad are underlined and marked in bold (PDF 63 kb)
Online resource 3
Alignment of amino acid sequences of carboxylesterases (PDF 1748 kb)
Rights and permissions
About this article
Cite this article
Billig, S., Oeser, T., Birkemeyer, C. et al. Hydrolysis of cyclic poly(ethylene terephthalate) trimers by a carboxylesterase from Thermobifida fusca KW3. Appl Microbiol Biotechnol 87, 1753–1764 (2010). https://doi.org/10.1007/s00253-010-2635-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-010-2635-y