
Abstract
Shot-wise supplementation of cysteine to a yeast culture is a common means of promoting glutathione (GSH) production. In the present work, we study the accumulation kinetics of cysteine, γ-glutamylcysteine, and GSH and the expression of genes involved in GSH and sulfur metabolism in ethanol-stat fed-batch cultures as a result of switching to a medium enriched with cysteine and glycine. Supplementation in this fashion resulted in a rapid but short-term increase in the rate of GSH synthesis, while the expression of GSH1 decreased. Expression of GSH1 and GSH synthesis rate were observed to revert close to the base level after a few hours. These results indicate that, under such conditions, the control of GSH synthesis at higher concentrations occurred at the enzymatic, rather than the transcriptional level. The incorporation of cysteine into GSH was limited to ∼40% of the theoretical yield, due to its requirement as a source of sulfur for protein synthesis under conditions whereby the sulfate assimilation pathway is down-regulated. This was supported by the expression profiles of genes involved in cysteine and homocysteine interconversion.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The non-protein thiol glutathione (GSH) is a physiologically important tripeptide, which is synthesized in most eukaryotes (Meister and Anderson 1983; Pócsi et al. 2004) and in several prokaryotes (Fahey et al. 1978). Under standard conditions, GSH is required for protein and DNA synthesis and amino acid transport (Douglas 1987). The role of GSH under adverse conditions is primarily related to the defense of cells in response to a variety of environmental challenges, such as oxidative stress, toxification with heavy metals and xenobiotics, as well as nutrient depletion (Grant and Dawes 1996; Penninckx 2000; Soltaninassab et al. 2000; Grant 2001). In addition to its physiological role, GSH is also, due its antioxidative properties, widely used as a pharmaceutical compound and also has the potential to be used by the cosmetic and food industries (Sies 1999). Consequently, there is an increasing demand for the production of this tripeptide on an industrial scale. Although GSH can be produced by chemical or enzymatic synthesis, microbiological production using the yeasts Saccharomyces cerevisiae and Candida utilis is currently the most common method for the commercial production of GSH (Murata and Kimura 1990; Li et al. 2004).
The concentration of GSH in wild-type S. cerevisiae strains has been reported to occur within the range of 0.1–1.0% of the dry weight (dwt) of cells (Li et al. 2004). This variation can be caused by several factors, such as growth conditions and the characteristics of individual strains. GSH is synthesized in two sequential ATP-dependent reactions, catalyzed firstly by γ-glutamylcysteine (γGCS) synthetase (Gsh1p) and secondly by GSH synthetase (Gsh2p). Analysis of GSH biosynthesis in different eukaryotes has suggested that cellular levels are controlled at both the transcriptional and post-translational levels (Richman and Meister 1975; Soltaninassab et al. 2000; Wheeler et al. 2002; Pócsi et al. 2004) and that the first reaction, catalyzed by Gsh1p, is the rate-limiting step (Huang et al. 1988; Meister 1988; Murata and Kimura 1990; Grant et al. 1997). Post-translational regulation is a non-allosteric feedback inhibition of Gsh1p activity by GSH (Soltaninassab et al. 2000). At the transcriptional level, the expression of GSH1 is regulated by coupling of Met4p, a transcription factor which governs the MET gene network in the biosynthesis of the sulfur-containing amino acids (Thomas and Surdin-Kerjan 1997) and a redox-sensitive Yap1p transcription factor (Wheeler et al. 2002, 2003). It has been suggested that Met4p could be regulated in response to the availability of methionine and GSH (Wheeler et al. 2003). More recently, Menant et al. (2006) proposed that the intracellular cysteine, and not GSH or methionine, is responsible for Met4p regulation. In addition to transcriptional and post-translational regulation of the Gsh1p reaction step, cysteine has been shown to be the major limiting precursor for GSH accumulation (Alfafara et al. 1992a; Wen et al. 2004) and thus, the regulation of sulfur assimilation pathways also becomes important from the perspective of cysteine biosynthesis.
Efficient GSH production using yeast cells relies on high-density cultivation of biomass with maximum possible GSH content at the point of harvesting. Different cultivation conditions for increasing the intracellular GSH concentration using fed-batch cultures have been studied (Alfafara et al. 1992b; Sakato and Tanaka 1992; Wen et al. 2006; Zhang et al. 2007; Wang et al. 2007; Liang et al. 2008a, b). With few exceptions, these works rely on the addition of precursor amino acids of GSH, particularly cysteine, to the yeast culture. However, the primary aim of these studies was to determine the optimum conditions required to maximize the GSH content in yeast and not to investigate the metabolic changes resulting from increased cysteine concentration in cells. To the best of our knowledge, the kinetics of GSH accumulation under conditions of increased cysteine concentration in S. cerevisiae has not been studied to date. The aim of the present work is to describe the physiological capacity of baker's yeast to promote GSH synthesis in high biomass density fed-batch cultures with continuous cysteine and glycine addition.
Materials and methods
Yeast strain and cultivation media composition
A commercial S. cerevisiae baker's yeast (strain #7048, Lallemand Yeast Culture Collection, Lallemand, Inc.), kindly provided by AS Salutaguse Pärmitehas (Tallinn, Estonia) was used in all experiments. Fed-batch cultivations were performed using mineral media. The feeding media composition, described by van Hoek et al. (2000) and Walker (2004) was used with some modifications. Starting medium (1.9 L) comprised 17 g L-1 KH2PO4, 6 g L-1 MgSO4·7H2O and 7 g L-1 NH4Cl. The basic feeding medium contained 400 g L-1 sucrose, 30 mg L-1 CaCl2·2H2O, 150 mg L-1 ZnSO4·7H2O, 150 mg L-1 FeSO4·7H2O, 50 mg L-1 CuSO4·5H2O, 40 mg L-1 MnSO4·H2O, 10 mg L-1 CoSO4·7H2O, 2 mg L-1 (NH4)6Mo7O24·4H2O, 250 mg L-1 myo-inositol, 10 mg L-1 thiamine, 10 mg L-1 Ca-pantothenate, 10 mg L-1 pyridoxine, 10 mg L-1 nicotinamide, and 5 mg L-1 biotin. Microelements and vitamins were sterilized separately by microfiltration and added aseptically to the autoclaved feeding medium prior to experimentation. The pH of the feeding medium was adjusted to pH 4.6, using 1 N H2SO4. Precursor amino acids (cysteine and glycine) containing feeding media (56, 112, and 225 μmol of each per gram of sucrose) were prepared by dissolving the amino acids in deoxygenated (flushed with N2 for at least 20 min) basic feeding medium. Notably, glycine was included in equimolar concentrations with cysteine to prevent its possible limitation and any effects of this possible limitation on the experimental results.
Cultivation system and cultivation process routines
A series of fed-batch cultivations using different cysteine and glycine concentrations in the feeding (exp. 1, exp. 2, and exp. 3) and a reference experiment without cysteine addition were carried out in a 7 L “BioBench” fermenter (Applikon, The Netherlands), equipped with pO2, pH, O2, CO2, and temperature sensors. Ethanol concentration was measured online using a “Chemgard” infrared gas monitor (Mine Safety Appliances, USA). Cultivation was controlled using an ADI 1010 bio-controller (Applikon, The Netherlands) and cultivation control software “BioXpert” (Applikon, The Netherlands). Variable speed pumps (Cole Parmer, USA) were used to supply the feeding media with or without the added cysteine and glycine. The feeding media vessels were each placed on a balance (Sartorius, Germany) to precisely determine the feeding rates of substrate and precursor amino acids. The pH control (pHset-point = 4.6) was carried out by titration with 1 M NH4OH, which also served as the nitrogen source. The dO2 was controlled at min 20% of air saturation by adjusting the ratio of air and oxygen in the aeration gas mixture (aeration rate 10 L min-1), while the reactor was stirred continuously at 800 rpm. One hundred milliliters of seed cream yeast (∼20 g dwt) was inoculated into 1.9 L of starting medium. An in-house developed adaptive proportional–integral algorithm for controlling the ethanol concentration in the exhaust gas at a set-point of 40 ppm (corresponding to a concentration of 0.2 g L-1 in the culture medium) by adjusting the feeding rate was used to grow the cells at near-maximum respiratory growth rate μ ≈ μ crit (ethanol-stat). In exp. 1, exp. 2, and exp. 3, the culture was grown overnight in fed-batch, and a shift from the basic feeding medium to one containing cysteine and glycine was carried out at a biomass concentration of ∼40 g dwt L-1, followed by cultivation a further 5 h as described above.
Analytical methods
Culture samples (6–7 mL) were collected and rapidly transferred into 15-mL tubes maintained on ice; 1-mL aliquots were distributed into six Eppendorf microcentrifuge tubes and centrifuged at 14,000 rpm for 2 min to wash and recover the biomass. The biomass was washed once with ice-cold RNase-free [dimethyl pyrocarbonate-treated] water. Total processing of the samples did not take more than 5 min. The washed biomass samples and the culture supernatant were frozen in liquid nitrogen and stored at −80°C until required. All measurements were performed in duplicate.
Biomass concentration was determined by weighing the cell pellets after drying in the oven at 105°C for 24 h. Intracellular thiols were extracted using 0.1 N formic acid (70°C, 1 h). Cell debris was removed by centrifugation, and the supernatant was derivatized with 5,5’-dithiobis(2-nitrobenzoic acid) (DTNB). An ACQUITY UPLC system (Waters, USA), equipped with a C18 column (BEH C18, 100 × 2.1 mm, 1.7 μm, Waters, USA) and a photo diode array (PDA) detector ACQUITY PDA 2996 was used for the chromatographic determination of GSH, γGCS, and cysteine. A two-gradient mobile phase (A: water + 0.1% formic acid and B: acetonitrile + 0.1% formic acid) and a flow rate of 0.3 mL min-1 was used. Concentration of cysteine in the cultivation medium was expressed as the sum of the concentration of reduced and oxidized cysteine, determined simultaneously by the two UPLCs using derivatization with DTNB and an AccQFluor kit (Waters, USA), respectively. The standards (cysteine, γGCS, and GSH) used for external calibration were obtained from Sigma (USA). Concentration of hydrogen sulfide (H2S) in the culture medium was determined as described by Moest (1975).
Expression of nine genes (GSH1, GSH2, ECM38, SUL1, MET17, CYS3, CYS4, STR2, and STR3; Fig. 1) was monitored by a solution hybridization method termed “Transcript Analysis with the aid of Affinity Capture” (TRAC) using a pool of oligonucleotide probes designed for the respective target genes, separable and quantifiable by capillary electrophoresis (Rautio et al. 2006, 2007). Yeast samples were prepared for the TRAC analysis by adding 25 to 50 mg (dwt) of harvested yeast to 500 µL of lysis buffer (PlexPress, Finland) to give a final concentration of 120–240 µg µL-1. The yeast cells were disrupted twice with 500 μL acid-washed glass beads (Sigma) in a FastPrep cell homogenizer (ThermoSavant, Germany), using 6 m/s for 45 s. Lysed yeast samples (120–240 µg dwt/reaction) were added to hybridization buffer (PlexPress, Finland) containing target-specific probes and control ssDNA (PlexPress sequence ID 418) strand as an internal hybridization control. Affinity capture of hybrids, washing, and elution were carried out in 96-well plates using a magnetic bead particle processor KingFisher Flex (Thermo Electron, Finland) as follows: (1) affinity capture of hybridized RNA targets to 50 μg of streptavidin-coated TRACPACK® Beads (PlexPress, Finland) for 30 min at room temperature; (2) washing of the beads five times for 1 min in 150 μL of wash buffer (PlexPress, Finland) at room temperature; (3) elution of probes with 10 μL formamide (Applied Biosystems, USA) for 20 min at 37°C. The eluates were analyzed by capillary electrophoresis using an ABI PRISM 3100 Genetic Analyser (Applied Biosystems, USA).

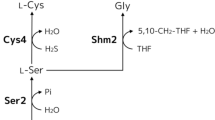
Schematic representation of the main biosynthetic pathways involved in the metabolism of cysteine and related compounds. Q Gsh1 , Q Gsh2 , specific rates of γGCS and GSH biosynthetic reactions, respectively (micromole per gram per hour); SUL1 high-affinity sulfate permease, MET17 homocysteine synthase, CYS4 cystathionine beta-synthase, CYS3 cystathionine gamma-lyase, STR3 cystathionine beta-lyase, STR2 cystathionine gamma-synthase, GSH1 gamma-glutamylcysteine synthetase, GSH2 glutathione synthetase, ECM38 gamma-glutamyltranspeptidase
To calibrate the separation of the detection probes by size, GeneScan-120LIZ size standard (Applied Biosystems, USA) was added to each sample. The identity of the probes was determined by the migration speed and the quantity by the peak area. To minimize non-biological variation in the TRAC assay, the signal intensities measured for the target genes were normalized between samples, using the signal measured for the internal hybridization control. The resulting signal intensities were either normalized to the biomass content in the hybridization mixture or related to the average signal of ACT1 (YFL039C) and SHR3 (YDL212W) signal level in the respective samples.
System equations
The specific biomass growth rate (μ, per hour) and biomass yield based on the substrate (sucrose) consumption (Y XS , gram dry weight per gram) were calculated as follows:
where V is the current fermenter volume (liters); X is biomass concentration in the fermenter (gram dry weight per liter); F in is the addition rate of the feeding medium (liters per hour); S S is the sucrose concentration in the feeding medium (grams per liter) and S is the sucrose concentration in the culture medium (grams per liter).
The specific rates of Gsh1p and Gsh2p reactions (Q Gsh1 and Q Gsh2 , micromole per gram per hour) were expressed as follows:
where GSH X and γGCS X are concentrations of glutathione and γ-glutamylcysteine in the biomass (micromole per gram). Thus, the specific GSH accumulation rate in the biomass was equal to Q Gsh2 and that of γGCS to Q Gsh1 −Q Gsh2 .
The GSH molar yield based on the consumption of cysteine fed into the fermenter (mole per mole) was calculated as follows:
where CYS S is the concentration of cysteine in the feeding medium (micromolars); CYS is the concentration of cysteine in the culture medium (micromolars).
Results
GSH biosynthesis in high-density fed-batch culture without cysteine addition
The specific GSH and γGCS synthesis rates, as well as the mRNA expression levels of the relevant genes were first determined under conditions of fed-batch culture using the basic feeding medium (i.e., without added cysteine and glycine). The ethanol-stat cultivation strategy (EtOH set-point = 0.2 g L-1) was used to grow the cells at near-maximum respiratory capacity (i.e., μ ≈ μ crit ). Sucrose concentration corresponding to the given ethanol set-point in the culture medium was consistently very low (data not shown). A constant specific growth rate (μ ≈ 0.24 h-1) was observed up to a biomass concentration of ∼50 g L-1. At higher biomass concentrations, a slow and consistent decrease in the specific growth rate was observed (Fig. 2b). As no decrease in the biomass yield occurred with this decreasing growth rate, the phenomenon is suggested to be related to growth rate inhibition rather than to nutrient limitation and decoupling of energy metabolism. With a few exceptions (notably GSH1 and GSH2), the inhibitory effect of higher biomass concentrations was also observed on the mRNA concentrations of most of the studied target genes (Fig. 3a). The expression of GSH1 and GSH2 relative to the expression of housekeeping genes ACT1 and SHR3 increased with decreasing growth rate (Fig. 3b).

Ethanol-stat fed-batch cultivation of S. cerevisiae LYCC7048 without cysteine and glycine addition. EtOH ethanol concentration in the cultivation medium (milligrams per milliliter); X biomass concentration in the cultivation medium (grams per liter); γGCS X , GSH X concentration of γGCS and GSH in the cells, respectively (micromoles per gram), Y XS biomass yield based on substrate (sucrose) consumption (grams per gram), μ specific growth rate (per hour), Q Gsh1 , Q Gsh2 specific γGCS and GSH synthesis rates, respectively (micromoles per gram per hour)

Fold change of the expression of the target and reference genes (ACT1 and SHR3) compared with the value at 4 h in the ethanol-stat fed-batch cultivation of S. cerevisiae LYCC7048 without cysteine and glycine addition: a relative to internal hybridization control and biomass content in the hybridization mixture and b relative to internal hybridization control and the expression of reference genes. Error bars represent the average absolute deviation of samples from their mean, the numbers under the columns represent the cultivation time in hours of sampling. For gene designations, see Fig. 1
Concentration of γGCS in the cells was almost constant (∼2 μmol g-1) during the entire cultivation while a slight decrease of GSH concentration (from ∼18 to 16 μmol g-1) was observed (Fig. 2a). The initial rate of γGCS biosynthesis (Q Gsh1 ) in the biomass was ∼4.7 μmol g-1 h-1 and that of GSH (Q Gsh2 ) was ∼4.5 μmol g-1 h-1 (Fig. 2b). Along with the decreasing growth rate at higher biomass concentrations, the specific rates of Gsh1p and Gsh2p reactions decreased in almost direct proportion.
Fed-batch cultivations with the shift to continuous addition of cysteine and glycine
Continuous addition of cysteine and glycine in ethanol-stat (EtOH set-point = 0.2 g L-1) fed-batch culture was initiated by changing the basic feeding medium to one containing both cysteine and glycine at different concentrations of 56 μmol g-1 sucrose (exp. 1), 112 μmol g-1 sucrose (exp. 2), or 225 μmol g-1 sucrose (exp. 3). This change in media composition resulted in a significant accumulation of intracellular cysteine and a decrease in the specific growth rate in exp. 2 and exp. 3 (Fig. 4e, f). However, in exp. 1, a decrease in the specific growth rate was only observed at cell densities higher than 60 g L-1 (Fig. 4d).

Ethanol-stat fed-batch cultivation of S. cerevisiae LYCC7048 with a shift to a feeding medium containing cysteine and glycine: 56 μmol g-1 sucrose in exp. 1; 112 μmol g-1 sucrose in exp. 2; 225 μmol g-1 sucrose in exp. 3. X biomass concentration in the cultivation medium (grams per liter); Y XS biomass yield based on substrate (sucrose) consumption (grams per gram); μ specific growth rate (per hour); cys X , γGCS X , GSH X concentration of cysteine, γGCS, and GSH in the cells, respectively (micromoles per gram); Q Gsh1 , Q Gsh2 hourly averages of specific γGCS and GSH synthesis rates, respectively (micromoles per gram per hour); Y GSH/cys hourly average of cysteine accumulation yield into intracellular GSH (micromoles per micromole cys). The arrows denote the switch to the cultivation media containing cysteine and glycine
In each experiment, the shift to medium enriched with cysteine and glycine caused an immediate increase in the concentration of GSH in yeast. This effect was more pronounced at higher cysteine to sucrose ratios in the feed (exp. 2 and exp. 3; Fig. 4e, f). The average specific rates of γGCS and GSH biosynthesis (Q Gsh1 and Q Gsh2 , respectively) and molar yields per consumed cysteine (Y GSH/cys ) after the shifts of the feeding media were calculated at intervals of 1 h (Fig. 4g–i). The highest specific accumulation rate of GSH in all experiments was observed during the first 2 h following the media shift. The maximum accumulation rate in exp. 1 (14 μmol g-1 h-1) was somewhat lower than that in exp. 2 and exp. 3 (17 μmol g-1 h-1) where two and four times higher cysteine concentrations were used. This can be explained by the lower concentration of intracellular cysteine than was required for the saturation of Gsh1p enzymatic activity in exp. 1. Due to the accumulation of γGCS in cells, a higher reaction rate of Gsh1p compared with that of Gsh2p was calculated for all three experiments (Fig. 4g–i). The maximum GSH yields (0.35–0.4 mol mol-1) based on cysteine consumption were similar in all experiments (Fig. 4g–i). Notably, both the GSH yield as well as the specific reaction rates Q Gsh1 and Q Gsh2 started to decrease when GSH reached its maximum value.
Results of the mRNA expression of the target genes relative to the expression of the housekeeping genes ACT1 and SHR3 for all three experiments are given on Fig. 5. Although an increase in intracellular GSH concentration was observed during the first 2 h following the shift to medium containing cysteine and glycine, up-regulation of the genes involved in the GSH biosynthetic pathway (GSH1 and GSH2) was not detected. Instead, a 25–50% down-regulation in the expression of GSH1 was observed after the shift. The extent of this down-regulation increased with the increasing cysteine/sucrose ratio in the feed. This suggests that the observed initial increase in the reaction rates Q Gsh1 and Q Gsh2 is most likely to be independent of control at the transcriptional level. The most remarkable down-regulation was observed in the expression of genes encoding the enzymes involved in homocysteine and cysteine biosynthesis, homocysteine synthase (MET17) and cystathionine gamma-lyase (CYS3). In exp. 2 and exp. 3 the expression of these genes ceased almost completely. Interestingly, an increased production of H2S was observed under conditions where MET17 expression was restricted (Fig. 6). Expression of cystathionine beta-synthase (CYS4) decreased more than 50% once cysteine addition was initiated; however, unlike CYS3 and MET17, it was subsequently observed to maintain a stable relative expression. A similar behavior (down-regulation by at least 50%) was also observed for high-affinity sulfate permease (SUL1) under each set of conditions. Interestingly, the expression of STR2 (cystathionine gamma-synthase) was observed to increase in conjunction with an increasing intracellular cysteine concentration during exp. 2 and exp. 3. A similar behavior would have been expected for cystathionine beta-lyase (STR3); however, the fluorescence signal intensity of STR3 was observed to be close to the detection threshold (400 U) and consequently, the missing expression signal in some data points of exp. 2 and exp. 3 is believed to be a result of detection issues, rather than down-regulation of the gene. While no changes in the expression of gamma-glutamyltranspeptidase (ECM38), responsible for the degradation of GSH, were observed in exp. 1, the expression increased remarkably under the conditions encountered during exp. 2 and exp. 3. Although this suggests an increase in the concentration of Ecm38p, we were not able to determine any traces of cysteinylglycine, the intermediate of GSH catabolism in the cells.

Expression of the target genes (in fluorescence intensity units), relative to internal hybridization control and the expression of reference genes (ACT1 and SHR3) in the ethanol-stat fed-batch cultivation of S. cerevisiae LYCC7048 with a shift to a feeding medium containing cysteine and glycine: 56 μmol g-1 sucrose in exp. 1; 112 μmol g-1 sucrose in exp. 2; 225 μmol g-1 sucrose in exp. 3. Error bars represent the average absolute deviation of samples from their mean values, the numbers under the columns represent the cultivation time (in hours from the switch of the feeding). For gene designations, see Fig. 1

Concentration of H2S in the culture medium (micrograms per liter) in ethanol-stat fed-batch cultivation of S. cerevisiae LYCC7048 (exp. 3) after the switch to the feeding medium containing cysteine and glycine (225 μmol g-1 sucrose)
Discussion
In the present work, we have studied the accumulation of GSH and the expression of related genes in ethanol-stat fed-batch fermentations, which allowed starting the experiments with a well-defined high-density culture. The gradual addition of cysteine prevented its excessive accumulation, which has been reported to result in prompt inhibition of growth when introduced using a shot-wise addition protocol (Alfafara et al. 1992a, b; Wang et al. 2007).
Prior to switching the feeding medium, the culture characteristics were virtually identical in all experiments (X = 39 ± 1 g L-1, μ = 0.24 h-1, GSH X = 17 ± 1 µmol g-1 and Q Gsh1 = 4.5 ± 0.5 μmol g-1 h-1). The switch to enriched medium resulted in a rapid increase in intracellular cysteine, γGCS, and GSH concentrations (Fig. 4d–f). Surprisingly, along with increasing γGCS and GSH synthesis rates, a decrease in GSH1 expression, relative to that of the housekeeping genes ACT1 and SHR3, was observed (Fig. 4g–i; Fig. 5). However, this decrease was only temporary and was followed by a full recovery at higher GSH concentrations in conditions of decreasing GSH synthesis rate. Thus, the extent of repression of GSH1 expression is not directly related to the concentration of intracellular GSH, but may depend on the rate at which the GSH, γGCS, or cysteine concentration changesFootnote 1. Based on the results of Menant et al. (2006), the rapidly increasing cysteine concentration in the cells could be the cause of the temporary down-regulation of GSH1. While the exact mechanisms causing this down-regulation remain to be elucidated, the results show that, under the studied conditions, the transcriptional regulation has a relatively minor role in GSH accumulation. The increase of γGCS and GSH synthesis rates and concentrations is most likely to be caused by the rapid increase of substrate concentrations for Gsh1p and Gsh2p reactions. The decrease in GSH synthesis rate at higher GSH concentrations (Fig. 4) suggests that the feedback control of Gsh1p activity by GSH was significant in determining its maximum concentration in the cells. Notably, similar GSH accumulation and gene expression patterns were also observed in preliminary studies with a different baker's yeast strain under the conditions identical to those of exp.1 and exp. 2 (data not illustrated).
In each experiment, the cellular uptake of the total added cysteine was higher than 85%. However, only ∼50% of the consumed cysteine was identified in the composition of γGCS, GSH, and as free intracellular cysteine. The up-regulation of STR2 and STR3 and the down-regulation of SUL1, MET17, CYS4, and CYS3 in the two experiments with the higher cysteine content in the feed (Fig. 5) indicate that cells are switching from sulfate to cysteine as a preferred sulfur source. Considering the average methionine and cysteine content in yeast protein fractions (Halasz and Lásztity 1991), our mass balance calculations showed that the total cysteine utilization for GSH and protein synthesis was close to 100% (data not illustrated) in case of complete halt of de novo synthesis of cysteine.
A high cysteine concentration in the feeding resulted in an accumulation of H2S (Fig. 6), repression of respiration, and a decrease in specific growth rate (Fig. 4). H2S as well as sulfite have been reported to inhibit cellular respiration (Schimz 1980; Marzluf 1997; Grieshaber and Völkel 1998; Sohn et al. 2000). If Met17p reaction is completely blocked, the incomplete inhibition of Sul1p, Met3p (ATP sulfurylase), Met14p (APS kinase), Met16p (PAPS reductase), and/or Met10p (sulfite reductase) reactions may cause an accumulation of sulfite and sulfide in the cells. On the other hand, operation of this pathway under conditions of excess cysteine would result in a significant waste of metabolic energy, and it is likely to be down-regulated through the degradation of Met4p transcription factor (Menant et al. 2006). Therefore, catabolization of cysteine can also be considered a possible source of sulfite and thus H2S. Unfortunately, no comprehensive data is available on cysteine catabolism in S. cerevisiae. Thus, further studies are required to elucidate the pathways of sulfur metabolism in the presence of cysteine.
In conclusion, under conditions of continuous cysteine addition, an initial increase in GSH synthesis rate is caused by the increased substrate (cysteine) availability for Gsh1p reaction. Our results indicate that the subsequent feedback inhibition of Gsh1p activity resulting from elevated intracellular GSH concentrations is the main factor in determining the maximum attainable GSH concentration in S. cerevisiae.
Notes
We have recently shown that the magnitude of the expression of stress response protein Hsp12p in S. cerevisiae is similarly dependent on the rate of change of the concentration of stressor compounds in the cultivation medium (Nisamedtinov et al. 2008).
References
Alfafara CG, Kanda A, Shioi T, Shimizu H, Shioya S, Suga K (1992a) Effect of amino acids on glutathione production by Saccharomyces cerevisiae. Appl Microbiol Biotechnol 36:538–540
Alfafara CG, Miura K, Shimizu H, Shioya S, Suga K (1992b) Cysteine addition strategy for maximum glutathione production in fed-batch culture of Saccharomyces cerevisiae. Appl Microbiol Biotechnol 37:141–146
Douglas KT (1987) Mechanism of action of glutathione-dependent enzymes. Adv Enzymol Relat Areas Mol Biol 59:103–167
Fahey RC, Brown WC, Adams WB, Worsham MB (1978) Occurrence of glutathione in bacteria. J Bacteriol 133:1126–1129
Grant CM (2001) Role of the glutathione/glutaredoxin and thioredoxin systems in yeast growth and response to stress conditions. Mol Microbiol 39:533–541
Grant CM, Dawes IW (1996) Synthesis and role of glutathione in protection against oxidative stress in yeast. Redox Rep 2:223–229
Grant CM, MacIver FH, Dawes IW (1997) Glutathione synthetase is dispensable for growth under both normal and oxidative stress conditions in the yeast Saccharomyces cerevisiae due to an accumulation of the dipeptide gamma-glutamylcysteine. Mol Biol Cell 8:1699–1707
Grieshaber MK, Völkel S (1998) Animal adaptations for tolerance and exploitation of poisonous sulfide. Annu Rev Physiol 60:33–53
Halasz A, Lásztity R (1991) In vitro data of yeast protein quality based on amino acid composition. In: Halasz A (ed) Use of yeast biomass in food production, 1st edn. CRC-Press, Boca Raton, pp 147–149
Huang CS, Moore WR, Meister A (1988) On the active site thiol of gamma-glutamylcysteine synthetase: relationships to catalysis, inhibition, and regulation. Proc Natl Acad Sci U S A 85:2464–2468
Li Y, Wei G, Chen J (2004) Glutathione: a review on biotechnological production. Appl Microbiol Biotechnol 66:233–242
Liang G, Du G, Chen J (2008a) Enhanced glutathione production by using low-pH stress coupled with cysteine addition in the treatment of high cell density culture of Candida utilis. Lett Appl Microbiol 46:507–512
Liang G, Du G, Chen J (2008b) A novel strategy of enhanced glutathione production in high cell density cultivation of Candida utilis—cysteine addition combined with dissolved oxygen controlling. Enzyme Microb Technol 42:284–289
Marzluf GA (1997) Molecular genetics of sulfur assimilation in filamentous fungi and yeast. Annu Rev Microbiol 51:73–96
Meister A (1988) Glutathione metabolism and its selective modification. J Biol Chem 263:17205–17208
Meister A, Anderson ME (1983) Glutathione. Annu Rev Biochem 52:711–760
Menant A, Baudouin-Cornu P, Peyraud C, Tyers M, Thomas D (2006) Determinants of the ubiquitin-mediated degradation of the Met4 transcription factor. J Biol Chem 28:11744–11754
Moest RR (1975) Hydrogen sulfide determination by the methylene blue method. Anal Chem 47:1204–1205
Murata K, Kimura A (1990) Overproduction of glutathione and its derivatives by genetically engineered microbial cells. Biotechnol Adv 8:59–96
Nisamedtinov I, Lindsey GG, Karreman R, Orumets K, Koplimaa M, Kevvai K, Paalme T (2008) The response of the yeast Saccharomyces cerevisiae to sudden vs. gradual changes in environmental stress monitored by expression of the stress response protein Hsp12p. FEMS Yeast Res 8:829–838
Penninckx M (2000) A short review on the role of glutathione in the response of yeasts to nutritional, environmental, and oxidative stresses. Enzyme Microb Technol 26:737–742
Pócsi I, Prade RA, Penninckx MJ (2004) Glutathione, altruistic metabolite in fungi. Adv Microb Physiol 49:1–76
Rautio JJ, Huuskonen A, Vuokko H, Vidgren V, Londesborough J (2007) Monitoring yeast physiology during very high gravity wort fermentations by frequent analysis of gene expression. Yeast 24:741–760
Rautio JJ, Kataja K, Satokari R, Penttilä M, Söderlund H, Saloheimo M (2006) Rapid and multiplexed transcript analysis of microbial cultures using capillary electrophoresis-detectable oligonucleotide probe pools. J Microbiol Methods 65:404–416
Richman PG, Meister A (1975) Regulation of gamma-glutamylcysteine synthetase by nonallosteric feedback inhibition by glutathione. J Biol Chem 250:1422–1426
Sakato K, Tanaka H (1992) Advanced control of glutathione fermentation process. Biotechnol Bioeng 40:904–912
Schimz KL (1980) The effect of sulfite on the yeast Saccharomyces cerevisiae. Arch Microbiol 125:89–95
Sies H (1999) Glutathione and its role in cellular functions. Free Radic Biol Med 27:916–921
Sohn HY, Murray DB, Kuriyama H (2000) Ultradian oscillation of Saccharomyces cerevisiae during aerobic continuous culture: hydrogen sulphide mediates population synchrony. Yeast 16:1185–1190
Soltaninassab SR, Sekhar KR, Meredith MJ, Freeman ML (2000) Multi-faceted regulation of gamma-glutamylcysteine synthetase. J Cell Physiol 182:163–170
Zhang T, Wen S, Tan T (2007) Optimization of the medium for glutathione production in Saccharomyces cerevisiae. Process Biochem 42:454–458
Thomas D, Surdin-Kerjan Y (1997) Metabolism of sulfur amino acids in Saccharomyces cerevisiae. Microbiol Mol Biol Rev 61:503–532
van Hoek P, de Hulster E, van Dijken JP, Pronk JT (2000) Fermentative capacity in high-cell-density fed-batch cultures of baker's yeast. Biotechnol Bioeng 68:517–523
Walker GM (2004) Metals in yeast fermentation processes. Adv Appl Microbiol 54:197–229
Wang Z, Tan T, Song J (2007) Effect of amino acids addition and feedback control strategies on the high-cell-density cultivation of Saccharomyces cerevisiae for glutathione production. Process Biochem 42:108–111
Wen S, Zhang T, Tan T (2004) Utilization of amino acids to enhance glutathione production in Saccharomyces cerevisiae. Enzyme Microb Technol 25:501–507
Wen S, Zhang T, Tan T (2006) Maximizing production of glutathione by amino acid modulation and high-cell-density fed-batch culture of Saccharomyces cerevisiae. Process Biochem 41:2424–2428
Wheeler GL, Trotter EW, Dawes IW, Grant CM (2003) Coupling of the transcriptional regulation of glutathione biosynthesis to the availability of glutathione and methionine via the Met4 and Yap1 transcription factors. J Biol Chem 278:49920–49928
Wheeler GL, Quinn KA, Perrone G, Dawes IW, Grant CM (2002) Glutathione regulates the expression of γ-glutamylcysteine synthetase via the Met4 transcription factor. Mol Microbiol 46:545–556
Acknowledgements
The financial support for this research was provided by the Enterprise Estonia project EU22704, Estonian Ministry of Education and Research grant SF0140090s08, and by Estonian Science Foundation grant G7323. We would like to thank Mr. David Schryer and Dr. Chris Powell for critical revision of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Nisamedtinov, I., Kevvai, K., Orumets, K. et al. Glutathione accumulation in ethanol-stat fed-batch culture of Saccharomyces cerevisiae with a switch to cysteine feeding. Appl Microbiol Biotechnol 87, 175–183 (2010). https://doi.org/10.1007/s00253-010-2502-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-010-2502-x