
Abstract
Two alternative cell-surface display systems were developed in Pichia pastoris using the α-agglutinin and Flo1p (FS) anchor systems, respectively. Both the anchor cell wall proteins were obtained originally from Saccharomyces cerevisiae. Candida antarctica lipase B (CALB) was displayed functionally on the cell surface of P. pastoris using the anchor proteins α-agglutinin and FS. The activity of CALB displayed on P. pastoris was tenfold higher than that of S. cerevisiae. The hydrolytic and synthetic activities of CALB fused with α-agglutinin and FS anchored on P. pastoris were investigated. The hydrolytic activities of both lipases displayed on yeast cells surface were more than 200 U/g dry cell after 120 h of culture (200 and 270 U/g dry cell, respectively). However, the synthetic activity of CALB fused with α-agglutinin on P. pastoris was threefold higher than that of the FS fusion protein when applied to the synthesis of ethyl caproate. Similarly, the CALB displayed on P. pastoris using α-agglutinin had a higher catalytic efficiency with respect to the synthesis of other short-chain flavor esters than that displayed using the FS anchor. Interestingly, for some short-chain esters, the synthetic activity of displaying CALB fused with α-agglutinin on P. pastoris was even higher than that of the commercial CALB Novozyme 435.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In recent years, considerable progress has been made in the development of expression systems that display heterologous functional proteins on the surfaces of bacteria, phage, and yeast. Yeast surface display is a powerful method for the expression of functional proteins and can be used to increase the affinity, specificity, and stability of the expressed proteins (Boder and Wittrup 1997). Major advantages of yeast surface display are that the protein-folding and secretory machineries of yeast are strikingly homologous to those of mammalian cells. In this system, foreign proteins are displayed on the yeast cell surface in the form of a fusion with an anchoring protein. Quantitative comparisons can be performed by the use of fluorescence-activated cell analysis, and this allows the effectiveness of different anchor proteins for the expression of a particular protein to be compared. Recently, some yeast cell-surface display systems were developed in S. cerevisiae. Several functional proteins were displayed on yeast cell surface in the form of fusion proteins linked to the C-terminal half of α-agglutinin and N-terminal portion of Flo1p (FS; Kondo and Ueda 2004; Ueda 2004; Kato et al. 2006). Subsequently, Kato et al. (2007) expressed Candida antarctica lipase B (CALB) using the S. cerevisiae display system by α-agglutinin anchor protein. Tanino et al. (2007) used the FS anchor protein to display CALB on the S. cerevisiae cell surface. Yeast cell-surface engineering has been used to date in various industrial applications. CALB displayed on yeast has been used to catalyze many reactions successfully, for example, the alcoholysis of oil, including biodiesel production and the synthesis of esters (Tanino et al. 2009). We have used α-agglutinin to anchor CALB on the cell surface of S. cerevisiae and applied to the synthesis of ethyl hexanoate (Han et al. 2009). However, industrial applications are often limited by the cost of enzyme and efficiency of synthesis. To improve the efficiency of yeast cell-surface display, several groups have described the use of the yeast P. pastoris for the surface display of heterologous proteins, such as Rhizopus oryzae lipase, in conjunction with a FS anchor protein (Tanino et al. 2006; Ren et al. 2007). Two groups have used α-agglutinin from S. cerevisiae to anchor their protein of interest on the cell surface of P. pastoris (Mergler et al. 2004; Wang et al. 2007). We have used α-agglutinin to anchor CALB on the cell surface of S. cerevisiae. However, there have been no previous reports of the use on the commercially available α-agglutinin and FS systems to display CALB on the cell surface of P. pastoris.
Lipases (EC 3.1.1.3) are used as efficient catalysts in numerous industries, such as the food industry, detergent production, manufacture of paper, and pharmaceutical processing (Sharma et al. 2001). Lipases catalyze the hydrolysis of triacylglycerols at the interface between water and a hydrophobic substrate. In addition, they can catalyze the synthesis of esters due to their broad substrate specificity, in particular their specificity for different lengths of fatty acid chains. CALB is one of the most important lipases because it retains its enzymatic activity in organic media and possesses high enantioselectivity towards secondary alcohols.
In this study, to prepare highly active CALB and extend its application, we have developed and compared the use of α-agglutinin and the flocculation functional domain of FS as anchor proteins in the host P. pastoris. Furthermore, we compared the reaction characteristics of CALB displayed on the cell surface of P. pastoris and S. cerevisiae, especially with respect to the synthesis of flavor esters.
Materials and methods
Strains and materials
The gene-encoding FS domain was cloned from S. cerevisiae ATCC 60715. The coding sequences for α-agglutinin and CALB were subcloned from the plasmid pICAS-CALB (Han et al. 2009). The yeast display vector pICAS and S. cerevisiae strain MT8-1 (MATa, ade, his3, leu2, trp1, ura3), which was used to display CALB, were kind gifts from Professor Mitsuyoshi Ueda (Ueda and Tanaka 2000). S. cerevisiae MT8-1 was used for transformations and was cultured in SD medium (0.67% yeast nitrogen base [YNB] without amino acids, 2% glucose, 30 mg/L Leu, 20 mg/L His, 20 mg/L adenine, and 20 mg/L uracil) at 30°C for 96 h. P. pastoris GS115 was purchased from Invitrogen (Carlsbad, CA, USA) and cultured in complete medium (YPD: 1% yeast extract, 2% peptone, 2% glucose), selective medium (MD: 1.34% YNB, 4 × 10−5% biotin, 2% dextrose, 1.5% agar), BMGY (1% yeast extract, 2% peptone, 100 mM potassium phosphate pH 6.0, 1.34% YNB, 4 × 10−5% biotin, 1% glycerol), or BMMY (1% yeast extract, 2% peptone, 100 mM potassium phosphate pH 6.0, 1.34% YNB, 4 × 10−5% biotin, 0.5% methanol). The vector pPIC9K was purchased from Invitrogen. A mouse anti-FLAG monoclonal antibody was obtained from Sigma (St. Louis, MO, USA). Alexa Fluor 488 conjugated goat antimouse IgG was purchased from Molecular Probes (Eugene, OR, USA).
Construction of recombinant expression vectors for P. pastoris
The coding sequences for α-agglutinin and CALB were amplified from the plasmid vector pICAS-CALB by polymerase chain reaction using the following primers: EcoRI-CALB 5′TGTAGAATTCCTGCCTTCCGGTTCGGACCCTG and NotI-α-agglutinin 5′ATTAGCGGCCGCTTAGAATAGCAGGTACGACAAAAG.
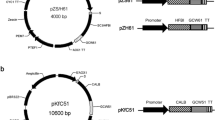
The amplified sequence, which encodes a CALB-α-agglutinin fusion protein together with a FLAG tag, was inserted into the vector pPIC9K. The vector's secretion signal peptide α-factor was used as secretion signal for cell surface displaying CALB on P. pastoris. The resulting plasmid was named pKNS-CALB (Fig. 1a). The coding sequences for the FS anchor protein and its secretion signal peptide was also cloned and inserted into the vector pPIC9K. The CALB coding sequence was amplified from pICAS-CALB using the following primers: EcoRI-CALB2 5′TGTAGAATTCGATTACAAGGATGACGACGATAAGCTGCCTTCCGGTTCGGACCCTG and NotI-CALB 5′ATTAGCGGCCGCTCAGGGGGTGACGATGCCG. The resulting plasmid was named pKFS-CALB (Fig. 1b). All the plasmids were verified by sequencing in Sangon (Shanghai, China).

a To construct the plasmid pKNS-CALB for cell-surface display, the coding sequence for CALB fused at the C terminus with a FLAG tag and α-agglutinin was inserted into pPIC9K after enzymatic digestion with EcoRI and NotI. b To construct the plasmid pKFS-CALB for cell-surface display, the coding sequence for CALB tagged at the N terminus with FLAG was inserted into the plasmid pKFS to form an open reading frame that encoded a CALB-FS fusion protein
Yeast transformation
P. pastoris GS115 was transformed with pKFS-CALB and pKNS-CALB that had been linearized with SacI using a Pichia Easy-Comp Transformation Kit (Invitrogen) according to the manufacturer's instructions. The transformants were isolated by incubation at 30°C for 48 h on MD plates.
Culture conditions and assay for CALB activity
The yeast transformants were precultured in BMGY medium at 30°C for 16 h and used to inoculate 50 ml of BMGY medium in a 500-ml baffled flask at an initial OD600 value of 0.1. After incubation for 24 h, the cultures were centrifuged at 6,000×g for 10 min and resuspended in BMMY medium that contained 1.0% methanol. To maintain the induction of the fusion proteins FS-CALB and CALB-α-agglutinin, methanol was added to the culture every 24 h to the final concentration mentioned above.
The activity of CALB was determined spectrophotometrically using p-nitrophenyl butyrate (pNPB).The substrate, pNPB, was emulsified by sonication in 50 mM potassium phosphate buffer (pH 8.0) that contained 1% Triton X-100. Yeast cells were collected by centrifugation at 6,000×g at room temperature for 10 min. After the cells had been washed twice with 20 ml of 50 mM potassium phosphate buffer (pH 7.5), the cell suspension was adjusted to an OD600 of 3.0. A 100-µl aliquot of the cell suspension and an equal volume of the substrate were mixed and allowed to react at 45°C for 5 min. The reaction was stopped by the addition of 1 ml of acetone. Then the reaction mixture was centrifuged at 16,000×g at room temperature for 1 min. A 200-µl aliquot of the resulting supernatant was placed in a 96-well plate, and the activity of CALB was assayed by measuring the absorbance of liberated p-nitrophenol (pNP) at 405 nm using a kinetic microplate reader (Molecular Devices, Sunnyvale, CA, USA). The activity was measured by the absorbance of liberated pNP at 405 nm. One unit of activity was defined as the amount of enzyme required to release 1 µmol pNP/min.
Preparation of lipase-displaying yeast cells
Yeast cells that displayed the lipase were prepared for use as a whole-cell biocatalyst in the following manner. S. cerevisiae that had been transformed with pICAS-CALB, as MT8-1/pICAS-CALB, was cultured in SD medium at 30°C for 96 h, whereas P. pastoris that had been transformed with pKNS-CALB or pKFS-CALB was cultured in BMMY medium at 30°C for 120 h as described above. The cells were then collected, washed twice with distilled water, and lyophilized for 24 h.
Immunofluorescence microscopy and flow cytometer analysis
The yeast cells were analyzed by immunofluorescence microscopy according to the method of Kobori et al. (1992). Induced cells were washed twice in ice-cold water and resuspended at 4°C in phosphate-buffered saline (PBS; pH 7.4) supplemented with 1 mg/ml bovine serum albumin. Immunostaining was carried out as follows: an antibody against FLAG was used as the primary antibody at a dilution of 1:1,000 in a total volume of 1 ml. The cells were incubated with the antibody on a rotator at room temperature for 2 h. The cells were then washed with PBS (pH 7.4) and exposed to the secondary antibody, Alexa Fluor™ 488 goat antimouse IgG (H + L), which was diluted 1:300 in a total volume of 300 µl, for 1 h at room temperature. After three washing steps, the cells were examined using a fluorescence microscope (IX71, Olympus, Tokyo, Japan) or a Cell Lab Quanta™ SC Flow Cytometer (Beckman-Coulter, Fullerton, CA, USA). For the latter, a total of 10,000 cells were analyzed for each sample, and the data were analyzed using the EXP032 software (Beckman-Coulter). Cells that had been transformed with pPIC9K or pICAS were also processed in the above manner to serve as the negative controls. The ratio between the intensity of the signal obtained from CALB-displaying and control yeast cells was calculated for each individual sample. A sample was considered to be positive if this ratio was ≥2.
Synthesis of flavor esters using yeast cells displaying CALB
All the reaction media for ester synthesis were dehydrated by gentle shaking with 3 Å molecular sieves overnight before being used. In a typical experiment for ethyl hexanoate synthesis, 10 ml of n-heptane containing 4 mmol of hexanoic acid and 6 mmol of ethanol were added to a 50-ml Erlenmeyer shaking flask capped with a septum. Then, the reaction was initiated by the addition of 0.1 g of lyophilized yeast cells displaying lipase or the commercial CALB Novozyme 435 to the reaction system and the mixture was incubated at 200 rpm and 55°C for 9 h. Samples of 50 µl were withdrawn periodically and centrifuged (10,000×g, 10 min), and 20 µl of the resulting upper layer were mixed with 1 ml of n-heptane that contained n-butyl acetate for analysis by gas chromatography (Agilent 7890A), which a hydrogen flame ionization detector and a DB-FFAP silica capillary column (0.25 mm × 30 m; Agilent, Santa Clara, CA, USA) were used. The column temperature was held at 50°C for 1 min, then increased to 60°C, 68°C, 95°C, and 200°C at a rate of 10°C/min, 40°C/min, 20°C/min, and 40°C/min, respectively, and kept at each of these temperatures for 2 min. The injector and detector temperatures were both set at 250°C. The average error for this determination was <0.7%. For the synthesis of ethyl acetate and ethyl propionate, it was carried out under the same reaction conditions. All reported data were the means of experiments performed at least in triplicate.
Results
Construction of a CALB yeast surface display system using P. pastoris
When CALB from C. antarctica LF 058 was displayed on S. cerevisiae MT8-1 using α-agglutinin as an anchor, the activity of the enzyme was about 25 U/g dry cell (Fig. 3b). It is well known that P. pastoris grows to a high cell density and efficiently secretes heterologous proteins (Cregg et al. 2007). To display CALB on the cell surface of P. pastoris, the sequence of the C-terminal portion of α-agglutinin was fused to the C-terminal sequence of CALB, and a FLAG tag and linker sequence encoding 18 amino acid residues (Gly and Ser) were inserted between the sequences of CALB and α-agglutinin (Fig. 1a). In addition, the N-terminal sequence of CALB was linked with the sequence for the N-terminal portion of the FS anchor protein. The FLAG tag and linker were also inserted between the sequences for CALB and FS (Fig. 1b). Both of the constructed plasmids were integrated in a stepwise manner into the genome of P. pastoris GS115, respectively. Transformants were isolated by incubation at 30°C for 48 h on MD plates that contained 1% tributyrin and methanol. On the MD plates, a halo was observed around the yeast colonies when CALB was displayed, but not around the control transformants (Fig. 2). The selected transformants that displayed FS-CALB and CALB-α-agglutinin were designated as GS115/KFS-CALB and GS115/KNS-CALB, respectively.

Halo formation of CALB-displaying yeast with different anchor protein on tributyrin agar plate. A P. pastoris yeast cells that harbored pKNS-CALB, B P. pastoris yeast cells that harbored pKFS-CALB, C S. cerevisiae yeast cells that harbored pICAS-CALB, CK host cell P. pastoris GS115 as a control, MT8-1 S. cerevisiae yeast cells as a control
Comparison of the α-agglutinin and FS anchor proteins for the display of CALB on P. pastoris
The time courses of lipase hydrolytic activity for the P. pastoris cells that displayed CALB with an FS (GS115/KFS-CALB) or α-agglutinin (GS115/KNS-CALB) anchor protein are shown in Fig. 3 and compared with that for S. cerevisiae that displayed CALB with α-agglutinin anchor protein. The lipase hydrolytic activity of the whole cells increased with culture time. The activities of both forms of CALB displayed on P. pastoris cells were higher than 200 U/g dry cell after 120 h of culture, and this was tenfold higher than that of CALB displayed on S. cerevisiae. The highest hydrolytic activity was obtained with GS115/KFS-CALB.

Time course of the lipase hydrolytic activities and growth curves of whole P. pastoris and S. cerevisiae yeast cells that displayed CALB fused with α-agglutinin or FS. In a, the lines represent the hydrolytic activity of P. pastoris yeast cells displaying CALB with an FS anchor protein (circles), the hydrolytic activity of P. pastoris yeast cells displaying CALB with α-agglutinin anchor (diamonds), the growth curve of P. pastoris cells displaying CALB with an FS anchor (squares), and the growth curve of P. pastoris cells displaying CALB with α-agglutinin anchor (triangles). In b, the hydrolytic activity of S. cerevisiae yeast cells displaying CALB with α-agglutinin anchor (diamonds) and the growth curve of S. cerevisiae yeast cells displaying CALB with α-agglutinin anchor protein (squares). The data points represent the mean of three independent experiments
The display of CALB on the yeast cell surface was evaluated further by immunofluorescence. Immunofluorescence labeling with a mouse anti-FLAG monoclonal antibody as the primary antibody and Alexa Fluor 488 goat antimouse IgG, which cannot penetrate the cell membrane, as the secondary antibody was performed to verify the presence and localization of CALB on the cell surface. Fluorescence was observed on the cell surface of the GS115/KFS-CALB, GS115/KNS-CALB, and MT8-1/pICAS-CALB strains by a fluorescence microscope, but not on that of the control cells (Fig. 4). It is confirmed that CALB was anchored on the cell surface of P. pastoris and S. cerevisiae.

Fluorescence and phase-contrast micrographs of yeast cells that displayed the FLAG-tagged CALB fusion proteins after immunofluorescent labeling. Cells were labeled with an anti-FLAG antibody and Alexa Fluor 488 antimouse IgG. Right column phase-contrast micrographs, left column labeling of CALB with anti-FLAG antibody and Alexa Fluor 488 antimouse IgG. P. pastoris cells GS115 (a), FS anchor on P. pastoris with CALB (b), CALB with α-agglutinin anchor on P. pastoris (c), S. cerevisiae cells MT8-1 (d), and CALB with α-agglutinin anchor on S. cerevisiae (e). The fluorescence emitted at 519 nm after excitation at 495 nm was observed by fluorescence microscopy
The expression of the CALB fusion proteins on P. pastoris was also analyzed by indirect immunofluorescence labeling using the FLAG tag and flow cytometry. For S. cerevisiae MT8-1/pICAS-CALB, <25% of cells expressed the CALB-α-agglutinin fusion protein. However, for both GS115/KFS-CALB and GS115/KNS-CALB, over 95% of the cells expressed the appropriate CALB fusion protein on their surface (Fig. 5). A small difference was detectable between the amounts of expression obtained with the two different anchor proteins. The data demonstrated that the FS anchor protein resulted in slightly more efficient expression of CALB in P. pastoris compared with that of α-agglutinin, and more efficient expression was obtained with either anchor protein in P. pastoris compared with S. cerevisiae. These results correlated with the amounts of hydrolytic lipase activity that were obtained with the different systems.

Flow cytometry analysis of cells transformed with the expression constructs. P. pastoris was cultured for 120 h, FS anchor on P. pastoris with CALB (a), CALB with α-agglutinin anchor on P. pastoris (b), and CALB with FS anchor on S. cerevisiae for 48 h (c). The flow cytometry histograms shown depict the mean fluorescent signal obtained after antibody labeling of P. pastoris or S. cerevisiae that displayed CALB using the FLAG tag on the CALB fusion protein (in gray). The control P. pastoris GS115 and S. cerevisiae MT8-1 cells (in white) did not express a FLAG tag and emitted very little fluorescence
Synthesis of flavor esters by yeast cells that displayed CALB
The catalytic synthesis ability of lyophilized yeast cells displayed on the different CALB fusion proteins was assayed. The reactions were carried out in batch reactions at 55°C using the different yeast cells displaying lipase described above. Figure 6 shows the time courses of the production of the ester for the three different yeast cells. The amount of product increased with reaction time and, in each case, the reaction was terminated after 9 h. CALB fused to α-agglutinin and displayed on the cell surface of P. pastoris was most active with respect to the synthesis of ethyl hexanoate. It gave a yield of more than 90% in only 1.5 h. CALB fused to the FS anchor and displayed on the P. pastoris cell surface required 6 h to produce the same amount of product. CALB that was fused with α-agglutinin and displayed on the cell surface of S. cerevisiae showed the lowest efficiency of synthesis. It only gave a yield of 20% after a reaction time of 9 h. Similar results were obtained for the synthesis of other flavor esters, such as ethyl propanoate, ethyl butyrate, ethyl heptylate, and so on. Interestingly, CALB that was fused with α-agglutinin and displayed on P. pastoris synthesized some short-chain esters more efficiently than the commercial CALB preparation Novozyme 435. In Fig. 7a, b, it shows the time courses for the esterification of ethyl acetate and ethyl propionate as mediated by CALB displaying on P. pastoris and Novozyme 435. The results indicated that 96% of the acetic acid was converted to ethyl acetate after a reaction time of 9 h by CALB displaying on P. pastoris using α-agglutinin and 96% of the propionic acid was converted to ethyl propionate after a reaction time of 6 h. In contrast, in the presence of Novozyme 435, 62% of the acetic acid and 61% of the propionic acid was converted after the same reaction time. These results suggested that CALB displayed on P. pastoris using the α-agglutinin anchor has potential commercial applications in the synthesis of short-chain flavor esters.

Time course of whole-cell biocatalyst-mediated esterification reactions. The reaction mixture, which consisted of 10 ml of n-heptane containing 4 mmol of hexanoic acid, 6 mmol of ethanol, and 0.1 g of lyophilized whole-cell biocatalyst, was incubated at 200 rpm and 55°C. Molecular sieves (3 Å) were added to the reaction system to remove the water produced in the reaction. The graph shows the percentage molar conversion of substrate that was obtained with CALB with α-agglutinin anchor on P. pastoris (squares) and S. cerevisiae (triangles) and CALB with an FS anchor on P. pastoris (diamonds) and Novo435 (circles). The percentage molar conversion of hexanoic acid was plotted against the reaction time. The data points represent the average of three independent experiments

Time course of whole-cell biocatalyst-mediated synthesis of ethyl acetate and ethyl propionate. Reaction conditions: 10 ml of n-heptane containing 4 mmol of acetic acid (a) or propionic acid (b), 6 mmol of ethanol, and 0.1 g of lyophilized whole-cell biocatalyst were incubated at 200 rpm and 55°C. Molecular sieves (3 Å) were added to the reaction system to remove the water produced in the reaction. The reaction was terminated after 3 h. The graph shows the percentage molar conversion of substrate that was obtained with CALB with α-agglutinin anchor on P. pastoris (squares), CALB with an FS anchor on P. pastoris (triangles), and the commercial CALB Novo435 (diamonds).The percentage molar conversion of acetic acid and propionic acid was plotted against the reaction time. The data bars represent the mean of three independent experiments
Discussion
P. pastoris as a cellular host for the expression of recombinant proteins has provided a great potential for producing soluble, correctly folded recombinant proteins that have undergone all the post-translational modifications required for functionality. In recent studies (Jiang et al. 2007), it has been indicated that the fermentation characteristics of P. pastoris, which include growth on an economical carbon source and a very high density of cell culture, are superior to those of the widely used yeast S. cerevisiae. Therefore, P. pastoris is well suited to applications that require large-scale fermentations. It is important to develop a P. pastoris cell display system for practical application. Recently, several groups described the use of P. pastoris for surface display of heterologous proteins and used the S. cerevisiae α-agglutinin to anchor their protein or S. cerevisiae FS as an anchor protein. And a novel P. pastoris cell-surface display system was based on the Pir1 cell wall protein of S. cerevisiae by Wang et al. (2008). In this study, we have demonstrated that the P. pastoris for displaying CALB on cells surface gives a higher efficiency of expression with both the α-agglutinin and FS anchor proteins than that of the S. cerevisiae system. The expression amount of CALB on P. pastoris cells surface was by indirect immunofluorescent labeling using the FLAG tag and flow cytometry analysis (Fig. 5). The results of the lipase assay showed that CALB-α-agglutinin was expressed at a lower level than that of FS-CALB, 200 and 270 U/g dry cell, respectively. The activity of CALB was tenfold higher when it was displayed on P. pastoris compared with S. cerevisiae.
Many functional enzymes have been genetically immobilized on the cell surface of yeast as a whole-cell biocatalyst. The industrial use of whole-cell biocatalysts with surface-displayed lipase has grown in recent years owing to unique advantages such as improved enzyme stability, simpler product purification, and cost-effective downstream processing. CALB has been displayed on S. cerevisiae cells surface as a whole-cell biocatalyst and used to synthesis reaction (Tanino et al. 2007, 2009). We applied P. pastoris displaying CALB as a whole-cell biocatalyst to catalyze the esterification of short-chain flavor esters in n-heptane. When the synthesis of ethyl hexanoate was analyzed using the yeast as whole-cell biocatalysts, the synthetic activity of CALB that was fused with α-agglutinin and displayed on P. pastoris was threefold higher than that of CALB fused with FS. In addition, the activity of the former was close to that of the commercial CALB Novozyme 435 (Fig. 6). The same results were obtained for the synthesis of other flavor esters under the same reaction conditions. Interestingly, for the catalytic synthesis reaction of some short-chain esters, the synthetic activity of P. pastoris that displayed CALB fused with α-agglutinin surpassed that of Novozyme 435 (Fig. 7). A number of explanations for this behavior in a non-aqueous system have been reported and include an increase in the stability of the enzyme structure and the improved solubility of lipophilic substrates and products. For example, Schilke and Kelly (2008) used long hydrophobic linkers to immobilize CALB and thus increased the activity of the enzyme in organic media. The hydrophobicity of every amino acid of the two anchor proteins was analyzed by the ExPASy ProtScale tool (http://www.expasy.org/cgi-bin/protscale.pl). These results indicated that the hydrophobicity of the α-agglutinin anchor protein was higher than that of the FS anchor protein. It may result to the hydrolytic side reactions that interfere with syntheses, which are restricted in organic media. CALB does not possess a hydrophobic “lid” covering the catalytic site, which does not exhibit “interfacial activation” caused by large structural changes. Therefore, the higher hydrophobicity of the α-agglutinin anchor protein could increase the synthetic efficiency in short-chain esters synthesis by affecting the active sites with the higher hydrophobic tethers.
In conclusion, CALB that was displayed on P. pastoris retained its original hydrolytic activity after lyophilization of the yeast and efficiently catalyzed the synthesis of flavor esters in non-aqueous media. Therefore, enzymes displayed on P. pastoris cells are expected to be effective whole-cell biocatalysts. Further studies to develop the application of yeast cells that display CALB to the synthesis of various flavor esters are underway.
References
Boder ET, Wittrup KD (1997) Yeast surface display for screening combinatorial polypeptide libraries. Nat Biotechnol 15:553–557
Cregg JM, Cereghino JL, Shi J, Higgins DR (2007) Recombinant protein expression in Pichia pastoris. Mol Biotechnol 16:23–52
Han SY, Pan ZY, Huang DF, Ueda M, Wang XN, Lin Y (2009) Highly efficient synthesis of ethyl hexanoate catalyzed by CALB-displaying Saccharomyces cerevisiae whole-cells in non-aqueous phase. J Mol Catal B Enzym 59:168–172
Jiang ZB, Song HT, Gupta N, Ma LX, Wu ZB (2007) Cell surface display of functionally active lipases from Yarrowia lipolytica in Pichia pastoris. Protein Expr Purif 56:35–39
Kato M, Kuzuhara Y, Maeda H, Shiraga S, Ueda M (2006) Analysis of a processing system for proteases using yeast cell surface engineering: conversion of precursor of proteinase A to active proteinase A. Appl Microbiol Biotechnol 72:1229–1237
Kato M, Fuchimoto J, Tanino T, Kondo A, Fukuda H, Ueda M (2007) Preparation of a whole-cell biocatalyst of mutated Candida antarctica lipase B (mCALB) by a yeast molecular display system and its practical properties. Appl Microbiol Biotechnol 75:549–555
Kobori H, Sato M, Osumi M (1992) Relationship of actin organization to growth in the two forms of the dimorphic yeast Candida tropicalis. Protoplasma 167:193–204
Kondo A, Ueda M (2004) Yeast cell-surface display—applications of molecular display. Appl Microbiol Biotechnol 64:28–40
Mergler M, Wolf K, Zimmermann M (2004) Development of a bisphenol A-adsorbing yeast by surface display of the Kluyveromyces yellow enzyme on Pichia pastoris. Appl Microbiol Biotechnol 63:418–421
Ren R, Jiang Z, Liu MY, Tao XY, Ma YS, Wei DZ (2007) Display of adenoregulin with a novel Pichia pastoris cell surface display system. Mol Biotechnol 35:103–108
Schilke KF, Kelly C (2008) Activation of immobilized lipase in non-aqueous systems by hydrophobic poly-DL-tryptophan tethers. Biotechnol Bioeng 101:9–18
Sharma R, Chisti Y, Banerjee C (2001) Production, purification, characterization, and applications of lipases. Biotechnol Adv 19:627–662
Tanino T, Fukuda H, Kondo A (2006) Construction of a Pichia pastoris cell-surface display system using Flo1p anchor system. Biotechnol Prog 22:989–993
Tanino T, Ohno T, Aoki T, Fukuda H, Kondo A (2007) Development of yeast cells displaying Candida antarctica lipase B and their application to ester synthesis reaction. Appl Microbiol Biotechnol 75:1319–1325
Tanino T, Aoki T, Chung WY, Watanabe Y, Ogino C, Fukuda H, Kondo A (2009) Improvement of a Candida antarctica lipase B-displaying yeast whole-cell biocatalyst and its application to the polyester synthesis reaction. Appl Microbiol Biotechnol 82:59–66
Ueda M (2004) Future direction of molecular display by yeast-cell surface engineering. J Mol Catal B Enzym 28:139–143
Ueda M, Tanaka A (2000) Cell surface engineering of yeast: construction of arming yeast with biocatalyst. J Biosci Bioeng 90:125–136
Wang QJ, Li L, Chen M, Qi QQ, Wang PG (2007) Construction of a novel system for cell surface display of heterologous proteins on Pichia pastoris. Biotechnol Lett 29:1561–1566
Wang Q, Li L, Chen M, Qi Q, Wang PG (2008) Construction of a novel Pichia pastoris cell-surface display system based on the cell wall protein Pir1. Curr Microbiol 56:352–357
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (No. 30670053) and the Ministry of Science and Technology of the People's Republic of China (National “863” Project No. 2006AA020203).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Su, GD., Huang, DF., Han, SY. et al. Display of Candida antarctica lipase B on Pichia pastoris and its application to flavor ester synthesis. Appl Microbiol Biotechnol 86, 1493–1501 (2010). https://doi.org/10.1007/s00253-009-2382-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-009-2382-0