
Abstract
Phage display with antibody libraries has been widely used with versatile applications. However, phage display with cDNA libraries is rare and inefficient. Because of uncontrollable reading frames and stop codons in cDNA repertoires, high percentage of phage clones identified from conventional cDNA libraries are non-open reading frames (non-ORFs) encoding unnatural short peptides with minimal implications in protein networks. Consequently, phage display has not been used as a technology of functional proteomics to elucidate protein–protein interactions like yeast two-hybrid system and mass spectrometry-based technologies. Several strategies, including C-terminal display and ORF cDNA libraries, have been explored to circumvent the technical problem. The accumulative endeavors eventually led to the efficient elucidation of a large number of tubby- and phosphatidylserine-binding proteins in recent studies by ORF phage display with minimal reading frame issue. ORF phage display inherits all the versatile applications of antibody phage display, but enables efficient identification of real endogenous proteins with efficiency, sensitivity, and accuracy comparable to other technologies of functional proteomics. Its ELISA-like procedure can be conveniently adapted by individual laboratories or fully automated for high-throughput screening. Thus, ORF phage display is an efficient, sensitive, versatile, and convenient technology of functional proteomics for elucidation of global and pathway-specific protein–protein interactions, disease mechanisms, or therapeutic targets.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
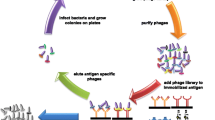
Phage display is a technology first described by Smith (1985) to identify polypeptides with specific bait-binding activity and subsequently evolved with many versatile applications (Paschke 2006; Sidhu 2001; Fig. 1). In phage display, foreign cDNA library is genetically fused to a phage capsid protein in phage genome, so that the library proteins are expressed as capsid fusion proteins and displayed on phage surface. Each phage displays multiple copies of the same foreign protein. Two unique features of phage display, the physical linkage of a polypeptide’s phenotype to its corresponding genotype and the rescue of bait-binding phages, enable enrichment of phages with specific bait-binding activity by multiple rounds of affinity selection (Fig. 1). Compared with yeast two-hybrid system (Y2H) and other cloning technologies, phage enrichment substantially improves the efficiency and sensitivity of identifying unknown bait-binding polypeptides.

General scheme of phage display with affinity selection or panning. Bait molecule is immobilized onto 96 well ELISA plates or bead surface, blocked, and incubated with phage library displaying different proteins (different colors). After washing, bait-binding phages (with blue color protein) are eluted. A small aliquot of eluted phages are used for phage quantification by plaque assay (for T7 phage). The remaining eluted phages is amplified in host bacteria and used as input for the next round of selection. After multiple rounds of selection, bait-binding phages are enriched and can be individually characterized for their bait-binding activity. Non-specific binding phages (with non-blue color proteins) are also co-eluted (not shown) and amplified, but will be marginalized by multiple rounds of affinity selection
Phage display systems can be classified into two categories: non-lytic phage display and lytic phage display. Non-lytic phage display systems use vectors derived from filamentous phages (M13, fl, or fd; Barbas et al. 2000; Paschke 2006; Sidhu 2005). The most popular strategy is to fuse the library proteins to the N-terminus of phage gene III capsid protein (pIII). Protein display on other phage capsid proteins, such as pVI, pVII, pVIII, and pIX, was also described (Kehoe and Kay 2005). Most filamentous phage display systems use phagemids, which are plasmids expressing only capsid fusion protein with a packaging signal and require a helper phage to provide wild-type pIII and other phage proteins to “rescue” the assembly of phagemids as phage articles with the displayed foreign proteins. Detailed strategies of filamentous phage display are covered by other excellent reviews (Kehoe and Kay 2005; Paschke 2006). Lytic phage display includes lambda phage and T7 phage (Danner and Belasco 2001; Santini et al. 1998; Zhang et al. 2005). Unlike filamentous phagemids, foreign cDNA library is directly inserted into lambda or T7 phage genome and expressed as capsid fusion proteins. A unique feature of lytic phage display is that it is not necessary for the proteins displayed on the surface of lambda and T7 phage to be secreted through the host bacterial membrane (Kruger and Schroeder 1981). However, this is an essential step in filamentous phage assembly (Russel 1991).
A popular strategy of phage display is affinity selection or phage panning with bait immobilized on plate or bead surface (Fig. 1). The bait molecules can be either proteins, such as antibodies (Zhang et al. 2005), or non-protein molecules, including fatty acids (Gargir et al. 2002), phospholipids (Nakai et al. 2005), polysaccharides (Deng et al. 1994), RNAs (Danner and Belasco 2001), DNAs (Cicchini et al. 2002), etc. The bait can also be multimolecular complexes, such as viruses (Lim et al. 2008), cells (Kehoe and Kay 2005; Zhang et al. 2007), tissues, or organs (Valadon et al. 2006). Phage affinity selection can be performed in either in vitro or in vivo settings (Li et al. 2006; Valadon et al. 2006). Moreover, various strategies of functional selection have also been described in literature. For example, phage display has been used to elucidate specific substrate motifs for proteases and kinases from random peptide libraries (i.e., substrate phage display; Deperthes 2002; Paschke 2006; Schmitz et al. 1996; Sidhu 2005), or to identify antibodies with cell internalization capacity (Becerril et al. 1999; Goenaga et al. 2007).
Phage display with cDNA library
Phage display has been widely used to identify bait-binding antibodies or short peptides from antibody libraries or random peptide libraries (Paschke 2006; Szardenings 2003). However, phage display with cDNA libraries is rare and inefficient. Among more than 4,000 literature citations related to phage display, only a few (∼5%) deal with cDNA libraries. The critical issue is possible reading frame shifts in the cDNA repertoires fused to the N-terminus of filamentous phage pIII. Antibody libraries with predictable reading frames can be conveniently fused to pIII in correct frames without problem, whereas cDNA repertoires with unpredictable reading frames and stop codons may interfere with pIII expression, resulting in only ∼6% of identified clone encoding real proteins (Faix et al. 2004). Majority of identified non-open reading frames (non-ORFs) encoding unnatural short peptides have minimal implications in protein interaction networks.
Several strategies have been developed to circumvent the problem. One strategy is to display polypeptides at the C-terminus of pIII, pVI, and pVIII (Jestin 2008; Paschke 2006). Moreover, to avoid the difficulties of displaying cDNA library proteins at the C-terminus of pIII, Crameri and Suter (1993) innovatively generated phagemid pJuFo, in which c-Jun leucine zipper domain was displayed on the N-terminus of pIII in frame. In addition, cDNA library was fused to the C-terminus of c-Fos leucine zipper domain and secreted with a PelB signal sequence at the N-terminus of c-Fos. Both leucine zipper domains were flanked by cysteine residues. The Fos-library fusion proteins were captured by displayed c-Jun domain with the formation of heterodimer and disulfide bonds. Furthermore, T7 phage display system with cDNA library proteins fused to the C-terminus of capsid 10B protein was also commercially developed by Novagen (Danner and Belasco 2001). Lambda phage display vector with foreign proteins fused to the C-terminus of capsid D protein was engineered (Santini et al. 1998).
However, C-terminal display cannot ensure that the cDNA library is expressed in the correct reading frames. This is well illustrated by a study in which only less than 10% (24/243) of clones identified from a conventional C-terminal cDNA library of T7 phage display were ORFs (Kalnina et al. 2008). Another study with a similar cDNA library showed only ∼6% (8/130) of identified clones encoding real proteins (Lin et al. 2007). Identified unnatural peptides encoded by non-ORFs have minimal implications in protein networks. As a result, more than a decade after the description of pJuFo phagemid and T7 phage display, literature citations about cDNA phage display remain rare and sporadic, mostly reporting merely one or two identified proteins without elaborating high frequencies of non-ORFs. In this regard, phage display with C-terminal cDNA libraries fails to identify protein–protein interactions with an efficiency comparable to Y2H or mass spectrometry-based technologies of functional proteomics.
ORF phage display
An alternative strategy to improve the efficiency to identify real proteins is ORF cDNA libraries. The principle to construct ORF cDNA libraries is based on the fact that non-ORF cDNA has high frequency of stop codon(s). Database analysis revealed that ∼96% of 200-bp non-ORF cDNAs have at least one stop codon (Garufi et al. 2005). This number drastically increases to 99.6% for non-ORF cDNAs with 300 bp. A C-terminal selection tag or marker is expressed only with ORF cDNA inserts (Fig. 2). Tag- or marker-based selection eliminates non-ORFs and generates ORF libraries. Several strategies have been explored for the C-terminal selection and are summarized here.

Illustration of ORF phage display cDNA library with a C-terminal biotin tag. The library proteins are fused to the C-terminus of phage capsid, followed by a biotin tag. If the library protein is an ORF, biotin tag is expressed and biotinylated. Otherwise, the biotin tag is not expressed in non-ORF phage clone. Biotinylated ORF phage clones can be enriched by binding to immobilized streptavidin to generate ORF cDNA library
C-terminal phage capsid selection
In theory, if cDNA library proteins were fused to the N-terminus of a phage capsid, non-ORF phage clones with stop codon(s) would not express the fusion capsid. As a result, non-ORF phages would be efficiently eliminated by phage panning. However, this strategy typically results in only ∼6% of identified phage clones encoding ORFs (Faix et al. 2004). One of the possible explanations for low percentage of ORFs is that filamentous phagemids encoding the library-pIII fusion protein require a helper phage carrying a predominant wild-type pIII gene to supply other proteins for the rescue of the phagemid assembly. It was speculated that avoiding the delivery of wild-type pIII during the phage packaging may solve this problem. Consequently, a new type of phage packaging system of hyperphage was developed to eliminate the packaging of wild-type pIII (Rondot et al. 2001). Approximately, 60% of cDNA library phages generated with the hyperphage had ORF inserts (Hust et al. 2006). However, phage panning with this ORF cDNA library was not reported. As a result, the efficiency of this system to identify ORF phage clones is unknown.
As short non-ORF cDNA inserts may also lack a stop codon, the quality of ORF library is determined by the size distribution of cDNA inserts. Pavoni et al. (2004) fused a cDNA repertoire to the N-terminus of lambda phage capsid D protein. Despite multiple fractionations of the cDNA repertoire for 300–1,000 bp fragments, including the final procedure of polyacrylamide gel electrophoresis (PAGE) purification, more than ∼57% of the library clones have cDNA inserts smaller than 300 bp with most of them between 100 and 200 bp. These data suggest that the size distribution of a cDNA library does not always mirror the fractionated cDNA repertoire before the library ligation. Shorter cDNA inserts are more efficient for DNA ligation. Consequently, it is important to characterize the actual size distribution of cDNA inserts in ORF libraries to ensure their quality. Although Pavoni et al. (2004) identified 21 clones encoding 18 different proteins from the lambda phage library with immobilized cancer-related antibodies, the percentage of ORFs was not described.
C-terminal ampicillin selection
The concept of C-terminal selection with an antibiotic resistant gene to remove deletion mutants from antibody library was originally described by Seehaus et al. (1992) with a plasmid in which antibody library was cloned upstream of a β-lactamase gene. Zacchi et al. (2003) further demonstrated a similar strategy with a phagemid, wherein cDNA inserts were followed by β-lactamase gene and pIII. The β-lactamase gene was flanked by two homologous lox sites. After ampicillin selection, the β-lactamase gene was removed by Cre recombinase-mediated recombination. The removal of β-lactamase gene was necessary for the efficient display of foreign polypeptides at pIII N-terminus. Faix et al. (2004) pre-selected ORFs with a C-terminal β-lactamase gene in a plasmid. The sequences of ORFs were extracted from ampicillin-resistant plasmids, re-cloned into a phagemid, and rescued by hyperphage. The library had ∼87% of ORF clones. Affinity selection with a monoclonal antibody (mAb) against human placental lactogen identified eight clones with six ORFs encoding lactogen. However, the technical challenge of this strategy is the complicated procedure of generating the ORF phage display cDNA library with a shuttle plasmid. In fact, the cDNA library in the ampicillin-resistant shuttle plasmid in this study had only limited representation of 1 × 106 clones (Faix et al. 2004), which restricted the quality of the subsequent phagemid cDNA library.
C-terminal biotin tag
Ansuini et al. (2002) generated ORF phage display cDNA library in lambda phage with a C-terminal 13-amino acid biotinylation epitope or biotin tag. cDNA library was fused to the C-terminus of capsid D protein, followed by the biotin tag. If a cDNA insert is an ORF, the C-terminal tag is expressed and efficiently biotinylated by biotin holoenzyme synthetase (BirA) endogenously present in Escherichia coli (Schatz 1993). As a result, only the ORF phage clones are labeled with biotin (Fig. 2) and enriched by binding to immobilized streptavidin. Affinity selection with anti-GAP-43 mAb was used as a model system to evaluate the library. After selection, a total of 34 clones were randomly chosen and analyzed with ∼79% of them in correct reading frames, including seven GAP-43-expressing clones.
ORF phage display as a technology for functional proteomics
Despite the limited success of various strategies, a critical question is whether phage display technology can identify protein–protein interactions with efficiency, sensitivity, and accuracy comparable to Y2H or mass spectrometry-based technologies of functional proteomics. To address this question, we recently engineered T7 phage-based system of ORF phage display with four technical improvements: high-quality ORF cDNA libraries, protease cleavage for specific phage elution, dual phage display for sensitive high-throughput screening and post-panning ORF re-selection (Caberoy et al. 2009b).
ORF phage display cDNA library in a T7 vector
We chose to engineer T7 bacteriophage for ORF phage display because of its C-terminal display and robust growth rate. A cleavage motif for human rhinovirus (HRV) 3C protease (Cordingley et al. 1990) was fused to the C-terminus of capsid 10B protein, followed by two GS flexible linkers and a biotin tag in T7Bio3C vector (Caberoy et al. 2009b). Two orientation-directed ORF cDNA libraries were generated from mouse eye and embryo by tagged random priming method and inserted at the multiple cloning site between the two GS linker sequences (Caberoy et al. 2009a, b). The initial titer immediately after phage packaging was 2 × 107 pfu for the eye library and 1 × 108 pfu for the embryo library. Considering three possible starting frames, three possible ending frames, 5′- and 3′-untranslated regions, it is estimated that only ∼1 out of 20–30 clones may be in correct reading frame with capsid 10B and the C-terminal biotin tag. In this regard, the library with the initial titer of 2 × 107 pfu after packaging can cover each of ∼28,900 ORFs in mouse genome in correct reading frames about ∼28 times on average. Given the length of average ORF in mouse genome, various abundance of mRNA transcripts and their differential processing, this coverage is probably a minimal requirement. Both libraries have more than 75% of cDNA inserts longer than 300 bp with most of the inserts between 300–500 bp (Caberoy et al. 2009b). The libraries have more than 90% ORF clones.
Specific phage elution with protease digestion
Although specific phage elution will minimize non-specific phage enrichment, most studies used non-specific elution methods in phage panning, including trypsin digestion of bait protein, pH alternation (Barbas et al. 2000), 1% SDS for T7 phages (Zhang et al. 2005) or direct infection with host bacteria (Pavoni et al. 2004). A more specific elution is the competitive elution with an excessive amount of the ligand or bait molecule. An alternative method to elute bound phage is trypsin digestion with a trypsin cleavage site inserted between phage capsid and library protein (Kristensen and Winter 1998). However, trypsin with broad substrate specificity may likely digest immobilized bait to release phages non-specifically bound through other phage surface proteins. To develop a broadly applicable strategy for specific elution, we inserted a consensus cleavage sequence of LEVLFQ↓GP (arrow for cleavage site) for 3C protease between capsid 10B and C-terminal library proteins in T7Bio3C vector (Caberoy et al. 2009b). 3C protease can efficiently cleave at 4°C and specifically elute phages bound to bait proteins through the displayed proteins rather than through the other phage surface proteins (Fig. 3). In fact, 3C protease digestion at 4°C for 1 h eluted ∼80 times more bound phages than the standard elution method of 1% SDS for T7 phage, suggesting that this is a more specific and efficient method for phage elution. This strategy should be broadly applicable to other phage vectors.

Specific phage elution by 3C protease cleavage. A 3C cleavage site is inserted between phage capsid and the library protein. 3C protease digestion specifically releases the phages (with blue protein) bound to immobilized bait through the displayed library proteins, but not the non-specific phages (non-blue protein) bound through other phage surface proteins. Non-specific phages may also be present in the eluate, but will be marginalized by multiple rounds of affinity selection
Dual phage display
In filamentous phage display, bound phages are conveniently quantified by ELISA using anti-phage capsid antibodies. Although anti-T7 tag mAb is commercially available from Novagen for Western blot and has been used for phage quantification by ELISA in the literature (Sheu et al. 2003), the mAb actually is not recommended by the company for ELISA due to limited sensitivity. As a result, T7 phage is traditionally quantified by tedious plaque assays. To efficiently quantify bound phages, we developed dual T7 phage display to replace the plaque assay. T7 phage consists of 415 copies of capsid 10A and/or 10B (Caberoy et al. 2009b). Each engineered T7 phage displays ∼5–15 copies of a library protein fused to the C-terminus of capsid 10B with the remaining copies of C-terminal FLAG-tagged capsid 10A provided by host bacteria. Capsid 10A-FLAG fusion protein can be conveniently switched to 10A-biotin by amplifying the same phage clone in a different bacterial strain expressing capsid 10A-biotin. Co-display of ∼5–15 copies of capsid 10B-library fusion proteins and >400 copies of FLAG-10A on the same phage surface not only allows the functional analysis of library proteins, but also enables the sensitive quantification of bound phages by ELISA using anti-FLAG mAb. Thus, the tedious T7 plaque assay is converted into convenient colorimetric assay for high-throughput screening and analysis of individual bait-binding clones.
Post-panning ORF re-selection
Unlike plate-based Y2H screening, non-ORF phage clones encoding unnatural short peptides tend to outgrow ORF clones through multiple rounds of liquid-based selection and amplification. Post-panning ORF re-selection before the isolation and analysis of individual phage clones will help elimination of emerged non-ORF clones (Caberoy et al. 2009b). Two previous studies with C-terminal selection of β-lactamase had to remove β-lactamase gene to generate ORF phage display cDNA libraries, because β-lactamase reduced the efficiency to display foreign proteins (Faix et al. 2004; Zacchi et al. 2003). Thus, post-panning ORF re-selection is not feasible for β-lactamase-based ORF libraries. In this regard, C-terminal biotin tag is preferred with the convenience of ORF library construction and post-panning ORF re-selection.
ORF phage display to efficiently elucidate protein–protein interactions
To demonstrate the feasibility of ORF phage display to efficiently identify protein–protein interactions, we chose mouse tubby as a bait protein. Tubby belongs to a well-characterized tubby protein family with four members (tubby, tubby-like proteins 1, 2, and 3), which share a highly conserved C-terminal “tubby domain” of ∼260 amino acids (Ikeda et al. 2002). Tubby has been demonstrated as a putative membrane-bound, G protein-activated transcription factor with unknown regulatory gene(s). A splice site mutation in tubby with the replacement of its C-terminal 44 amino acids leads to adult-onset obesity, progressive retinal and cochlear degeneration (Noben-Trauth et al. 1996), whereas a similar mutation in the highly conserved C-terminal end of tubby-like protein 1 (Tulp1) only causes retinal degeneration with no other clinical manifestation (Banerjee et al. 1998). The pathological mechanisms for both proteins are undefined. It is speculated that the diverse N-terminal domains of tubby and Tulp1 interact with different binding partners, which may impart their partially overlapping disease profiles (Caberoy et al. 2009b). Thus, identification of the proteins binding to the N-terminus of tubby (tubby-N) will help elucidate the pathological mechanisms.
We used tubby-N (1M-242P) as a bait to rapidly identify 28 tubby-N-binding phage clones by ORF phage display (Caberoy et al. 2009b). All identified clones were ORFs encoding 16 new tubby-N-binding proteins. We independently analyzed 14 binding proteins by Y2H and/or protein pull-down assay. The data suggest that the accuracy of ORF phage display was ∼71% (10/14). However, their biological relevance is yet to be investigated. Phage binding assay revealed that CCCTC-binding factor (Ctcf) specifically binds to tubby-N, but not Tulp1-N, suggesting that this protein is a likely candidate involved in adult-onset obesity, but not in retinal degeneration. All other verified tubby-binding proteins also bind to Tulp1-N with various activities, implicating that they may be involved in a common pathogenesis, such as retinal degeneration. This study illustrates that efficient identification of protein–protein interactions by ORF phage display may facilitate the elucidation of the disease mechanisms by comparative analysis of their binding specificity.
ORF phage display to identify protein interactions with non-protein molecules
To demonstrate the versatility of ORF phage display, we used phosphatidylserine (PS) as a non-protein bait to identify PS-binding proteins. PS not only serves as a major structural component of cellular membranes, but also functions as a signaling molecule regulating both intracellular and extracellular biological processes (Stace and Ktistakis 2006; Wu et al. 2006). Because of its importance, the search for PS-binding proteins is of considerable interest and should help define its regulatory roles. However, PS-binding proteins are traditionally identified on a case-by-case basis with daunting challenges (Miyanishi et al. 2007; Park et al. 2007). Because of the inefficiency of identifying real proteins from conventional phage display cDNA libraries, a previous study identified PS-binding peptides from a phage display random peptide library, delineated a consensus PS-binding motif, analyzed Drosophila genes for the possible match to the motif, identified and verified a new PS-binding protein (Nakai et al. 2005). This complicated process illustrates the challenge to identify a PS-binding protein.
We used ORF phage display to quickly identify 17 PS-binding phage clones (Caberoy et al. 2009a). All identified clones were ORFs encoding 13 proteins, including one known PS-binding protein and another protein with a known phospholipid-binding C2 domain (Rizo and Sudhof 1998). All except one preferentially bound to PS, but not phosphatidylcholine (PC). Moreover, we expressed three identified proteins, purified the recombinant proteins and independently verified their specific binding to PS liposomes, but not to PC liposomes, by liposome pull-down assay (Caberoy et al. 2009a). These data suggest that ORF phage display is an efficient and versatile technology, applicable not only to protein baits but also to non-protein molecules.
Different technologies of functional proteomics
With the advances in DNA sequencing technologies and possible realization of the $1,000 genome (Bennett et al. 2005), many genomes of different species have been completely sequenced and mapped. The Genome database at NCBI provides a variety of genome information for 5,779 different species. As a result, research focus is shifting from genomics to proteomics. Current proteomics investigations are essentially focused on two major areas: expression proteomics and functional proteomics. Expression proteomics aims to measure the upregulation and downregulation of protein levels, and is typically to investigate protein expression patterns in abnormal or treated cells in comparison with normal or control cells. Functional proteomics is to analyze protein function in large scales. A widely used strategy is to identify protein physical association. Given that proteins regulate nearly every biological process, efficient identification of protein physical association is expected to have major impacts on the elucidation of protein functions. Consequently, developing technologies of functional proteomics is of major importance. Protein affinity purification or tandem affinity purification coupled with 1D or 2D gel electrophoresis and mass spectrometry (AP-MS or TAP-MS) has been widely used to elucidate protein interaction complexes (i.e., interactomes; Collins and Choudhary 2008). Y2H is another popular technology to identify protein–protein interactions (Suter et al. 2008). Both AP/TAP-MS and Y2H have been used for identification of binding proteins for specific protein bait as well as for proteome-scale interactome mapping. The recent applications of ORF phage display to efficiently identify tubby-N-binding and PS-binding proteins with minimal reading frame issue (Caberoy et al. 2009a; Caberoy et al. 2009b) demonstrate that the accumulative efforts from various laboratories, including C-terminal phage display and ORF cDNA libraries, have successfully transformed phage display into a technology of functional proteomics.
Comparison of ORF phage display with Y2H
Because of yeast slow growth rate, Y2H usually takes several weeks or months to finish the screening and verification from 106–107 library clones. One of the advantages of ORF phage display is efficiency. T7 phage has a robust growth rate. Its liquid amplification requires only 1–3 h. As a result, one round of phage panning, including binding, washing, elution, and liquid amplification, can be completed within ∼4 h. Phage enrichment with 2–3 rounds of affinity selection can be finished in 1–2 days. Phage quantification by plaque assay only needs less than 3 h. Coupled with sensitive high-throughput screening by T7 dual phage display, bait-binding proteins can be efficiently identified from more than 1010–1011 pfu library clones in as fast as ∼4–7 days in a regular laboratory setting.
Another advantage of ORF phage display is versatility. Although yeast one-hybrid and three-hybrid systems are capable of identifying DNA- and RNA-binding proteins (Jaeger et al. 2004; Sieweke 2000), Y2H is applicable only to protein–protein interactions (Suter et al. 2008). Phage display with antibody libraries or random peptide libraries has been demonstrated with different strategies of affinity selection and functional selection (Jestin 2008; Paschke 2006; Sidhu 2005). For affinity selection, the bait molecules can be proteins, antibodies (Zhang et al. 2005), non-protein molecules, or even multimolecular complexes (Caberoy et al. 2009c). Affinity selection may be performed in in vitro or in vivo settings (Li et al. 2006; Valadon et al. 2006). Different strategies of functional selection have also been explored, including substrate phage display (Deperthes 2002; Schmitz et al. 1996; Sidhu 2005) and phagocytosis-based phage display (Caberoy et al. 2009c; Goenaga et al. 2007). However, the caveat is that most of these applications have been previously described only with antibody or random peptide libraries. Unfortunately, identified antibodies or unnatural short peptides have minimal implications in protein biological networks. ORF phage display inherits all the versatile applications of phage display with antibody libraries or random peptide libraries, but enables the efficient identification of real endogenous binding proteins or proteins with specific functions.
Comparison of ORF phage display with AP/TAP-MS
As a technology of functional proteomics, AP/TAP-MS has been well demonstrated for efficient mapping of protein interactomes for yeast, E. coli, and human (Collins and Choudhary 2008). A major advantage of AP/TAP-MS is to identify multicomponent protein complexes. The proteins identified by AP/TAP-MS may directly or indirectly bind to the bait protein. Additionally, MS technology is more versatile than Y2H. One of the examples is to elucidate protease substrates or degradomes by MS (Schilling and Overall 2007). However, AP/TAP-MS has several limitations (Table 1). First, single-tag affinity purification or AP may result in high level of promiscuous binding, whereas TAP substantially reduces non-specific interactions with relatively low yield for the materials obtained from TAP. As a result, TAP-MS requires large initial quantities of cells (∼5 × 107–109 cells; Burckstummer et al. 2006). Second, the technology has a limited sensitivity, including the current sensitivity of MS technology to detect proteins at subfemtomole level or 108 copies of the same proteins. Consequently, less abundant proteins may not be detected. Third, lack of ready available cDNA clones for the proteins identified by AP/TAP-MS as direct by-products of the technology itself is transiently inconvenient for independent verification or characterization. Finally, although co-immunoprecipitation coupled with Western blot or other detection methods is widely used to verify proteins identified by AP/TAP-MS, co-immunoprecipitation itself is an integral part of AP/TAP-MS. Thus, the validation in a sense is not truly independent.
Other advantages and limitations
Besides efficiency and versatility, sensitivity and accuracy are two other important parameters for the technologies of functional proteomics. A technology with low sensitivity will miss most of genuine binding proteins. On the other hand, a highly sensitive technology with poor accuracy will identify a large number of false positives, which not only are misleading but also may consume enormous amount of time and effort to verify genuine binding partners. Although in theory Y2H can detect single clone of bait-binding proteins, labor-intensive procedure of Y2H is a rate limiting step for screening more library clones. Yeast transformation efficiency may also influence the sensitivity of the technology. Less abundant clones with high binding activity could be overlooked because of variation in the screening and detection conditions. For example, a previous study identified only one tubby-binding protein from a Caenorhabditis elegans cDNA library by Y2H (Mukhopadhyay et al. 2005). It is unknown whether any other identified binding proteins were not reported. In contrast, ORF phage display identified 16 new tubby-binding proteins (Caberoy et al. 2009b). The unique phage enrichment process substantially improves the efficiency and sensitivity of identifying unknown bait-binding proteins from up to 1010–1011 pfu library clones. Once they survived the first round of selection, less abundant phage clones with high affinity will be amplified more than 1 million times in bacteria before the next round of selection. Thus, less abundant binding proteins will be much more efficiently and sensitively identified than other technologies.
While the estimated error rate is ∼15% for TAP-MS, ∼50% for AP-MS, and ∼45–80% for Y2H (Dziembowski and Seraphin 2004), the error rate for ORF phage display is ∼29% (Caberoy et al. 2009b). Although these initial studies suggest that ORF phage display has competitive sensitivity and accuracy, both parameters should be further analyzed with more diverse baits in large scales and compared to Y2H and AP/TAP-MS.
Although Y2H and AP/TAP-MS have been widely used to identify protein–protein interactions, they are limited by high cost, technical complexity, instrument requirements, labor, and time commitments (Table 1). Furthermore, their application is narrowly restricted to protein–protein interactions, but not to protein interactions with other non-protein molecules.
A major disadvantage of bacterium-based ORF phage display is that proteins displayed on phage surface lack appropriate post-translational modifications, such as glycosylation. Thus, glycosylation-dependent protein–protein interactions may not be identified by ORF phage display. Moreover, unlike Y2H and TAP-MS, ORF phage display requires pure bait proteins for affinity selection. As many recombinant proteins and non-protein molecules, such as PS, are already commercially available, this is a major advantage for ORF phage display. On the other hand, the requirement of pure bait proteins is also a major barrier for proteome-scale interactome mapping. Integration of the procedure of protein affinity purification or tandem affinity purification (Collins and Choudhary 2008) into ORF phage display, i.e., AP/TAP-ORF phage display, may eliminate the requirement for pure protein bait and facilitate the efficient mapping of protein–protein interactions in a large scale with an efficiency and sensitivity comparable to Y2H and TAP-MS.
Prospects
In summary, ORF phage display is an efficient, sensitive, versatile, and convenient technology for elucidation of proteins with specific binding activities and functions. Its application as a technology of functional proteomics is only at the beginning. One of the major advantages of ORF phage display over other technologies is its versatile applications (Table 1), such as the unique applications for eat-me signals and in vivo selection, which are yet to be explored. ORF phage display with a straightforward procedure can be conveniently adapted by individual laboratories with minimal technical requirement for bacterial culture, ELISA, and PCR. Once ORF libraries generated from various normal or diseased tissues are widely available, tens of thousands of research laboratories around the world will be able to conveniently perform ORF phage display to identify protein–protein interactions of their interest at a fraction of the costs for other technologies. Moreover, ORF phage display with an ELISA-like procedure can be fully automated for high-throughput screening. Therefore, ORF phage display has the potential to join Y2H and AP/TAP-MS as a major technology of functional proteomics for efficient elucidation of protein–protein interactions, disease mechanisms, and therapeutic targets.
References
Ansuini H, Cicchini C, Nicosia A, Tripodi M, Cortese R, Luzzago A (2002) Biotin-tagged cDNA expression libraries displayed on lambda phage: a new tool for the selection of natural protein ligands. Nucleic Acids Res 30:e78
Banerjee P, Kleyn PW, Knowles JA, Lewis CA, Ross BM, Parano E, Kovats SG, Lee JJ, Penchaszadeh GK, Ott J et al (1998) TULP1 mutation in two extended Dominican kindreds with autosomal recessive retinitis pigmentosa. Nat Genet 18:177–179
Barbas CF 3rd, Burton DR, Scott JK, Silverman GJ (2000) Phage display: a laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York
Becerril B, Poul MA, Marks JD (1999) Toward selection of internalizing antibodies from phage libraries. Biochem Biophys Res Commun 255:386–393
Bennett ST, Barnes C, Cox A, Davies L, Brown C (2005) Toward the 1,000 dollars human genome. Pharmacogenomics 6:373–382
Burckstummer T, Bennett KL, Preradovic A, Schutze G, Hantschel O, Superti-Furga G, Bauch A (2006) An efficient tandem affinity purification procedure for interaction proteomics in mammalian cells. Nat Methods 3:1013–1019
Caberoy NB, Zhou Y, Alvarado G, Fan X, Li W (2009a) Efficient identification of phosphatidylserine-binding proteins by ORF phage display. Biochem Biophys Res Commun 386:197–201
Caberoy NB, Zhou Y, Jiang X, Li W (2009b) Efficient identification of tubby-binding proteins by an improved system of T7 phage display. J Mol Recognit, in press
Caberoy NB, Zhou Y, Li W (2009c) Can phage display be used as a tool to functionally identify endogenous eat-me signals in phagocytosis? J Biomol Screen 14:653–661
Cicchini C, Ansuini H, Amicone L, Alonzi T, Nicosia A, Cortese R, Tripodi M, Luzzago A (2002) Searching for DNA–protein interactions by lambda phage display. J Mol Biol 322:697–706
Collins MO, Choudhary JS (2008) Mapping multiprotein complexes by affinity purification and mass spectrometry. Curr Opin Biotechnol 19:324–330
Cordingley MG, Callahan PL, Sardana VV, Garsky VM, Colonno RJ (1990) Substrate requirements of human rhinovirus 3C protease for peptide cleavage in vitro. J Biol Chem 265:9062–9065
Crameri R, Suter M (1993) Display of biologically active proteins on the surface of filamentous phages: a cDNA cloning system for selection of functional gene products linked to the genetic information responsible for their production. Gene 137:69–75
Danner S, Belasco JG (2001) T7 phage display: a novel genetic selection system for cloning RNA-binding proteins from cDNA libraries. Proc Natl Acad Sci USA 98:12954–12959
Deng SJ, MacKenzie CR, Sadowska J, Michniewicz J, Young NM, Bundle DR, Narang SA (1994) Selection of antibody single-chain variable fragments with improved carbohydrate binding by phage display. J Biol Chem 269:9533–9538
Deperthes D (2002) Phage display substrate: a blind method for determining protease specificity. Biol Chem 383:1107–1112
Dziembowski A, Seraphin B (2004) Recent developments in the analysis of protein complexes. FEBS Lett 556:1–6
Faix PH, Burg MA, Gonzales M, Ravey EP, Baird A, Larocca D (2004) Phage display of cDNA libraries: enrichment of cDNA expression using open reading frame selection. Biotechniques 36:1018–1029
Gargir A, Ofek I, Meron-Sudai S, Tanamy MG, Kabouridis PS, Nissim A (2002) Single chain antibodies specific for fatty acids derived from a semi-synthetic phage display library. Biochim Biophys Acta 1569:167–173
Garufi G, Minenkova O, Lo Passo C, Pernice I, Felici F (2005) Display libraries on bacteriophage lambda capsid. Biotechnol Annu Rev 11:153–190
Goenaga AL, Zhou Y, Legay C, Bougherara H, Huang L, Liu B, Drummond DC, Kirpotin DB, Auclair C, Marks JD, Poul MA (2007) Identification and characterization of tumor antigens by using antibody phage display and intrabody strategies. Mol Immunol 44:3777–3788
Hust M, Meysing M, Schirrmann T, Selke M, Meens J, Gerlach GF, Dubel S (2006) Enrichment of open reading frames presented on bacteriophage M13 using hyperphage. Biotechniques 41:335–342
Ikeda A, Nishina PM, Naggert JK (2002) The tubby-like proteins, a family with roles in neuronal development and function. J Cell Sci 115:9–14
Jaeger S, Eriani G, Martin F (2004) Results and prospects of the yeast three-hybrid system. FEBS Lett 556:7–12
Jestin JL (2008) Functional cloning by phage display. Biochimie 90:1273–1278
Kalnina Z, Silina K, Meistere I, Zayakin P, Rivosh A, Abols A, Leja M, Minenkova O, Schadendorf D, Line A (2008) Evaluation of T7 and lambda phage display systems for survey of autoantibody profiles in cancer patients. J Immunol Methods 334:37–50
Kehoe JW, Kay BK (2005) Filamentous phage display in the new millennium. Chem Rev 105:4056–4072
Kristensen P, Winter G (1998) Proteolytic selection for protein folding using filamentous bacteriophages. Fold Des 3:321–328
Kruger DH, Schroeder C (1981) Bacteriophage T3 and bacteriophage T7 virus-host cell interactions. Microbiol Rev 45:9–51
Li XB, Schluesener HJ, Xu SQ (2006) Molecular addresses of tumors: selection by in vivo phage display. Arch Immunol Ther Exp (Warsz) 54:177–181
Lim AP, Chan CE, Wong SK, Chan AH, Ooi EE, Hanson BJ (2008) Neutralizing human monoclonal antibody against H5N1 influenza HA selected from a Fab-phage display library. Virol J 5:130
Lin HS, Talwar HS, Tarca AL, Ionan A, Chatterjee M, Ye B, Wojciechowski J, Mohapatra S, Basson MD, Yoo GH et al (2007) Autoantibody approach for serum-based detection of head and neck cancer. Cancer Epidemiol Biomarkers Prev 16:2396–2405
Miyanishi M, Tada K, Koike M, Uchiyama Y, Kitamura T, Nagata S (2007) Identification of Tim4 as a phosphatidylserine receptor. Nature 450:435–439
Mukhopadhyay A, Deplancke B, Walhout AJ, Tissenbaum HA (2005) C. elegans tubby regulates life span and fat storage by two independent mechanisms. Cell Metab 2:35–42
Nakai Y, Nomura Y, Sato T, Shiratsuchi A, Nakanishi Y (2005) Isolation of a Drosophila gene coding for a protein containing a novel phosphatidylserine-binding motif. J Biochem 137:593–599
Noben-Trauth K, Naggert JK, North MA, Nishina PM (1996) A candidate gene for the mouse mutation tubby. Nature 380:534–538
Park D, Tosello-Trampont AC, Elliott MR, Lu M, Haney LB, Ma Z, Klibanov AL, Mandell JW, Ravichandran KS (2007) BAI1 is an engulfment receptor for apoptotic cells upstream of the ELMO/Dock180/Rac module. Nature 450:430–434
Paschke M (2006) Phage display systems and their applications. Appl Microbiol Biotechnol 70:2–11
Pavoni E, Vaccaro P, Pucci A, Monteriu G, Beghetto E, Barca S, Dupuis ML, De Pasquale Ceratti A, Lugini A, Cianfriglia M et al (2004) Identification of a panel of tumor-associated antigens from breast carcinoma cell lines, solid tumors and testis cDNA libraries displayed on lambda phage. BMC Cancer 4:78
Rizo J, Sudhof TC (1998) C2-domains, structure and function of a universal Ca2+-binding domain. J Biol Chem 273:15879–15882
Rondot S, Koch J, Breitling F, Dubel S (2001) A helper phage to improve single-chain antibody presentation in phage display. Nat Biotechnol 19:75–78
Russel M (1991) Filamentous phage assembly. Mol Microbiol 5:1607–1613
Santini C, Brennan D, Mennuni C, Hoess RH, Nicosia A, Cortese R, Luzzago A (1998) Efficient display of an HCV cDNA expression library as C-terminal fusion to the capsid protein D of bacteriophage lambda. J Mol Biol 282:125–135
Schatz PJ (1993) Use of peptide libraries to map the substrate specificity of a peptide-modifying enzyme: a 13 residue consensus peptide specifies biotinylation in Escherichia coli. Biotechnology (N Y) 11:1138–1143
Schilling O, Overall CM (2007) Proteomic discovery of protease substrates. Curr Opin Chem Biol 11:36–45
Schmitz R, Baumann G, Gram H (1996) Catalytic specificity of phosphotyrosine kinases Blk, Lyn, c-Src and Syk as assessed by phage display. J Mol Biol 260:664–677
Seehaus T, Breitling F, Dubel S, Klewinghaus I, Little M (1992) A vector for the removal of deletion mutants from antibody libraries. Gene 114:235–237
Sheu TJ, Schwarz EM, Martinez DA, O’Keefe RJ, Rosier RN, Zuscik MJ, Puzas JE (2003) A phage display technique identifies a novel regulator of cell differentiation. J Biol Chem 278:438–443
Sidhu SS (2001) Engineering M13 for phage display. Biomol Eng 18:57–63
Sidhu SS (2005) Phage display in biotechnology and drug discovery. CRC, Boca Raton, FL
Sieweke M (2000) Detection of transcription factor partners with a yeast one hybrid screen. Methods Mol Biol 130:59–77
Smith GP (1985) Filamentous fusion phage: novel expression vectors that display cloned antigens on the virion surface. Science 228:1315–1317
Stace CL, Ktistakis NT (2006) Phosphatidic acid- and phosphatidylserine-binding proteins. Biochim Biophys Acta 1761:913–926
Suter B, Kittanakom S, Stagljar I (2008) Two-hybrid technologies in proteomics research. Curr Opin Biotechnol 19:316–323
Szardenings M (2003) Phage display of random peptide libraries: applications, limits, and potential. J Recept Signal Transduct Res 23:307–349
Valadon P, Garnett JD, Testa JE, Bauerle M, Oh P, Schnitzer JE (2006) Screening phage display libraries for organ-specific vascular immunotargeting in vivo. Proc Natl Acad Sci USA 103:407–412
Wu Y, Tibrewal N, Birge RB (2006) Phosphatidylserine recognition by phagocytes: a view to a kill. Trends Cell Biol 16:189–197
Zacchi P, Sblattero D, Florian F, Marzari R, Bradbury AR (2003) Selecting open reading frames from DNA. Genome Res 13:980–990
Zhang Y, Davis JL, Li W (2005) Identification of tribbles homolog 2 as an autoantigen in autoimmune uveitis by phage display. Mol Immunol 42:1275–1281
Zhang Y, Chen J, Zhang Y, Hu Z, Hu D, Pan Y, Ou S, Liu G, Yin X, Zhao J et al (2007) Panning and identification of a colon tumor binding peptide from a phage display peptide library. J Biomol Screen 12:429–435
Acknowledgments
This work is supported in part by NIH grant R01EY016211. N. Caberoy is a recipient of a Fight for Sight postdoctoral fellowship. We thank Dr. Sanjoy Bhattacharya for discussion.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Li, W., Caberoy, N.B. New perspective for phage display as an efficient and versatile technology of functional proteomics. Appl Microbiol Biotechnol 85, 909–919 (2010). https://doi.org/10.1007/s00253-009-2277-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-009-2277-0