
Abstract
Prolidases hydrolyze the unique bond between X-Pro dipeptides and can also cleave the P–F and P–O bonds found in organophosphorus compounds, including the nerve agents, soman and sarin. The advantages of using hyperthermophilic enzymes in biodetoxification strategies are based on their enzyme stability and efficiency. Therefore, it is advantageous to examine new thermostable prolidases for potential use in biotechnological applications. Two thermostable prolidase homologs, PH1149 and PH0974, were identified in the genome of Pyrococcus horikoshii based on their sequences having conserved metal binding and catalytic amino acid residues that are present in other known prolidases, such as the previously characterized Pyrococcus furiosus prolidase. These P. horikoshii prolidases were expressed recombinantly in the Escherichia coli strain BL21 (λDE3), and both were shown to function as proline dipeptidases. Biochemical characterization of these prolidases shows they have higher catalytic activities over a broader pH range, higher affinity for metal and are more stable compared to P. furiosus prolidase. This study has important implications for the potential use of these enzymes in biotechnological applications and provides further information on the functional traits of hyperthermophilic proteins, specifically metalloenzymes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Pyrococcus horikoshii is a hyperthermophilic, anaerobic organism that was isolated from a deep (1,400-m depth) hydrothermal vent in the Okinawa Trough in the northeastern Pacific Ocean (Gonzalez et al. 1998). Closely related Pyrococcus furiosus was isolated from a shallow marine solfatara at Vulcano Island off the coast of Italy (Fiala and Stetter 1986). P. furiosus is one of the most studied hyperthermophiles to date (Adams 1993). Both Pyrococcus species grow optimally at extreme temperatures of 98–100 °C and at pH 7.0 (Gonzalez et al. 1998). They are both obligately heterotrophic anaerobes that are able to use selected peptides and proteins to produce organic acids, CO2, and H2 (Fiala and Stetter 1986; Gonzalez et al. 1998). P. furiosus grows on maltose, starch, and pyruvate, whereas P. horikoshii cannot and is only able to utilize proteinaceous media substrates such as yeast extract, tryptone, or a 21-amino-acid mixture supplemented with vitamins (Gonzalez et al. 1998; Schut et al. 2003). P. horikoshii, unlike other closely related species such as P. furiosus and Pyrococcus abyssi, requires tryptophan and histidine for growth and cannot grow on a nonpeptide carbon source (Gonzalez et al. 1998; Lecompte et al. 2001). The growth profile for these three organisms is similar (Gonzalez et al. 1998), with the main differences being the substrates they are able to use for energy, which could lead to subtle differences in the regulation of metabolism.
The genomes of P. furiosus and P. horikoshii have been compared. The genomes consist of 1.908 and 1.738 mbp for P. furiosus and P. horikoshii, respectively (Maeder et al. 1999). The missing 170-kbp gap between the two organisms contains sequences that code for amino acid biosynthetic pathways, specifically trp, his, aro, leu-ile-val, arg, pro, cys, thr, and mal operons (Maeder et al. 1999). However, P. horikoshii has many genes for chemotaxis that P. furiosus lacks (Lecompte et al. 2001; Maeder et al. 1999). The reliance of P. horikoshii on proteinaceous substrates for energy production and metabolism suggests that P. horikoshii may require more proteases that function to provide the organism with amino acids that it is unable to make on its own. This could explain why P. horikoshii possesses more than one functional proline dipeptidase.
Enzymes that are able to catalyze the hydrolysis of proteins into smaller peptide fractions and amino acids are defined as proteases. There are few proteases that are able to cleave a peptide bond adjacent to a proline residue. This is due to the conformational constraint that the cyclic structure of proline puts on a peptide bond (Cunningham and O’Connor 1997). Prolidase is one of these proteases that is able to hydrolyze dipeptides with proline in the C-terminus, X-Pro, and a nonpolar amino acid in the N-terminus (Lowther and Matthews 2002). Prolidases are ubiquitous in nature and can be found in archaea, bacteria, and mammals (Endo et al. 1989; Fernandez-Espla et al. 1997; Ghosh et al. 1998; Suga et al. 1995). It is unclear what role prolidase plays in archaea and bacteria, but it has been suggested to aid in protein degradation and could be responsible for the recycling of proline. Due to its reaction mechanism, it could also play a role in regulating biological processes (Cunningham and O’Connor 1997). In humans, however, prolidase has been shown to be involved in the final stage of the degradation of endogenous and dietary protein and is important in the breakdown of collagen. PD, or prolidase deficiency in humans, is a recessive disorder marked by mutations and or deletions in the human prolidase gene (Ledoux et al. 1996). PD is characterized by skin ulcerations, mental retardation, and recurrent infections of the respiratory tract (Endo et al. 1989; Forlino et al. 2002).
The specificity of prolidase reactions and the substrates that it is able to cleave may be dependent on the metal center it possesses. Prolidase belongs to a small class of metalloenzymes known as the “pita bread enzymes” because they contain the same pita bread fold encompassing a similar metal center and substrate-binding pocket (Lowther and Matthews 2002). Other enzymes within this class include methionine aminopeptidase, aminopeptidase P, and creatinase, each having slightly different substrate specificity, but the same conserved metal binding pocket suggesting they might have a conserved catalytic mechanism (Lowther and Matthews 2002). Most enzymes in this class require one or two divalent ions such as Co2+, Mn2+, or Zn2+ to be present in their active sites to catalyze a reaction, with one of the metal atoms being more tightly bound than the other (Lowther and Matthews 2002). The first prolidase structurally and biochemically characterized was from the hyperthermophilic archaeon P. furiosus (Ghosh et al. 1998; Grunden et al. 2001; Maher et al. 2004). P. furiosus prolidase showed maximum activity at 100 °C and pH 7.0 (Ghosh et al. 1998; Grunden et al. 2001) and a narrow substrate specificity, only hydrolyzing dipeptides with a proline in the C-terminus and nonpolar amino acid (Leu, Met, Val, Phe, or Ala) in the N-terminus (Ghosh et al. 1998). This dipeptidase is maximally active with the addition of divalent cations Co2+ and Mn2+ but cannot be substituted with other divalent cations (Mg2+, Ca2+, Ni2+, Cu2+, or Zn2+; Ghosh et al. 1998). It requires two cobalt atoms occupying the metal binding sites, one tight binding at residues E313 and H284 and one loose binding at D209, with a Kd of 0.24 mM (Du et al. 2005). P. furiosus prolidase has also been shown to be maximally active with Fe2+ (1,434 U/mg) and somewhat less active with Co2+ (573.6 U/mg) under anaerobic conditions, suggesting that the metal preference in vivo may be for Fe2+ rather than Co2+ (Du et al. 2005).
Recombinant prolidase has use in several biotechnological applications (Theriot et al. 2009). It is used by the food and dairy industry in the fermentation process, specifically in cheese taste and texture development (Bockelmann 1995). Recombinant prolidase is also being examined for enzyme replacement therapy for patients that have PD. However, it has also been shown to degrade organophosphorus (OP) nerve agents. OP nerve agents act by inhibiting acetylcholinesterase (AChE), which leads to a buildup of acetylcholine in the body and can lead to hypersecretion, convulsions, respiratory problems, coma, and finally, death. OPAAs or organophosphorus acid anhydrolases from Alteromonas have shown the ability to hydrolyze OPs because they are able to cleave the P–F, P–O, P–CN, and P–S bonds of the nerve agents, sarin and soman (Cheng et al. 1998). OPAA can also hydrolyze specific proline dipeptides characterized by [Xaa-Pro], specifically Leu-Pro and Ala-Pro, therefore it is now considered a prolidase. OPAA’s target substrates (soman and sarin) mimic that of the prolidase specific substrates [Xaa-Pro] in shape, size, and surface charges (Cheng and DeFrank 2000). Prolidase could be a potential biodecontaminant for detoxification of OP nerve agents in the field. An activity of 30 U mg−1 was seen when purified P. furiosus prolidase was tested at 55 °C against G-series OP nerve agent diisopropylfluorophosphate (DFP; Theriot et al. 2009). Currently, Alteromonas OPAA-1 and OPAA-2 prolidases are used in a foam formulation DS2 for biodecontamination, although metal is still required for enzyme activity (Cheng et al. 1999). It is necessary to study other candidate prolidases in the hopes of identifying an enzyme that will be more efficient in detoxifying OP nerve agents under harsh conditions that exist with field applications. To further evaluate new enzymes that could potentially be used for decontamination of OP nerve agents, two putative prolidases from P. horikoshii have been expressed and biochemically characterized.
Materials and methods
Identification and cloning of the P. horikoshii prolidase-encoding genes
The Pfprol accession number (AAL81467) was entered into the Basic Local Alignment Search Tool (BLAST) or blastp to look for similar structures based on Pfprol. The results included homologous protein structures Phprol (BAA30249) and Ph1prol (BAA30071).
P. horikoshii genomic DNA was obtained from ATCC, strain 700860D-5. P. horikoshii prolidase (PH1149), prolidase homolog 1 (PH0974), and prolidase homolog 2 (PH1902) genes were amplified by PCR for subsequent cloning of these genes into the T7-polymerase-driven expression vector pET-21b (Novagen). For the PCR amplification of P. horikoshii prolidase, prolidase homolog 1, and prolidase homolog 2 genes, two primers were designed for each: Phprol primer 1 (5′-AAGATCAAGGAGGTCATATGGACATAA-3′; forward, containing an NdeI restriction site shown in bold) and primer 2 (5′-CCTACTAAAGCTTGCTAGATGAGTTCTC-3′; reverse, containing an HindIII restriction site shown in bold); Ph1prol primer 1 (5′AACCATATGAGGCTTGAAAAGTTCATTCAC-3′; forward, containing an NdeI restriction site shown in bold), and primer 2 (5′-TGAGTCGACAGTAGTAGAATAATAACA-3′; reverse, containing a SalI restriction site shown in bold); Ph2prol primer 1 (5′-GAACTCATATGGTTATGAGAGGGAACA-3′; forward, containing an NdeI restriction site shown in bold) and primer 2 (5′-CACTGGTCGACATAGACATTCTAATAA-3′; reverse, containing a SalI restriction site shown in bold). All primers were designed using MacVector (Accelrys, San Diego, CA) computer software.
PCR amplification was performed using native P. furiosus DNA polymerase (0.2 µl) in a 50 µl reaction solution containing 5.0 µl 10× Taq buffer, 0.4 µl dNTP (25 mM), 0.5 µl forward primer (40 µM), 0.5 µl reverse primer (40 µM), 0.5 µl Taq polymerase, and 1.0 µl P. horikoshii genomic DNA (100 ng/µl). The following PCR protocol was run on the thermocycler (Biorad, Hercules, CA): Two initial cycles for 4 min at 94 °C for denaturation, 1 min at 55 °C for annealing, 1 min at 72 °C for extension; followed by 39 cycles of 94 °C for 1 min, 55 °C for 1 min, 72 °C for 1 min; and one final cycle at 72 °C for 7 min. The prolidase PCR product sizes were 1.08 kb, 1.07 kb, and 1.09 kb for Phprol, Ph1prol, and Ph2prol, respectively. PCR products were electrophoresed through a 1% agarose gel for visual inspection.
The amplified prolidase genes were subsequently cloned into the EcoRV site of plasmid pCR-Script (Stratagene) to yield plasmids phprol-script, ph1prol-script, and ph2prol-script. Plasmids were transformed into Escherichia coli strain XL1-Blue (Novagen), and the transformed cells were plated on Luria–Bertani (LB) plates supplemented with ampicillin (100 µg/ml) and X-gal (40 μg/ml) and incubated at 37 °C overnight. Blue-white screening was used to select colonies for plasmid isolation.
Plasmids with insert were isolated from white colonies and digested with NdeI and HindIII for the phprol gene and NdeI and SalI to excise prolidase homolog 1 and 2 genes, respectively. The excised prolidase and homolog genes were subsequently cloned into the NdeI and HindIII or NdeI and SalI (NEB) sites in expression vector pET-21b (Novagen), resulting in plasmids pET-Phprol, pET-Ph1prol, and pET-Ph2prol. All plasmids were sent to MWG Biotech (High Point, NC) for sequencing to ensure that no sequence changes occurred in the cloning process.
Overexpression of P. horikoshii prolidases
P. horikoshii prolidase expression plasmids and the rare arginine, leucine, and isoleucine tRNA encoding plasmid pRIL (Stratagene) were transformed into E. coli BL21(λDE3) cells (Novagen), which has isopropyl-β-D-thiogalactopyranoside inducible expression of T7-RNA polymerase encoded on the chromosome. Transformants were selected on LB–Ampicillin–Chloramphenicol plates after incubation at 37 °C overnight.
Large-scale protein expression was done for Phprol and Ph1prol by inoculating 1-L cultures of autoinduction media (Studier 2005) supplemented with 100 µg/ml of ampicillin and 34 µg/ml chloramphenicol for plasmid maintenance. Cells were grown with shaking (200 rpm) at 37 °C for 14 h. Cells were harvested by centrifugation and stored at −80 °C until broken. Recombinant protein expression was evaluated throughout this process using SDS-PAGE analysis.
Purification of recombinant P. horikoshii prolidases
Cell pellets containing the Phprol and Ph1prol protein were suspended in 50 mM Tris–HCl, pH 8.0 containing 1 mM benzamidine–HCl, and 1 mM DTT. The cell suspension was passed through a French pressure cell (20,000 lb/in2) two times. The lysed suspension was centrifuged at 38,720 g for 30 min to remove cell debris. The supernatant was made anaerobic and was heated at 80 °C for 30 min. E. coli cell debris and denatured protein were removed by centrifugation of the heated supernatant at 38,720 g for 30 min. The clarified extract was applied to a 20-ml phenyl sepharose column. Before adding the extract to the column, ammonium sulfate, [NH4]2SO4, was added to the heat-treated extract gradually to a final concentration of 1.5 M. For phenyl sepharose chromatography, the binding buffer used was 50 mM Tris–HCl, 1.5 M [NH4]2SO4, pH 8.0, and the elution buffer was 50 mM Tris–HCl, pH 8.0. The fractions with active enzyme were applied to a 5 ml Q-column, which used a binding buffer of 50 mM Tris–HCl, pH 8.0 and an elution buffer of 50 mM Tris–HCl, 1 M NaCl, pH 8.0. All fractions were visualized on 12.5% SDS-polyacrylamide gels, and enzyme assays were performed as well to ensure activity after each purification step.
Prolidase enzyme assay
The enzyme reaction mixture and assay is based on a previously described method from Ghosh et al. 1998 and Du et al. 2005. The (500 µl) reaction mixture contains 50 mM MOPS (3-[N-morpholino]propanesulfonic acid) buffer (pH 7.0), 200 mM NaCl, 5% (v/v) glycerol, 0.1 mg/ml BSA protein and 1.2 mM CoCl2 (metal), and finally the enzyme. The reaction mixture was heated for 5 min at 100 °C in order for the metal and enzyme to interact. The reaction was initiated with the addition of the substrate, Met-Pro (final concentration of 4 mM). The reaction was heated for an additional 10 min at 100 °C. To stop the reaction, glacial acidic acid (500 µl) was added, and then (3% [wt. vol]) ninhydrin reagent (500 µl) was added. The mixture was heated again for 10 min at 100 °C and then cooled to 23 °C. The absorbance was determined at 515 nm with an extinction coefficient of 4,570 M−1 cm−1 for the ninhydrin-proline complex. One unit of prolidase activity is defined as the amount of enzyme that liberates one micromole of proline per minute (Ghosh et al. 1998). For assays conducted at different pH values, the following buffers were used at final concentrations of 100 mM: pH 5.0, Sodium Acetate; pH 6.0–8.0, MOPS; pH 9.0, CHES; pH 10.0, CAPS. For assays that evaluate metal preference, the following metals were substituted for Co2+: Mn2+, Fe2+, Ni2+, Zn2+, and Cu2+ at final concentrations of 1.2 mM. Also, when evaluating substrate specificity the following dipeptides: Leu-Pro, Gly-Pro, Ala-Pro, Arg-Pro, Phe-Pro, and Pro-Ala, were substituted for Met-Pro at a final concentration of 4 mM. Enzyme kinetic assays were done with increasing concentrations of Met-Pro and a metal concentration of Co2+ of 0.2 mM.
Anaerobic enzyme assay to determine metal preference
The active site metals were stripped from the purified prolidases by dialyzing 1 ml of 0.1 mg/ml of purified protein in 1 L of buffer (50 mM MOPS pH 7.0, 0.5 mM EDTA) for 1 h, followed by two more washes in 1 L of 50 mM MOPS pH 7.0 (repeated twice for 1 h each) to remove any EDTA. The metal centers were reconstituted by incubating the apo-proteins with 7.5 µM (this corresponds to threefold molar equivalents of metal/prolidase monomer) of the following metals, CoCl2, FeSO4, ZnCl2, under anaerobic conditions for 15 min at 80 °C. A final concentration of 1 mM of DT or sodium dithionite was added to the FeSO4 in order to keep the iron reduced to Fe (II). The reconstituted enzyme preparations were assayed following the same protocol described above. All assays were done under anaerobiosis in a Coy Anaerobic Chamber.
Metal content analysis of purified P. horikoshii prolidases
For preparation of protein samples for metal content determinations, 5 ml of 1 mg/ml purified prolidase protein was dialyzed in 2 L 50 mM MOPS, pH 7.0 overnight at 4 °C. The dialyzed protein sample and a dialysis buffer sample were collected. Metal content of the prolidases was determined by ICP emission spectrometry at the North Carolina State University Analytical Service Laboratory.
Metal competition experiments
The active site metals were stripped from the purified prolidases by dialyzing 1 ml of 1 mg/ml of protein in 1 L of buffer (50 mM MOPS pH 7.0, 5 mM EDTA) for 1 h, followed by two more washes in 1 L of 50 mM MOPS pH 7.0 (repeated twice for 1 h each) to remove any residual EDTA. The metal centers were reconstituted by incubating the apo-proteins with 125 µM (this corresponds to fivefold molar equivalents of metal/prolidase monomer) of the following metals, CoCl2, MnCl2, and ZnCl2, and 62.5 µM (this corresponds to 2.5-fold molar equivalents of metal/prolidase monomer) of the following metal mixtures: CoCl2 and MnCl2 and CoCl2 and ZnCl2 under anaerobic conditions for 15 min at 80 °C. Metal reconstituted enzyme samples were dialyzed in 50 mM MOPS pH 7.0 at 4 °C overnight. Enzyme assays were done following metal removal and after overnight dialysis.
Thermostability assay
Each enzyme (0.2 mg/ml in 100 mM MOPS, pH 7.0) was incubated anaerobically in a sealed vial in a 100 °C water bath for 8 h. Samples were taken every hour, and an enzyme assay was done to monitor activity. To calculate the thermal half-life t 50%, enzymes were prepared as described above but were incubated in a 90 °C oven for 48 h. Samples were taken at specific time points to monitor activity. Enzyme assays contained 0.2 mM CoC12 and 4 mM Met-Pro.
Amino acid sequence accession numbers
The prolidase amino acid sequences in the Clustal alignment (Fig. 1) can be accessed in the GenBank database with accession numbers: P. furiosus prolidase (AAL81467), P. horikoshii prolidase (BAA30249), P. horikoshii prolidase homolog 1 (BAA30071), P. horikoshii prolidase homolog 2 (NP 143731), Alteromonas OPAA-2 (AAB05590), prolidases from Lactobacillus delbrueckii (CAB07978), human (AAA60064), and E. coli (P15034).

Clustal alignment of P. horikoshii prolidase (PH1149) and prolidase homolog 1 (PH0974) and 2 (PH1902) along with previously characterized P. furiosus prolidase (PF1343) and other characterized prolidases from Alteromonas OPAA-2 (AAB05590), Lactobacillus delbrueckii (CAB07978), human (AAA60064), and E. coli (P15034). Gray shading designates identical and similar residues. Asterisks indicate the five-conserved residues in the prolidases identifying the dinuclear cobalt metal-binding sites
Results
Identification of P. horikoshii prolidase homolog genes
P. horikoshii prolidase or Phprol (PH1149), P. horikoshii prolidase homolog 1 or Ph1prol (PH0974) and P. horikoshii prolidase homolog 2 or Ph2prol (PH1902) each show 88%, 55%, and 27% similarity to previously characterized P. furiosus prolidase, respectively (Fig. 1). Phprol and Ph1prol both contain all five conserved metal center-liganding amino acid residues that are required for catalysis (Asp-209, Asp-220, His-192, Glu-313, Glu-327; Pfprol numbering; Maher et al. 2004). Ph2prol only contains two out of the five conserved residues and has different spacing between the residues signifying that it may be a different class of enzyme. National Center for Biotechnology Information (NCBI) lists the following annotations for each protein as follows: PH1149, X-Pro dipeptidase; PH0974, dipeptidase, and PH1902, hypothetical protein.
Expression and purification of recombinant prolidases from P. horikoshii
Prolidases from P. horikoshii were expressed in BL21 (λDE3) E. coli cells using autoinduction media (Studier 2005). Maximum recombinant protein expression levels were obtained after incubation of 14 h with autoinduction cultures. Assays were done using crude cell extract and heat-treated cell extract (heated at 80 °C for 20 min) to determine whether the recombinant prolidases had activity.
Although there was an overexpressed protein (41.78 kDa) that could be seen by SDS-gel analysis for the PH1902 expressed cell extract, there was no enzymatic activity observed for Ph2prol (PH1902) using the nonheat treated or heat-treated crude extracts and the entire battery of metals and dipeptide substrates. Therefore, the recombinantly expressed PH1902 protein was determined not to be a prolidase and was not subjected to further purification and characterization.
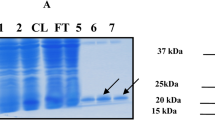
Phprol (PH1149) and Ph1prol (PH0974) showed high activity in crude and heat-treated cell extract, and multicolumn purification was performed to purify both enzymes. The overexpressed prolidases were identified using SDS-PAGE analysis, and protein bands of 39.27 kDa (Phprol) and 40.04 kDa (Ph1prol) were followed throughout the purification process, as was prolidase activity (Supplemental Fig. 1a and 1b).
To evaluate the metal content of each enzyme after purification, ICP emission spectrometry was used. The metal present in the highest amounts for purified Phprol and Ph1prol was zinc (0.268-g atoms of Zn/mol of subunit and 1.136-g atoms of Zn/mol of subunit, respectively; Supplemental Table 1). Cobalt was detected at levels less than 0.001-g atoms of Co/mol of subunit for both enzymes.
Catalytic properties of recombinant prolidases Phprol (PH1149) and Ph1prol (PH0974)
The temperature profile showed maximal activity of 1,938 U/mg for Phprol and 2,355 U/mg for Ph1prol at 100 °C (Supplemental Fig. 2). The optimum pH for both enzymes was at pH 7.0, although the second highest activity was recorded at pH 6.0 closely followed by pH 5.0 (Fig. 2). Previously characterized Pfprol was shown to have the highest activity with the substrate Met-Pro and the next highest activity with Leu-Pro (Ghosh et al. 1998). Phprol and Ph1prol showed the same trends as Pfprol (1,350 U/mg) with these dipeptides, having 100% relative activity with Met-Pro, correlating to specific activities of 1,824 U/mg and 2,751 U/mg, respectively (Table 1). All substrate specificity assays were done using 1.2 mM CoCl2 and 4 mM of each substrate. There were slight differences in activities for the following dipeptide substrates: Gly-Pro, Ala-Pro, Arg-Pro, and Phe-Pro. Both Phprol (10% relative activity) and Ph1prol (8% relative activity) showed slightly higher activity with Gly-Pro as the substrate compared to Pfprol (1% relative activity; Ghosh et al. 1998). While Ph1prol appeared to have more than twice the relative activity with Ala-Pro, Arg-Pro, and Phe-Pro compared to Phprol, all the prolidases showed very little to no activity with the substrate Pro-Ala.

Activity of P. horikoshii prolidases over a pH range of 4.0–10.0. Prolidase assays contained 14.8 ng for Phprol and 6.2 ng Ph1prol, Met-Pro (4 mM), and CoCl2 (1.2 mM). The following buffers were used for each pH at a final concentration of 100 mM: pH 5.0, Sodium Acetate; pH 6.0–8.0, MOPS; pH 9.0, CHES; pH 10.0, CAPS. One hundred percent specific activity corresponds to 2,321 U/mg for Phprol and 3,357 U/mg for Ph1prol
Phprol and Ph1prol are both very thermostable, showing no significant change in activity after 8 h at 100 °C. The t50% value at 90 °C for both Phprol and Ph1prol was 21.5 and 21 h, respectively.
Aerobic activity assay conditions
P. furiosus and P. horikoshii both require an anaerobic environment to grow optimally. Therefore, prolidases from these anaerobic organisms should be assayed anaerobically to evaluate their enzymatic properties under physiologically relevant conditions. However, for prolidases to be useful in biotechnological applications, they will need to be active under aerobic conditions. As such, it is important in this case to screen the P. horikoshii prolidases under both aerobic and anaerobic conditions.
For the aerobic assays, both prolidases were prepared with different divalent metal cations (1.2 mM), and the highest activity was seen with Co2+ followed by Mn2+, while Cu2+, Fe2+, Zn2+, and Ni2+ could not restore activity (Fig. 3). When assayed with different concentrations of Co2+ and Mn2+, the enzymes performed optimally at 0.2 mM Co2+ for Phprol with an activity of 2,075 U/mg and for Ph1prol with 0.15 mM Co2+ and an activity of 4,901 U/mg (Fig. 4). With Mn2+, the optimum activity was almost 724 U/mg for Phprol, and for Ph1prol, the activity reached 2,139 U/mg at a final concentration of 1.6 mM.

Activity of P. horikoshii prolidases with different divalent cations: Co2+, Mn2+, Cu2+, Fe2+, Zn2+, and Ni2+. Prolidase assays contained 14.8 ng for Phprol and 6.2 ng Ph1prol, Met-Pro (4 mM), and metals (1.2 mM). One hundred percent specific activity correlates to 2,321 U/mg for Phprol and 2,751 U/mg for Ph1prol

The effects of metal ion concentrations of Co2+ (circles) and Mn2+ (squares) on activities of purified Phprol (a) or Ph1prol (b). The reactions contained Met-Pro (4 mM) and 7.4 ng of enzyme when assayed with Co2+ and 14.8 ng of enzyme with Mn2+
The kinetic parameters of Phprol and Ph1prol were determined using lower metal concentrations (0.2 mM) than were used for Pfprol (1.2 mM; Ghosh et al. 1998). The kinetic analysis was done with Met-Pro and 0.2 mM of CoCl2 because their inclusion in the assay reactions provided the highest activity. The affinity or K m of Phprol for the substrate Met-Pro, reported here as 3.4 mM is in line with Pfprol, both the native (2.8 mM) and recombinant (3.3 mM) versions, whereas Ph1prol has a K m of around 1.9 mM. The V max of both enzymes was very high (3,997 and 5,714 µmol min−1 mg−1 for Phprol and Ph1prol, respectively), which were over threefold higher than that of recombinant Pfprol (Table 2). The k cat /K m for Phprol and Ph1prol were high as well (2,617 and 3,812 s−1 for Phprol and Ph1prol, respectively).
Anaerobic activity assay conditions
Although aerobic metal analysis demonstrated that Co2+ supported the highest activity, when the assays were conducted under anaerobic conditions, very different results were seen. The Phprol and Ph1prol purified enzymes were stripped of metals using EDTA, followed by metal reconstitution under anaerobiosis with Fe2+, Co2+, and Zn2+ at 80 °C for 20 min. The highest activity for the anaerobic assays was observed with Fe2+, not Co2+. With Fe2+, Phprol showed the highest activity of 2,371 U/mg while the activity for Ph1prol was 1,357 U/mg. The specific activity with Co2+ remained around 30% of the activity seen with Fe2+ (Supplemental Fig. 3 and Supplemental Table 2). Zn2+ was shown to only provide 1% of the relative activity in Ph1prol and did not support any activity in Phprol.
Metal competition experiment
Other than Pfprol, which has a solved Co–Co binuclear metal center, prolidases and metalloenzymes from different organisms and under varying conditions have been found to incorporate alternate divalent metal ions in their active sites (Besio et al. 2009; Lupi et al. 2006; Maher et al. 2004). Since, as purified, the P. horikoshii prolidases were shown to have been bound with zinc rather than with metals that support catalysis such as Co2+, Mn2+, and Fe2+; it became necessary to evaluate how different metals and or metal combinations affect catalysis of these enzymes. Phprol and Ph1prol were dialyzed in 50 mM MOPS buffer containing EDTA to strip the enzyme of metal. Metal reconstitution was done under the following metal conditions, no metal addition (apo-protein) and the addition of the following metal or metal combinations, Co2+, Mn2+, Zn2+, Co2+/Mn2+, Co2+/Zn2+. The activity of the WT-Ph1prol and WT-Phprol, pre-EDTA treatment, showed the normal activity seen with the addition of Co2+ (0.2 mM), Mn2+, or Zn2+ (1.2 mM) to an enzyme assay conducted at 100 °C (Tables 3 and 4). Prolidase activity with the addition of cobalt in the assay yielded the highest activity at 4,778 U/mg and 2,174 U/mg for WT-Ph1prol and WT-Phprol, respectively. Apo-protein represents protein that had EDTA treatment but contained no metal in the metal reconstitution process. Without the addition of metals to the enzyme assay, the apo-protein had very little to nondetectable activity (data not shown). When Co2+ ions were added to the assay with apo-protein, only 55% of the enzyme activity was restored for Ph1prol, and the activity was almost twofold higher for Phprol. When adding Mn2+ to the assay, both Ph1prol and Phprol apo-protein activity were almost fully restored. The highest activities for Ph1prol were observed when Co2+, Mn2+, Co2+/Mn2+, and Co2+/Zn2+ were used in the reconstitution of the metal center and Co2+ was added to the assay mix (Table 3). The same trend was seen with Phprol, except the difference was almost a twofold increase in activity when reconstituting with Co2+, Mn2+, Co2+/Mn2+, and Co2+/Zn2+, when the assay contained Co2+ (Table 4). When adding Mn2+ to the assay conditions with Phprol and Ph1prol reconstituted with different metals, the activity was restored but not to the high levels detected when Co2+ was added. The addition of Zn2+ to assay conditions resulted in minimal activity (less than 1% relative activity) for Ph1prol and little to no detected activity for Phprol (Tables 3 and 4).
Discussion
Biochemical characterization of both Phprol (PH1149) and Ph1prol (PH0974) supports their classification as prolidases or X-Pro dipeptidases. Although, Ph2prol possesses similar motifs to Xaa-Pro aminopeptidases when using pBLAST for protein comparison, no activity was seen when assays were done with Ph2prol crude cell extract when prolidase specific substrates and metals were supplied. Closer inspection of the P. furiosus genome shows that it also contains two annotated prolidase genes, one being PF1343, which is the prolidase designated Pfprol, and PF0747, which we have classified as Pf2prol. Pf2prol is similar to Ph2prol in that it only contains two out of five conserved metal-binding residues and shows low percent similarity to Pfprol. Pf2prol and Ph2prol cannot be confirmed as prolidases, and their function is still unknown at this time.
The catalytic properties of P. horikoshii prolidases are fairly similar to those observed for P. furiosus prolidase; however, there are key differences that could make them more ideal candidates for use in biotechnological applications. The temperature optima results for Phprol and Ph1prol were very similar to Pfprol in that the highest activity was seen at 100 °C. This is not surprising since optimal growth of Pyrococci is at 98–100 °C (Gonzalez et al. 1998). This would suggest that proteins isolated from these organisms should be very stable at high temperatures. Previously, when testing the thermal half-life of Pfprol under similar pH (7.0) and temperature conditions, Ghosh et al. found the t 50% of Pfprol (0.3 mg/ml) was 3 h. P. horikoshii prolidases (0.2 mg/ml) are even more thermostable with t 50% values of over 20 h at 90 °C, which is significantly higher than recombinant Pfprol and other characterized prolidases. Even after 48 h at 90 °C, Phprol and Ph1prol were still active with a relative activity of 28% and 20%, respectively.
The P. horikoshii prolidases are more active over a broader pH range than Pfprol, showing significantly higher activity at the lower pH range, specifically pH 5.0–7.0, and they continue to be active at pH 8.0 to a greater extent than is Pfprol. The enhanced stability of P. horikoshii prolidases and catalysis over a larger pH range are qualities that could help these enzymes withstand other potential denaturants in OP nerve agent decontamination formulations. The kinetic parameters for the recombinant P. horikoshii prolidases are more promising for biotechnological applications compared to those determined for Pfprol. The affinity for the substrate Met-Pro is highest in Ph1prol with a K m of 1.9 mM, and the reaction turnover rates are significantly higher than for Pfprol. The substrate profile is similar to other prolidases in that they show high specificity to dipeptides with proline in the C-terminus and a nonpolar amino acid in the N-terminal region and rarely are able to cleave dipeptides with proline in the N-terminus, Pro-X. The differences in relative activities between Phprol and Ph1prol could suggest differences in their roles in vivo. P. horikoshii, unlike P. furiosus, lacks many genes and operons that are responsible for de novo synthesis of amino acids; therefore, it is important that this organism obtain them from their environment. P. horikoshii may have two functional prolidases to aid in their amino acid metabolism requirements.
When examining the metal content of purified prolidases from P. horikoshii, it is not surprising that the incorporation of zinc, not cobalt was found in significant amounts. Previously in Ghosh, et al., both the native and recombinant Pfprol contained one Co atom per molecule. When further chemical analysis was done, Zn was found in variable to significant amounts, but was attributed to nonspecific binding due to its inability to restore enzyme catalysis. In addition, during the crystallization process used to solve the structure of Pfprol, Zn replaced Co, the metal needed for catalysis, in the active site of the crystallized prolidase enzyme (Ghosh et al. 1998; Maher et al. 2004). The Maher study (Maher et al. 2004) warned when purifying or crystallizing a metalloenzyme such as prolidase, other metals could be introduced based on the crystallization medium used, and this metal may not necessarily be physiologically or catalytically relevant to the enzyme.
The highest activity of Phprol and Ph1prol when assayed aerobically was observed when lower concentrations of Co2+ were introduced into the assay reaction than was used for Pfprol activity assays (0.2 mM Co2+ for Phprol and Ph1prol versus 1.2 mM for Pfprol). For both Phprol and Ph1prol, the association constant for Co2+ appears to be much lower, between 10–50 µM, while for Pfprol, it was previously projected to be 0.5 mM (500 µM) for recombinant P. furiosus prolidase (Ghosh et al. 1998). For Mn2+, the association constant was determined to be around 0.6 mM for Ph1prol, which is also consistent with Pfprol, which was shown to be 0.66 mM (Ghosh et al. 1998). The addition of metal for enzyme catalysis is one of the limitations in using this enzyme in applications because adding metal to the environment can be harmful. The P. horikoshii prolidases appear to be more active and more specific for their substrate and require less metal for full catalysis than Pfprol, making them good candidates in the future for biotechnological applications.
Metal competition experiments with Phprol and Ph1prol also show variations in the preferred metal or mixed metals needed for optimum activity. Interestingly, the highest activities were seen with the enzyme reconstituted with Co2+, Mn2+, or a combination of Co2+/Mn2+, with the addition of Co2+ to the assay conditions. This may suggest optimum activities could be achieved by a mixed metal center using Co and Mn ions in the active sites, rather than solely Co ions. Recently, in Besio et al. (2009), for the first time, structural ICP-MS and XAS data were presented of the recombinant human prolidase, showing that an active prolidase can use a heterogeneous dimeric metal site, employing both Zn and Mn ions (Besio et al. 2009). Recombinant human prolidase had previously been characterized and was shown to contain a fully loaded Mn active site for maximum catalysis. However, the Besio et al. study indicated that human prolidase is also enzymatically active when one of two active sites is occupied by two Zn ions and the other with one Mn and one Zn ion (Besio et al. 2009). It has been suggested that zinc could play a structural role, while other divalent cations play a role in catalysis. The incorporation of zinc, whether nonspecific or structural, may be present due to the host system used in the expression process. By using the E. coli host system to express recombinant proteins, zinc is more prevalent in vivo than any other divalent cations, which support prolidase activity (Maher et al. 2004; Willingham et al. 2001).
Although, cobalt is the preferred metal for catalysis aerobically, Fe2+ shows the highest activity anaerobically with Phprol and Ph1prol. In Du et al., it was reported that Pfprol also had the highest activity with Fe2+ followed by Co2+ under anaerobic conditions (Du et al. 2005). These findings are not particularly surprising considering the questions arising in the reported literature about the metal center in the closely related enzyme methionine aminopeptidase (Chai et al. 2008; Copik et al. 2005; Cosper et al. 2001; D’Souza et al. 2000, 2002; D’Souza and Holz 1999; Walker and Bradshaw 1998).
Methionine aminopeptidases share very similar structures with prolidases, as they both contain the same unique pita-bread fold and the same five metal ligands in the C-terminal region of the enzyme (Lowther and Matthews 2002). Other enzymes that are members of this family include aminopeptidase P and creatinase, and they all require metal for catalysis (Lowther and Matthews 2002). Purified MetAP-I apoenzyme from E. coli can be activated by divalent cations such as Co2+, Mn2+, Fe2+, Ni2+, and Zn2+, with cobalt supporting the highest activity, but it is considered unlikely that Co2+ is the native cofactor in vivo based on whole cell metal analysis and MetAP inhibitor studies (Chai et al. 2008; D’Souza and Holz 1999; Ye et al. 2006). In 1998, Walker and Bradshaw first demonstrated that Zn2+ was the native metal required in yeast MetAP-I, not Co2+ (Walker and Bradshaw 1998). Under physiologically relevant conditions and with inhibitors, E. coli MetAP-I (EcMetAP-I) was found to use Fe2+ or Mn2+ for maximal activity, while Fe2+ was also found to maximally activate P. furiosus MetAP-II or PfMetAP-II at its physiologically relevant temperature of 80 °C (D’Souza et al. 2000, 2002; D’Souza and Holz 1999; Meng et al. 2002). These two enzymes have also been suggested to function in vivo as mononuclear enzymes (Chai et al. 2008; Copik et al. 2005). More recently, a study by Chai et al. reports that Fe2+ is the native metal for MetAPs in both E. coli and two other Bacillus strains (Chai et al. 2008). Studies of prolidases and other related metallopeptidases continue to provide much needed information on the relationship and interactions between proteins and metals.
Many different prolidases are being studied for use in biotechnological applications including nerve agent detoxification, the development of cheese ripening and flavor in fermented foods, and most recently in enzyme therapy and drug delivery studies for patients with PD, and even cancer (Theriot et al. 2009). Currently, prolidases are being characterized from lactic acid bacteria evaluating their role in the food fermentation process (Yang and Tanaka 2008). Enzyme therapy and drug delivery studies are underway using both recombinant human and prokaryotic prolidases (Lupi et al. 2006; Mittal et al. 2005, 2007). By studying new prolidases from different organisms, we are able to explore the structure-function relationship of these enzymes and their interactions with metal.
Current biodecontamination formulations that incorporate Alteromonas prolidases have limited utility when used under harsh field conditions. The main limitations are in the long-term stability of the enzyme in a formulation mixture that includes other solvents and denaturing solutions and the need to add excess metal to reach full activity. An enzyme is needed with higher activity at a lower pH, over a broader temperature range, long-term stability under harsh conditions, and low metal requirement (Cheng and DeFrank 2000). Both prolidases characterized from P. horikoshii show promising enzymatic properties that make these enzymes potential candidates for future optimization studies for OP degradation. Compared to Pfprol, Phprol and Ph1prol show higher activity at a lower pH range, long-term stability, higher affinity for the substrate, and significantly lower metal requirement for catalysis. Future studies will focus on broadening the enzyme catalysis temperature range to include those suitable for application conditions. The differences in the structures of Pfprol compared to Phprol and Ph1prol could also provide insight into the properties needed for generating an enzyme with a higher affinity for metal. By utilizing genetic engineering strategies, we may be able to make a more catalytically active enzyme for detoxification of OP nerve agents.
References
Adams MW (1993) Enzymes and proteins from organisms that grow near and above 100 degrees C. Annu Rev Microbiol 47:627–658
Besio R, Alleva S, Forlino A, Lupi A, Meneghini C, Minicozzi V, Profumo A, Stellato F, Tenni R, Morante S (2009) Identifying the structure of the active sites of human recombinant prolidase. Eur Biophys J. doi:10.1007/s00249-009-0459-4
Bockelmann W (1995) The proteolytic system of starter and non-starter bacteria: components and their importance for cheese ripening. Int Dairy J 5:977–994
Chai SC, Wang WL, Ye QZ (2008) FE(II) is the native cofactor for Escherichia coli methionine aminopeptidase. J Biol Chem 283:26879–26885
Cheng TC, DeFrank JJ (2000) Hydrolysis of organophosphorus compounds by bacterial prolidases. In: Zwanenburg B, Mikolajczyk M, Kielbasinski P (ed) Enzymes in action green solutions for chemical problems, Vol 33. Kluwer Academic Publishers, Dordrecht, pp 243–261
Cheng TC, Rastogi VK, DeFrank JJ, Sawiris GP (1998) G-type nerve agent decontamination by Alteromonas prolidase. Ann N Y Acad Sci 864:253–258
Cheng TC, DeFrank JJ, Rastogi VK (1999) Alteromonas prolidase for organophosphorus G-agent decontamination. Chem Biol Interact 119–120:455–462
Copik AJ, Waterson S, Swierczek SI, Bennett B, Holz RC (2005) Both nucleophile and substrate bind to the catalytic Fe(II)-center in the type-II methionyl aminopeptidase from Pyrococcus furiosus. Inorg Chem 44:1160–1162
Cosper NJ, D’Souza VM, Scott RA, Holz RC (2001) Structural evidence that the methionyl aminopeptidase from Escherichia coli is a mononuclear metalloprotease. Biochemistry 40:13302–13309
Cunningham DF, O’Connor B (1997) Proline specific peptidases. Biochim Biophys Acta 1343:160–186
D’Souza VM, Holz RC (1999) The methionyl aminopeptidase from Escherichia coli can function as an iron(II) enzyme. Biochemistry 38:11079–11085
D’Souza VM, Bennett B, Copik AJ, Holz RC (2000) Divalent metal binding properties of the methionyl aminopeptidase from Escherichia coli. Biochemistry 39:3817–3826
D’Souza VM, Swierczek SI, Cosper NJ, Meng L, Ruebush S, Copik AJ, Scott RA, Holz RC (2002) Kinetic and structural characterization of manganese(II)-loaded methionyl aminopeptidases. Biochemistry 41:13096–13105
Du X, Tove S, Kast-Hutcheson K, Grunden AM (2005) Characterization of the dinuclear metal center of Pyrococcus furiosus prolidase by analysis of targeted mutants. FEBS Lett 579:6140–6146
Endo F, Tanoue A, Nakai H, Hata A, Indo Y, Titani K, Matsuda I (1989) Primary structure and gene localization of human prolidase. J Biol Chem 264:4476–4481
Fernandez-Espla MD, Martin-Hernandez MC, Fox PF (1997) Purification and characterization of a prolidase from Lactobacillus casei subsp. casei IFPL 731. Appl Environ Microbiol 63:314–316
Fiala G, Stetter KO (1986) Pyrococcus furiosus sp. nov. represents a novel genus of marine heterotrophic archaebacteria growing optimally at 100 °C. Arch Microbiol 145:56–61
Forlino A, Lupi A, Vaghi P, Icaro Cornaglia A, Calligaro A, Campari E, Cetta G (2002) Mutation analysis of five new patients affected by prolidase deficiency: the lack of enzyme activity causes necrosis-like cell death in cultured fibroblasts. Hum Genet 111:314–322
Ghosh M, Grunden AM, Dunn DM, Weiss R, Adams MW (1998) Characterization of native and recombinant forms of an unusual cobalt-dependent proline dipeptidase (prolidase) from the hyperthermophilic archaeon Pyrococcus furiosus. J Bacteriol 180:4781–4789
Gonzalez JM, Masuchi Y, Robb FT, Ammerman JW, Maeder DL, Yanagibayashi M, Tamaoka J, Kato C (1998) Pyrococcus horikoshii sp. nov. a hyperthermophilic archaeon isolated from a hydrothermal vent at the Okinawa Trough. Extremophiles 2:123–130
Grunden AM, Ghosh M, Adams MW (2001) Proline dipeptidase from Pyrococcus furiosus. Methods Enzymol 330:433–445
Lecompte O, Ripp R, Puzos-Barbe V, Duprat S, Heilig R, Dietrich J, Thierry JC, Poch O (2001) Genome evolution at the genus level: comparison of three complete genomes of hyperthermophilic archaea. Genome Res 11:981–993
Ledoux P, Scriver CR, Hechtman P (1996) Expression and molecular analysis of mutations in prolidase deficiency. Am J Hum Genet 59:1035–1039
Lowther WT, Matthews BW (2002) Metalloaminopeptidases: common functional themes in disparate structural surroundings. Chem Rev 102:4581–4608
Lupi A, Della Torre S, Campari E, Tenni R, Cetta G, Rossi A, Forlino A (2006) Human recombinant prolidase from eukaryotic and prokaryotic sources. Expression, purification, characterization and long-term stability studies. FEBS J 273:5466–5478
Maeder DL, Weiss RB, Dunn DM, Cherry JL, Gonzalez JM, DiRuggiero J, Robb FT (1999) Divergence of the hyperthermophilic archaea Pyrococcus furiosus and P. horikoshii inferred from complete genomic sequences. Genetics 152:1299–1305
Maher MJ, Ghosh M, Grunden AM, Menon AL, Adams MW, Freeman HC, Guss JM (2004) Structure of the prolidase from Pyrococcus furiosus. Biochemistry 43:2771–2783
Meng L, Ruebush S, D’Souza VM, Copik AJ, Tsunasawa S, Holz RC (2002) Overexpression and divalent metal binding properties of the methionyl aminopeptidase from Pyrococcus furiosus. Biochemistry 41:7199–7208
Mittal S, Song X, Vig BS, Landowski CP, Kim I, Hilfinger JM, Amidon GL (2005) Prolidase, a potential enzyme target for melanoma: design of proline-containing dipeptide-like prodrugs. Mol Pharmacol 2:37–46
Mittal S, Song X, Vig BS, Amidon GL (2007) Proline prodrug of melphalan targeted to prolidase, a prodrug activating enzyme overexpressed in melanoma. Pharm Res 24:1290–1298
Schut GJ, Brehm SD, Datta S, Adams MW (2003) Whole-genome DNA microarray analysis of a hyperthermophile and an archaeon: Pyrococcus furiosus grown on carbohydrates or peptides. J Bacteriol 185:3935–3947
Studier FW (2005) Protein production by auto-induction in high density shaking cultures. Protein Expr Purif 41:207–234
Suga K, Kabashima T, Ito K, Tsuru D, Okamura H, Kataoka J, Yoshimoto T (1995) Prolidase from Xanthomonas maltophilia: purification and characterization of the enzyme. Biosci Biotechnol Biochem 59:2087–2090
Theriot CM, Tove SR, Grunden AM (2009) Biotechnological applications of recombinant microbial prolidases. In: Laskin A, Gadd G, Sariaslani S (ed) Advances in applied microbiology, Vol 68. Academic Press, New York, pp 99–132
Walker KW, Bradshaw RA (1998) Yeast methionine aminopeptidase I can utilize either Zn2+ or Co2+ as a cofactor: a case of mistaken identity? Protein Sci 7:2684–2687
Willingham K, Maher MJ, Grunden AM, Ghosh M, Adams MW, Freeman HC, Guss JM (2001) Crystallization and characterization of the prolidase from Pyrococcus furiosus. Acta Crystallogr D Biol Crystallogr 57:428–430
Yang SI, Tanaka T (2008) Characterization of recombinant prolidase from Lactococcus lactis—changes in substrate specificity by metal cations, and allosteric behavior of the peptidase. FEBS J 275:271–280
Ye QZ, Xie SX, Ma ZQ, Huang M, Hanzlik RP (2006) Structural basis of catalysis by monometalated methionine aminopeptidase. Proc Natl Acad Sci USA 103:9470–9475
Acknowledgments
The authors thank Tatiana Quintero-Varca, Zeltina O’Neal, Prashant Joshi, and Eli Tiller for their help in analyzing Pyrococcus prolidase homologs. A special thanks to Dr. Jimmy Gosse for helping with the operation of the Coy Anaerobic Chamber. Also Dr. Robarge and Kim Hutchison for their assistance with analyzing metal samples via ICP. Support for these studies was provided by the Army Research Office (contract number 44258LSSR).
Author information
Authors and Affiliations
Corresponding author
Additional information
An erratum to this article can be found at http://dx.doi.org/10.1007/s00253-009-2300-5
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplemental Fig. 1a
Representative Phprol and Ph1prol specific activities throughout the purification process (GIF 52 kb)
Supplemental Fig. 1b
BSDS 12.5% polyacryalmide gel electrophoresis showing purified P. horikoshii prolidases, Phprol, and Ph1prol (1 µg). Molecular marker includes (kilodalton): myosin (200), β-galactosidase (116), phosphorylase b (97), serum albumin (66.2), ovalbumin (45), carbonic anhydrase (31), and trypsin inhibitor (21.5; GIF 27 kb)
Supplemental Fig. 2
Activity of P. horikoshii prolidases resulting from incubating the reaction assays at temperatures ranging from 40–100 °C. Prolidase assays contained 14.8 ng for Phprol and 6.2 ng Ph1prol, Met-Pro (4 mM), and CoCl2 (1.2 mM). 100% specific activity corresponds to 1,938 U/mg for Phprol and 2,355 U/mg for Ph1prol (GIF 29 kb)
Supplemental Fig. 3
Activity of P. horikoshii prolidases under anaerobic conditions with different divalent cations. Prolidase assays contained 5 ng of each enzyme when assayed with Co2+ and Fe2+ or 500 ng for the assay with Zn2+ (GIF 73 kb)
Supplemental Table 1
Metal content of purified prolidases from P. horikoshii (DOC 20 kb)
Supplemental Table 2
Metal reconstitution assay under anaerobic conditions (DOC 21 kb)
Rights and permissions
About this article
Cite this article
Theriot, C.M., Tove, S.R. & Grunden, A.M. Characterization of two proline dipeptidases (prolidases) from the hyperthermophilic archaeon Pyrococcus horikoshii . Appl Microbiol Biotechnol 86, 177–188 (2010). https://doi.org/10.1007/s00253-009-2235-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-009-2235-x