
Abstract
Two-stage culture was efficient in enhancing total ganoderic acid (GA) production by Ganoderma lucidum (Fang and Zhong, Biotechnol Prog 18:51–54, 2002). As different GAs have different bioactivities, it is critical to understand the kinetics of individual GA production during fermentation, but no related information is yet available. To understand the regulation of GA biosynthesis, investigation of the accumulation of intermediate (lanosterol) and by-product (ergosterol) and of the expression of three important biosynthetic genes was also conducted in liquid shaking and static cultures of G. lucidum. The results showed that the content of individual GAs increased rapidly in the liquid static culture, and their maximum value was 6- to 25-fold that of shaking culture while lanosterol content in the former was lower than the latter. The transcript of squalene synthase (SQS), lanosterol synthase and 3-hydroxy-3-methylglutaryl coenzyme A reductase in liquid static culture was 4.3-, 2.1-, and 1.9-fold that of the shaking culture, respectively. Higher GA content in liquid static culture was related to increased transcription of those genes especially SQS. The work is helpful to the production of individual GAs and provided an insight into why the liquid static culture was superior to the shaking culture in view of biosynthetic gene expression.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Ganoderma lucidum, Ling-zhi in Chinese and Reishi in Japanese, has been used as traditional medication for the prevention and treatment of various human diseases for several thousand years in East Asia. Ganoderic acids (GAs), a kind of secondary metabolite produced by this medicinal mushroom, have a number of important pharmacological activities including cytotoxicity to hepatoma cells, inhibition of histamine release, inhibition of cholesterol synthesis, stimulation of platelet aggregation, antitumor effect, and anti-HIV-1 and anti-HIV-protease activities (Shiao et al. 1994; Zhong and Tang 2004; Tang et al. 2006b; EI-Mekkaway et al. 1998; Akihisa et al. 2007; Chen et al. 2008).
GA is synthesized via the mevalonate/isoprenoid pathway, which involves the sequential conversion of farnesyl diphosphate to squalene and then to 2,3-oxidosqualene, as confirmed by isotope labeling experiments (Yeh et al. 1989; Hirotani et al. 1990; Shiao 1992). The enzyme 3-hydroxy-3-methyglutaryl coenzyme A reductase (HMGR) catalyzes the first step specific to isoprenoid biosynthesis; squalene synthase (SQS) catalyzes the first enzymatic step from the central isoprenoid pathway toward sterol and triterpenoid biosynthesis (Abe et al. 1993); and lanosterol synthase (LS) catalyzes the cyclization of 2,3-oxidosqualene to form lanosterol, which is the lanostane ring skeleton of GA (Shiao 1992). Although lanosterol was converted to GA in G. lucidum as reported by 13C NMR experiments (Shiao 1992), the later steps of GA biosynthetic pathway including a series of cyclization, oxidation, and reduction reactions are yet unclear in detail.
Due to the newly discovered important pharmacological functions of GAs such as antitumor and anti-HIV activities, the interest in G. lucidum has increased around the world in recent years, and many researchers have focused on studying fermentation conditions to accelerate mycelial growth (Yang et al. 2009; Liu and Zhang 2007) and optimize the GA production (Xu et al. 2008; Fang and Zhong 2002; Tang et al. 2009; Tang and Zhong 2003a; Li et al. 2006; Wagner et al. 2003) by mycelia fermentation. However, all those previous reports were about the production/content of total crude GAs based on UV absorbance measurement. In fact, different individual GAs have different bioactivities, e.g., GA-S stimulates platelet aggregation (Wang et al. 1989), GA-T induces apoptosis of lung cancer cells (Tang et al. 2006b), GA-Me inhibits tumor growth and lung metastasis (Wang et al. 2007), and GA-β inhibits the HIV-1 protease activity (Min et al. 1998). Therefore, from the viewpoint of heterogeneity engineering as well as for practical application, it is important to understand the kinetic accumulation of individual GAs during fermentation.
In addition, time course studies on individual GA accumulation may provide information for the elucidation of the biosynthetic pathway and transformation among various GAs. In a study on ginsenoside production by Panax notoginseng cells, the culture conditions were reported to affect both the amount and type of ginsenosides (Wang et al. 2006). During the formation of fruiting bodies of G. lucidum (Hirotani and Furuya 1990), the alteration of triterpenoid patterns was also reported. But there is no information on the formation of individual GAs, not to mention their kinetic study in mycelia cultures of G. lucidum.
Recently, Zhao and his co-workers (Shang et al. 2008; Ding et al. 2008; Zhao et al. 2007) have cloned three genes of farnesyl-diphosphate synthase, HMGR, and SQS involved in the biosynthesis of triterpenoid in G. lucidum. But to our knowledge, there is yet no information on the response of GA biosynthesis-related gene transcription to environmental conditions in fermentation processes in spite of the great need for such knowledge in understanding the secondary metabolism regulation. In another aspect, our previous work has shown that the two-stage culture process which combines liquid shaking culture with static culture was an efficient strategy to enhance the production of total GAs (Fang and Zhong 2002). However, it is yet unknown whether or not the biosynthetic gene transcription level was different in the liquid static culture compared to the shaking culture.
In this work, considering the role of LS in the biosynthesis of GAs which have the lanostane ring skeleton, we first cloned a LS partial complementary DNA (cDNA) clone from G. lucidum, the same as for HMGR and SQS. Then, the accumulation of GAs, ergosterol, and lanosterol along with the expression of key enzymes (HMGR, SQS, and LS) of the isoprenoid and GA biosynthetic pathway was analyzed and compared in liquid shaking and static fermentation of G. lucidum. The work will be helpful for further investigation on the metabolite biosynthesis regulation as well as for the development of a more efficient fermentation process.
Materials and methods
Maintenance and preculture of G. lucidum
The strain of G. lucidum CCGMC 5.616 from China General Microbiological Fermentation Center was maintained on potato dextrose agar slants. The details of preculture medium and preculture conditions were described earlier (Fang and Zhong 2002).
Liquid shaking culture and static liquid culture process
In liquid shaking culture process, a 45-mL culture medium (Fang and Zhong 2002) in a 250-mL flask was inoculated with 5 mL of second-stage preculture broth (with about 600–700 mg dry weight per liter). The culture was incubated in the dark at 30°C on a rotary shaker at 120 rpm. For liquid static culture process, the culture was moved to a 9-cm diameter plate for static incubation (30°C) after 2 days of liquid shaking fermentation.
Sampling, analyses of dry weight, medium sugar, crude total GAs, and individual GAs
For sampling, three flasks or plates were taken each time. Dry cell weight (DW) and residual sugar concentration was measured by gravimetric method and phenol–sulfuric acid method, respectively (Tang and Zhong 2002). Crude GAs were extracted and measured according to the method described elsewhere (Fang and Zhong 2002). For individual GAs, the dried mycelia (100 mg) were extracted by 50% (v/v) ethanol (3 mL), ultrasonic treatment for 1 h (three times). The supernatants were dried at 50°C under vacuum and redissolved in 500 µl absolute ethanol for high-performance liquid chromatography (HPLC) analysis by an Agilent 1200 series (5 µm Agilent Zorbax SB-C18 column, 250 × 4.6 mm) at 245 nm. The elution was performed at a flow rate of 1.0 mL/min with a linear gradient of solvent A (methanol/acetic acid, 100:0.5, v/v) and solvent B (water). The gradient, starting at sample injection, was linear from 80% to 100% A in 20 min and the elution continued for an additional 10 min at 100% A. Chromatographic peaks were identified by comparing the retention times and spectra against the known standards. The standards of GA-T, GA-Me, GA-S, and GA-Mk were extracted and purified from mycelia with preparative liquid chromatography in our lab with purity over 99% (Tang et al. 2006a and unpublished results).
Extraction and analysis of lanosterol and ergosterol
The sterols were extracted three times with methanol and ethanol (60:40, v/v) and the total extracts were further saponified according to Yuan et al. (2006) with 0.1 M methanolic NaOH at 50°C for 2 h. The hydrolyzed samples were mixed with 2 mL of distilled deionized water and extracted twice with 5 mL petroleum ether (boiling point range, 60–90°C). The petroleum ether layer was pooled and evaporated to dryness under a stream of nitrogen.
The dry samples were redissolved in 100 µl of methanol and were later injected into an Agilent 1200 series HPLC with an Agilent Zorbax SB-C18 column (250 × 4.6 mm, 5 µm) using methanol as the mobile phase (flow rate, 1 mL/min), and the detector was set at 210 and 282 nm. Chromatographic peaks were identified by comparing the retention times and spectra against the standards of lanosterol (97%, Sigma, St. Louis, MO, USA) and ergosterol (>95%, Sigma).
Total RNA extraction and cDNA synthesis
The total RNA from G. lucidum was extracted using TriZol (Invitrogen, Carlsbad, CA, USA) as an extracting solution. Residual genomic DNA was removed using RNase-free DNase I (MBI Fermentas, Canada) according to the manufacturer's protocol. RNA concentration was determined using a Biophotometer Plus (Eppendorf, Germany). Reverse transcription was achieved using total RNA as the starting material and the Superscript RNAase H− First-strand synthesis kit (Invitrogen).
Isolation of cDNA of GA biosynthesis genes from G. lucidum
The G. lucidum LS partial cDNA was isolated by polymerase chain reaction (PCR) amplification using degenerate forward/reverse primers designed on the basis of the regions conserved among fungi in the previous reported sequences: forward-LS, 5′-GAAGTYTTTGGRGACATCATG-3′R(A,G),Y(C,T); reverse-LS, 5′-YTCMCCCCATCCACCATC-3′ Y(C,T), M(A,C). At the same time, the cDNAs for HMGR and SQS were also isolated by reverse transcriptase PCR (RT-PCR) on the basis of the known sequences (Shang et al. 2008; Zhao et al. 2007). The amplified fragments were fused into the pMD19-T vector with a TA cloning system (Takara, Dalian, China) according to the manufacturer's instructions and sequenced by the fluorescence detection method using both M13+ and M13− as primers (Shanghai Sangon Biological Engineering Technology & Services Co., Ltd.). The LS amino acid sequence showed 62% and 62% identity with the available Aspergillus fumigatus LS amino acid sequence (XP_747936.1) and Neurospora crassa LS amino acid sequence (XP_961026.1), respectively. The LS nucleotide sequence was submitted to Genbank and its accession number was FJ195972.
Measurement of HMGR, SQS, and LS genes expression by conventional RT-PCR and real-time quantitative RT-PCR
The transcript levels were analyzed by conventional RT-PCR and real-time quantitative PCR. Primers, whose sequences are shown in Table 1, were designed using Primer Express software (Applied Biosystems) based on sequences present in databases (LS, GeneBank accession number FJ195972; SQS, GeneBank accession number DQ494675; HMGR, GeneBank accession number: EU263990; 18S rRNA, GeneBank accession number Z37049).
For conventional RT-PCR analysis, identical amounts of the RNA samples were subjected to reverse transcriptions with RevertAidTM First Strand cDNA synthesis kit (MBI Fermentas) and the PCR amplification was done under the standard condition by Taq DNA Polymerase (TaKaRa Ex TaqTM). All conventional RT-PCRs were optimized to ensure that amplification was terminated in the linear range. PCR products were electrophoresed on a 1.5% agarose gel, stained with ethidium bromide, and visualized with Tanon-3500 GIS (Tanon, Shanghai, China). All RT-PCRs were repeated at least two times.
For real-time quantitative RT-PCR, the cDNA was quantified using a Mastercycler® ep realplex 2S detection system (Eppendorf). PCR reactions were carried out according to MaximaTM SYBR Green qPCR Master Mix (MBI Fermentas) manufacturer's procedure. After denaturation at 95°C for 10 min, amplification occurred in three steps: 15 s of denaturation at 95°C, 30 s of annealing at 55°C, and 30 s of extension at 72°C, with a total of 40 cycles. Identical thermal cycling conditions were used for all targets. The gene for 18S rRNA was used as the reference gene because its expression was found to be stable under our experimental conditions. The expression level of the different genes was normalized with respect to the G. lucidum 18S rRNA expression level. For each gene, the reference sample (liquid shaking sample) was defined as the expression level 1.0, and results were expressed as the fold of mRNA level (liquid static sample) over the reference sample. Post quantitative real-time RT-PCR (qRT-PCR) calculations to analyze relative gene expression were performed according to the 2−△△CT method as described by Livak and Schmittgen (2001).
Results
Kinetic profile of individual GA (GA-Me, GA-T, GA-S, and GA-Mk) accumulation in liquid shaking and static cultures
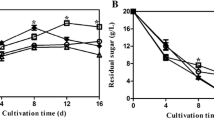
Figure 1a shows the kinetics of cell growth and sugar consumption in static cultures and shaking cultures. A higher final cell density of 12.5 g/L (by dry weight, DW) was obtained in static cultures and the consumption rate of sugar was lower compared to that in shaking cultures. The accumulation of total crude GAs was shown in Fig. 1b. The maximum content of total crude GAs was 3.25 mg/100 mg DW on day 10 in liquid static cultures, but in shaking cultures, its value was about 1.23 mg/100 mg DW and did not change substantially during the entire fermentation. The kinetic profiles of crude GA content in both culture systems were almost the same as reported earlier (Fang and Zhong 2002).

Kinetics of the cell growth and sugar consumption (a), the content of total GAs (b), and accumulation of GA-T, GA-Me (c) and GA-Mk, GA-S (d) in liquid static and shaking cultures of G. lucidum in shake flasks. The error bars in the figure indicate the standard deviations from three independent samples
The time profiles of individual GA formation in liquid static culture were also compared to those in shaking cultivation. The results were shown in Fig. 1c, d. In shaking cultures, the content of GA-T, GA-Me, and GA-S increased slightly during the fermentation, and the GA-Mk content increased until day 6 and then remained approximately constant thereafter. In static cultures, the content of GA-T, GA-Me, GA-S, and GA-Mk showed a similar profile, and their individual GA content increased rapidly during the fermentation and reached a maximal value at the end of fermentation.
In static cultures, individual GA content was much higher than that in shaking cultivation. The GA-S content was 55.8 µg/100 mg DW on day 14, which was 28-fold higher than that of shaking cultures (1.97 µg/100 mg DW). The maximum content of GA-T, GA-Me, and GA-Mk was 205.6, 96.3, and 42.6 µg/100 mg DW, respectively, about 6.2, 9.5, and 6.3 times that in the shaking cultures.
Analysis of intermediate and by-product accumulation and detection of HMGR, SQS, and LS gene expression in liquid shaking and static cultures
Lanosterol was an important intermediate while ergosterol is a by-product in the GA biosynthetic pathway, so the content of lanosterol and ergosterol was also analyzed in two different culture systems. As shown in Fig. 2, the lanosterol and ergosterol level was lower compared with that in the shaking cultures. In static cultures, lanosterol content decreased from 6.55 to 2.58 mg/g DW during the logarithmic phase, but there were no significant changes in the level of lanosterol content during the logarithmic phase in shaking fermentation.

Lanosterol and ergosterol content of G. lucidum during liquid static and shaking fermentation
To explore the molecular mechanism underlying the higher GA production observed in liquid static culture in contrast to shaking culture, as our initial efforts here, the expression kinetics of three important genes, i.e., HMGR, SQS, and LS, were examined by conventional RT-PCR and qRT-PCR in both systems.
As shown in Fig. 3a, b, LS gene showed a similar expression profile in both culture systems and the maximum mRNA abundance of LS was at day 6, followed by a decrease to the level near the beginning until the end of the experiment. For SQS gene, there was a clear increase in transcription at growth phase starting on day 6 until the end of the shaking culture (day 18). But in static culture, the SQS transcript level reached the highest level on day 14 and then gradually decreased, still maintaining a higher level than the beginning even after day 18. In the case of HMGR gene, the mRNA level increased after 4 days of cultivation and the elevated level continued in the shaking cultures. The highest expression of HMGR was observed at day 6; afterwards, there was a slight decrease in static cultures.

Detection of HMGR, SQS, and LS gene expression at different cultivation times in liquid shaking culture (a) and liquid static culture (b) of G. lucidum as well as their qRT-PCR results (c)
The three gene expression levels were also determined using qRT-PCR as described in “Materials and methods.” Gene expression was normalized against the expression level of the 18S rRNA gene. As shown in Fig. 3c, SQS was the most highly expressed gene, followed by LS and HMGR. The mRNA level of SQS in static cultures was about 4.3-fold that of shaking cultures on days 10 and 14. The expression of LS in static cultures was 2.1 times higher on day 6 than that in shaking cultures. For HMGR gene, the transcription level in static cultures was 1.5- and 1.9-fold that of the control on days 6 and 14, respectively.
Discussion
Ganoderic acids are a kind of highly oxygenated and pharmacologically active lanostane-type tritepenoids. Investigation of the individual GA formation during fermentation is critical to the practical application, as each of them has different bioactivities, and it is also the first step in understanding the molecular mechanism of GA biosynthesis in G. lucidum. Although it has been reported that GA accumulation was influenced by culture conditions, all the previous work was about total crude GA production based on UV absorbance measurement, while the total GAs are a complicated mixture which usually has other organic acids (Wagner et al. 2003) and the content may not accurately reflect the intracellular GA accumulation. Our previous work showed that total GAs production was enhanced in a two-stage process and the liquid static culture was a significant process (Fang and Zhong 2002), but the mechanism was unknown. Therefore, this work was focused on the investigation of the kinetic profiles of individual GA accumulation, intermediate metabolite formation, and related biosynthetic gene transcription in both liquid shaking and static cultures.
In our systems, GA-T, GA-Me, GA-S, and GA-Mk were detected as major GA components from the cultured mycelia of G. lucidum. Time profiles of GA formation in static culture were compared with those in shaking cultivation (as the control), and the content of four individual GAs was enhanced about 6–25 times; this was consistent with our previous work about total GA production in the two culture modes (Fang and Zhong 2002; Tang and Zhong 2003b). The content of total crude GAs maintained at a similar level during liquid shaking fermentation, but for individual GAs, they slightly increased from day 2 to day 18, although their accumulation levels were relatively low. In static culture, the level of four individual GAs showed a similar profile with a dramatic increase throughout the entire fermentation process. The results suggested that the biosynthetic pathway of individual GAs was mainly activated in liquid static culture mode. The high level of individual GA production might be due to the limitation of oxygen or nutrients in static culture. It is well known that oxygen and nutrients play an important role in mycelia growth and secondary metabolite production during the fermentation of higher fungi (Rau et al. 1992; Hansson and Seifert 1987). Previously, Tang and Zhong (2003a) reported that the limitation of oxygen was beneficial to the GA biosynthesis in submerged fermentation of G. lucidum. For polysaccharide production by G. lucidum, it was found that the nitrogen source limitation had a promoting effect (Hsieh et al. 2006).
Ganoderic acids are highly oxygenated C30 lanostane-type triterpenoids, and lanosterol is one of the important intermediates common to the biosynthesis of both ganoderic acid and ergosterol. Our results showed that both lanosterol and ergosterol levels in static culture were lower than that in the shaking culture during the whole fermentation process, which suggested that lanosterol may be metabolized faster to GAs and ergosterol in static culture and that the ergosterol biosynthetic pathway may be a major one for lanosterol metabolism in shaking culture. Those results were in agreement with the observation on higher GA accumulation in liquid static culture. Until now, the detailed biosynthetic steps from lanosterol to GA are yet unclear and the enzymes involved in those steps have not been identified; after the biosynthetic pathway is completely revealed, a better understanding on the association of intermediates and GA accumulation would be possible.
To gain insight into the molecular events responsible for the difference in ganoderic acid content between shaking culture and static culture, we analyzed the transcript levels of three genes HMGR, SQS, and LS, which are involved in the GA biosynthetic pathway. HMGR gene encodes a 3-hydroxy-3-methylglutaryl coenzyme A reductase, the rate-limiting enzyme of the MVA pathway. SQS gene encodes the squalene synthase, which catalyzes the first enzymatic step in sterol and triterpenoid biosynthesis. LS gene encodes lanosterol synthase that catalyzes the formation of triterpene skeleton. In this work, the three biosynthetic genes showed different transcription accumulation patterns or levels in two cultivation modes. In contrast to shaking culture, nutrient and oxygen gradients could be more easily present in liquid static culture. The difference in the gene expression may be caused by the difference in the environmental conditions. As a similar case, recently, Barrios-González et al. (2008) reported that lovastatin biosynthetic genes of Aspergillus terreus were expressed differentially in different fermentation modes. In Aspergillus oryzae, it was reported that different control mechanisms regulated glucoamylase and protease gene transcription in solid-state and submerged fermentations (Te Biesebeke et al. 2004). In addition, the SQS, LS, and HMGR transcript in liquid static culture was 4.3-, 2.1-, and 1.9-fold higher compared with the shaking culture, respectively. Those results showed that the differences in GA content between the two cultivation modes were in agreement with differences in transcription levels of genes involved in the biosynthesis pathway. Our results showed, for the first time, that higher content of GA in liquid static culture was at least partially due to a higher transcription level of its biosynthetic genes.
Among the three genes monitored, the transcript of SQS increased over the cultivation time in both modes, which corresponded to the increase in GA content during fermentation. Moreover, SQS gene also showed a higher transcription level compared with HMGR and LS genes. Those results were in accordance with the report by Zhao et al. (2004). They analyzed the squalene synthase expression by anti-SQ-4-ployclonal antibody during the development of the fruiting body of G. lucidum, and the results suggested that the low expression level of this enzyme was correlated with the low triterpene content of the fruiting body. It is known that squalene synthase is a key regulatory enzyme for phytosterol and triterpene biosynthesis in plants. For example, an addition of fungal elicitors into tobacco cell cultures induced the decline in sterol biosynthesis which correlated with the suppression of SQS activity (Devarenne et al. 1998), and the overexpression of squalene synthase in Panax ginseng roots resulted in a remarkable increase of phytosterol and triterpene saponins (Lee et al. 2004; Seo et al. 2005). Our results suggested that higher transcription level of SQS might be an important underlying cause of the higher GA content in static culture. Further work will be required to clarify the role of SQS and coordinately regulated enzymes.
References
Abe I, Rohmer M, Prestwich GD (1993) Enzymatic cyclization of squalene and oxidosqualene to sterols and triterpenes. Chem Rev 93:2189–2206
Akihisa T, Nakamura Y, Tagata M, Tokuda H, Yasukawa K, Uchiyama E, Suzuki T, Kimura Y (2007) Anti-inflammatory and anti-tumor-promoting effects of triterpene acids and sterols from the fungus Ganoderma lucidum. Chem Biodivers 4:224–231
Barrios-González J, Baños JG, Covarrubias AA, Garay-Arroyo A (2008) Lovastatin biosynthetic genes of Aspergillus terreus are differentially expressed in solid-state and in liquid submerged fermentation. Appl Microbiol Biotechnol 79:179–186
Chen NH, Liu JW, Zhong JJ (2008) Ganoderic acid Me inhibits tumor invasion through down-regulating matrix metalloproteinases 2/9 gene expression. J Pharmacol Sci 108:212–216
Devarenne TP, Shin DH, Back K, Yin SH, Chappell J (1998) Molecular characterization of tobacco squalene synthase and regulation in response to fungal elicitor. Arch Biochem Biophys 349:205–215
Ding YX, Ou-Yang X, Shang CH, Ren A, Shi L, Li YX, Zhao MW (2008) Molecular cloning, characterization, and differential expression of a farnesyl-diphosphate synthase gene from the basidiomycetous fungus Ganoderma lucidum. Biosci Biotechnol Biochem 72:1571–1579
El-Mekkaway SR, Meselhy M, Nakamura N, Tezuka Y, Hattori M, Kakiuchi N, Shimotohno K, Kawahata T, Otake T (1998) Anti-HIV-1 and anti-HIV-protease substances from Ganoderma lucidum. Phytochemistry 49:1651–1657
Fang QH, Zhong JJ (2002) Two-stage culture process for improved production of ganoderic acid by liquid fermentation of higher fungus Ganoderma lucidum. Biotechnol Prog 18:51–54
Hansson G, Seifert G (1987) Effects of cultivation techniques and media on yields and morphology of the basidiomycete Armillaria mellea. Appl Microbiol Biotechnol 26:468–473
Hirotani M, Furuya T (1990) Changes of the triterpenoid patterns during formation of the fruit body in Ganoderma lucidum. Phytochemistry 29:3767–3771
Hirotani M, Asaka I, Furuya T (1990) Investigation of the biosynthesis of 3-hydroxy triterpenoids, ganoderic acids T and S by application of a feeding experiment using [1, 2-13C2]acetate. J Chem Soc, Perkin Trans 1:2751–2754
Hsieh C, Tseng MH, Liu CJ (2006) Production of polysaccharides from Ganoderma lucidum (CCRC 36041) under limitation of nutrients. Enzyme Microb Technol 38:109–117
Lee MH, Jeong JH, Seo JW, Shin CG, Kim YS, In JG, Yang DC, Yi JS, Choi YE (2004) Enhanced triterpene and phytosterol biosynthesis in Panax ginseng overexpressing squalene synthase gene. Plant Cell Physiol 45:976–984
Li N, Liu XH, Zhou J, Li YX, Zhao MW (2006) Analysis of influence of environmental conditions on ganoderic acid content in Ganoderma lucidum using orthogonal design. J Microbiol Biotechnol 16:1940–1946
Liu GQ, Zhang KC (2007) Enhancement of polysaccharides production in Ganoderma lucidum by the addition of ethyl acetate extracts from Eupolyphaga sinensis and Catharsius molossus. Appl Microbiol Biotechnol 74:572–577
Livak K, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2−△△CT method. Methods 25:402–408
Min BS, Nakamura N, Mirashiro H, Bae KW, Hattori M (1998) Triterpenes from the spores of Ganoderma lucidum and their inhibitory activity against HIV protease. Chem Pharm Bull 46:1607–1612
Rau U, Gura E, Olszewski E, Wagner F (1992) Enhanced glucan formation of filamentous fungi by effective mixing, oxygen limitation and fed-batch processing. J Ind Microbiol 9:19–26
Seo JW, Jeong JH, Shin CG, Lo SC, Han SS, Yu KW, Harada E, Han JY, Choi YE (2005) Overexpression of squalene synthase in Eleutherococcus senticosus increases phytosterol and triterpene accumulation. Phytochemistry 66:869–877
Shang CH, Zhu F, Li N, Ou-Yang X, Shi L, Zhao MW, Li YX (2008) Cloning and characterization of a gene encoding HMG-CoA reductase from Ganoderma lucidum and its functional identification in yeast. Biosci Biotechnol Biochem 72:1333–1339
Shiao MS (1992) Triterpenoid natural products in the fungus Ganoderma lucidum. J Chin Chem Soc 39:669–674
Shiao MS, Lee KR, Lee JL, Cheng TW (1994) Natural products and biological activities of the Chinese medicinal fungus Ganoderma lucidum. Food Phytochemicals for Cancer Prevention II; ACS Symposium Series; American Chemical Society: Washington, DC, vol 547, pp 342–354
Tang YJ, Zhong JJ (2002) Fed-batch fermentation of Ganoderma lucidum for hyperproduction of polysaccharide and ganoderic acid. Enzyme Microb Technol 31:20–28
Tang YJ, Zhong JJ (2003a) Role of oxygen supply in submerged fermentation of Ganoderma lucidum for production of Ganoderma polysaccharide and ganoderic acid. Enzyme Microb Technol 32:478–484
Tang YJ, Zhong JJ (2003b) Scale-up of a liquid static culture process for hyperproduction of ganoderic acid by the medicinal mushroom Ganoderma lucidum. Biotechnol Prog 18:51–54
Tang W, Gu TY, Zhong JJ (2006a) Separation of targeted ganoderic acids from Ganoderma lucidum by reversed phase liquid chromatography with ultraviolet and mass spectrometry detections. Biochem Eng J 32:205–210
Tang W, Liu JW, Zhao WM, Zhong JJ (2006b) Ganoderic acid T from Ganoderma lucidum mycelia induces mitochondria mediated apoptosis in lung cancer cells. Life Sci 80:205–211
Tang YJ, Zhang W, Zhong JJ (2009) Performance analyses of a pH-shift and DOT-shift integrated fed-batch fermentation process for the production of ganoderic acid and Ganoderma polysaccharides by medicinal mushroom Ganoderma lucidum. Bioresour Technol 100:1852–1859
Te Biesebeke R, van Biezen N, de Vos WM, van den Hondel CA, Punt PJ (2004) Different control mechanisms regulate glucoamylase and protease gene transcription in Aspergillus oryzae in solid-state and submerged fermentation. Appl Microbiol Biotechnol 67:75–82
Wagner R, Mitchell DA, Sassaki GL, De Almeida Amazonas MAL, Berovic M (2003) Current techniques for the cultivation of Ganoderma lucidum for the production of biomass, ganoderic acid and polysaccharides. Food Technol Biotechnol 41:371–382
Wang CN, Chen JC, Shiao MS, Wang CT (1989) The aggregation of human platelet induced by ganoderic acid S. Biochim Biophys Acta 986:151–160
Wang W, Zhao ZJ, Xu YF, Qian XH, Zhong JJ (2006) Efficient induction of ginsenoside biosynthesis and alteration of ginsenoside heterogeneity in cell cultures of Panax notoginseng by using chemically synthesized 2-hydroxyethyl jasmonate. Appl Microbiol Biotechnol 70:298–307
Wang G, Zhao J, Liu JW, Huang Y, Zhong JJ, Tang W (2007) Enhancement of IL-2 and IFN-γ expression and NK cells activity involved in the anti-tumor effect of ganoderic acid Me in vivo. Int Immunopharmacol 7:864–870
Xu P, Ding ZY, Qian Z, Zhao CX, Zhang KC (2008) Improved production of mycelial biomass and ganoderic acid by submerged culture of Ganoderma lucidum SB97 using complex media. Enzyme Microb Technol 42:325–331
Yang FC, Yang MJ, Cheng SH (2009) A novel method to enhance the mycelia production of Ganoderma lucidum in submerged cultures by polymer additives and agitation strategies. J Taiwan Inst Chem Eng 40:148–154
Yeh SF, Chou CS, Lin LJ, Shiao MS (1989) Biosynthesis of oxygenated triterpenoids in Ganoderma lucidum. Proc Natl Sci Council B ROC 13:119–127
Yuan JP, Wang JH, Liu X, Kuang HC, Huang XN (2006) Determination of ergosterol in Ganoderma spore lipid from the germinating spores of Ganoderma lucidum by high-performance liquid chromatography. J Agric Food Chem 54:6172–6176
Zhao MW, Zhong JY, Liang WQ, Wang N, Chen MJ, Zhang DB, Pan YJ, Jong SC (2004) Analysis of squalene synthase expression during the development of Ganoderma lucidum. J Microbiol Biotechnol 14:116–120
Zhao MW, Liang WQ, Zhang DB, Wang N, Wang CG, Pan YJ (2007) Cloning and characterization of squalene synthase (SQS) gene from Ganoderma lucidum. J Microbiol Biotechnol 17:1106–1112
Zhong JJ, Tang YJ (2004) Submerged cultivation of medicinal mushrooms for production of valuable bioactive metabolites. Adv Biochem Eng Biotechnol 87:25–59
Acknowledgment
Financial support from the National Natural Science Foundation of China (NSFC project no. 20776084, no. 30821005), the National High Technology R&D Program (863 project no. 2007AA021506), and the Shanghai Leading Academic Discipline Project (project nos. B203 and B505) is gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Xu, JW., Xu, YN. & Zhong, JJ. Production of individual ganoderic acids and expression of biosynthetic genes in liquid static and shaking cultures of Ganoderma lucidum . Appl Microbiol Biotechnol 85, 941–948 (2010). https://doi.org/10.1007/s00253-009-2106-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-009-2106-5