
Abstract
Chitinases are listed as one class of pathogenesis-related proteins, and they have become a popular research topic because of their resistance to plant-pathogenic diseases. A chitinase with antifungal activity was isolated from the Canadian cranberry beans (Phaseolus vulgaris). The procedure included extraction, ammonium sulfate precipitation, affinity chromatography on Affi-gel blue gel, CM-Sephadex C-50, and Sephadex G-75. There was an almost 108-fold increase in specific activity of the purified chitinase compared with that of the crude extract. The enzyme exhibited a molecular mass of 30.6 kDa in sodium dodecyl sulfate-polyacrylamide gel electrophoresis both under reducing and non-reducing conditions, indicating that it was a monomeric protein. The pI was determined to be 7.6 by isoelectric-focusing electrophoresis. The optimum pH and the optimum temperature for activity towards N-acetyl-glucosamine was 5.4 and 40–55°C, respectively. It exerted a potent inhibitory action toward fungal species including Botrytis cinerea, Physalospora piricola, Fusarium oxysporum, and Pythium aphanidermatum. The chitinase was thermostable up to 58°C in both enzymatic reaction and antifungal activity. The present findings demonstrated a thermostable chitinase from cranberry beans with potentially exploitable significance.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Chitinases (EC 3.2.1.14) are glycosyl hydrolases that catalyze the hydrolytic degradation of chitin, an insoluble linear β-1,4-linked polymer of N-acetyl-glucosamine (GlcNAc). Chitin is a β-1,4-linked homopolymer of N-acetyl-glucosamine that is widely distributed in nature and forms a major constituent of the shells of crustaceans such as crabs and shrimps, the exoskeletons of insects, and cell walls of a variety of fungi (Konagaya et al. 2006). Chitin is structurally identical to cellulose except that the hydroxyl (–OH) group in cellulose at C2 is replaced by an acetamide group (NH.CO.CH3). After cellulose, chitin is the most abundant organic compound. During the previous decades, chitinases have received increased attention because of their wide range of applications. Practical applications of chitinases include use in the preparation of protoplasts from fungi as a protective agent against plant-pathogenic fungi (Ye and Ng 2002, 2003; Taira et al. 2005; Wang et al. 2007) and in the production of oligosaccharides as biologically active substances (Usui et al. 1990).
Chitinase is a widespread enzyme occurring in a wide range of organisms including viruses, bacteria, fungi, insects, higher plants, and animals and participates in a variety of bio-functions, including defense, nutrient digestion, morphogenesis, and pathogenesis (Kabir et al. 2006). Chitinases are classified according to the characteristics of hydrolyzing chitin; chitinases are classified into two types, exochitinase and endochitinase. The most extensively studied plant chitinases are endochitinases, which randomly hydrolyze internal β-1,4-linkages of chitin to release N,N′-diacetyl-chitobiose and cut them into shorter segments (Nielsen and Sdpresen 1999).
The function of plant chitinases appears to be defense against attack by chitin-containing fungal pathogens and insect pests. Synthesis of these enzymes, which are listed as the class of pathogenesis-related proteins (PR proteins), increases upon viral, bacterial, or fungal infection (Shih et al. 2001). In vitro experiments have shown that chitinases can inhibit the growth of many fungal species by causing lysis of the hyphal tip, which occurs, presumably, through the hydrolysis of chitin in the fungal cell wall (Taira et al. 2005; Kabir et al. 2006; Wang et al. 2007). In addition to pathogen stimulation, synthesis of chitinases can be induced by other environmental factors. They include wounding, heat shock, the phyto-hormone ethylene, fungal cell wall hydrolysate, and chemicals such as mercuric chloride, salicylic acid, and lead nitrate (Shih et al. 2001).
Chitinases are listed as the class of pathogenesis-related proteins, and they have become a popular research topic because of their resistance to plant-pathogenic disease. Up to date, chitinases have been isolated from various species and sources of plants, and their properties have been investigated. A number of plant chitinases have been purified from different seeds, especially cowpea (Gomes et al. 1996), chickpea (Vogelsang and Barz 1993), pearl millet (Radhajeyalakshmi et al. 2000), rice bean (Ye and Ng 2002), pinto bean (Ye and Ng 2002), and lima bean (Wang et al. 2008b). Plant chitinases are generally endochitinases; they have molecular masses in the range of 25–45 kDa and are acidic and basic isoelectric points in nature (Sahai and Manocha 1993). The individual chitinases often different in their physical–chemical and biological properties represent the diversity of chitinases, even from the same species.
The cranberry bean used in this study is a common dietary component, well known as a source of nutrients and proteins. During the growth period, cranberry beans are exposed to many phytopathogens including fungi and bacteria. It is to be expected that PR proteins containing chitinases are induced and expressed in certain quantities and exhibit unique characters when compared to those derived from other beans. As a result, this study herein was undertaken to investigate the properties of the purified chitinase previously not reported from cranberry beans. In particular, the results showed that it was a thermostable enzyme with potentially exploitable activities in agricultural and industrial applications.
Materials and methods
Materials
Dry Canadian cranberry bean seeds were purchased from a local supermarket, which were imported from Canada and ranked as the first-market class.
Affi-gel blue gel, CM-Sephadex C-50, and Sephadex G-75 were purchased from BIO-RAD Co. (Hercules, CA, USA), Amersham Biosciences (Piscataway, NJ, USA), and TOSOH America Co. (Grove city, OH, USA), respectively. The fungal species Botrytis cinerea, Physalospora piricola, Fusarium oxysporum, Pythium aphanidermatum, and Mycosphaerella arachidicola were kindly provided by the Microorganisms Center of Biochemistry Department, The Chinese University of Hong Kong, Hong Kong, China. Standard proteins for molecular weight determination were purchased from Gibco-BRL (Life Tech., San Francisco, CA, USA). All chemicals were of the highest purity available.
Sample preparation
Exactly 100 g of Canadian cranberry bean seeds were soaked in 1,000 mL distilled water for several hours and homogenized in 0.01 M Tris-HCl buffer (pH 7.2) using an electric homogenizer produced by Philips Electric Co. (Shanghai, China). The homogenate was centrifuged at 10,000×g for 20 min at 4°C. The supernatant was designated as the crude extract for the further investigations.
Isolation and purification
All isolation and purification steps were conducted at 4°C in the cold room. The crude chitinase sample was first fractionated by ammonium sulfate precipitation, in which the crude chitinase solution was treated with ammonium sulfate to 20% saturation. The resulting supernatant was then adjusted to 85% saturated ammonium sulfate. After centrifugation at 10,000×g for 20 min, the supernatant was discarded while the precipitate was collected, dissolved, and dialyzed against 0.01 M Tris-HCl buffer (pH 7.2) and then applied to an open column of an Affi-gel blue gel column (ϕ2.5 × 10 cm) previously equilibrated with the starting buffer, 0.01 M Tris-HCl buffer (pH 7. 2). Following removal of a large amount of unadsorbed proteins, the column was eluted with a linear gradient of NaCl (0–0.5 M) in the same buffer. The adsorbed fraction demonstrating chitinase activity was pooled, dialyzed against 0.01 M Tris-HCl buffer, pH 7.2 at 4°C for 24 h, and subsequently chromatographed on an open column of CM-Sephadex C-50 (ϕ2.5 × 25 cm) and the Sephadex G-75 column (ϕ2 × 100 cm) previously equilibrated with the starting buffer. Chitinase activity was determined for all the fractions obtained. The antifungal activity of each fraction was determined. The fraction Gm represents chitinase, the purified cranberry bean antifungal protein.
Characterization of the purified chitinase
Enzyme assays
Chitinase activity was determined by measuring the reducing end group N-acetamino-glucose produced from colloidal chitin according to the method of Boller et al. (1983). In a typical reaction, the reaction mixture, consisting of 0.1 mL 3 mM sodium azide, 1 mL enzyme solution, and 1 mL of 1% (w/v) colloidal chitin (pH 5.4), was incubated at 50°C for 60 min. The reaction was terminated by adding 2 mL dinitrosalicylic acid reagent and heating in boiling water for 5 min then cooled to room temperature and centrifuged at 8,000×g for 10 min. The supernatant was subjected to spectrophotometric measurement at 530 nm. One unit of chitinase activity was defined as the amount of enzyme that liberates 1 μmol N-acetamino-glucose per minute at pH 5.4 and 50°C.
Protein determination
Protein concentrations were determined by the method of Lowry et al. (1951) using bovine serum albumin as a standard.
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE; 12.5% T, 4% C) was performed according to the method of Laemmli and Favre (1973). The sample was mixed at 1:1 (v/v) ratio with sample buffer. The mixture was boiled for 3 min and centrifuged at 10,000 rpm for 5 min to remove undissolved debris. The concentration of dithiothreitol was 7% (v/v). After completion of the run, the gels were stained in 0.1% (w/v) Coomassie blue 30% (v/v) methanol 10% (v/v) acetic acid in water. The destaining solution was 30% (v/v) methanol 10% (v/v) acetic acid in water.
Isoelectric-focusing electrophoresis
The isoelectric-focusing (IEF)-PAGE was performed using a two-dimensional electrophoresis and Data Analysis System (Investigator™ 5000, Tokyo, Japan). The PhastGel IEF for standard proteins was bought from BIO-RAD Company, USA, covering the pH range 3–10. Proteins on the gel were stained with Coomassie blue R-250.
Determination of optimum pH and temperature
The effects of pH and temperature on the enzymatic activity of the purified chitinase preparation were investigated within a pH range between pH 4.0 and pH 8.0 using 0.02 M sodium acetate buffer (pH 4.0, 5.0, and 5.4), sodium citrate buffer (0.02 M, pH 6.0 and 6.6), and 0.02 M Tris-HCl buffer (pH 7.2 and 8.0) and a temperature range from 28 to 80°C.
Thermostability
According to the method described by Wang et al. (2008a, b), the thermostability of the purified chitinase was estimated by determining the residual activity of the enzyme solution after incubation for 30 min at various temperatures from 50 to 85°C.
Effect of metal ions and ethylenediaminetetra acetic acid on chitinase activity
Chitinase activity was determined by the standard assay method in the presence of Na+, K+, Mg2+, Ca2+, Zn2+, Fe2+, Cu2+, Hg2+, Mn2+, Pb2+, and ethylenediaminetetra acetic acid (EDTA) at 1 and 2 mM. The relative activity was calculated with respect to the control where the reaction was carried out in the absence of any additive.
Chitin-binding assay
Binding assay mixtures in 1-mL glass microtubes containing various concentrations of enzyme and 1 mg of binding substrate of chitin in 1 mL of 20 mM buffer were incubated on ice with occasional mixing. Each mixture was centrifuged at 4°C for 20 min at 10,000×g to separate supernatant and substrate with bound enzyme, the supernatant containing free enzyme was collected, and the protein concentration was determined. The regenerated chitin used in this experiment as binding substrate was prepared by acetylation of chitosan according to the method of Molano et al. (1977), and the degree of acetylation was more than 95%. The amount of bound protein was calculated from the difference between the initial protein concentration and the free protein concentration after binding. The relative equilibrium association constants Kr (l·g−1) was determined from double-reciprocal plots of binding data by the method described by Gilkes et al. (1992). The binding assay for these experiments was carried out at pH 5.4, based on the optimum pH of chitinase for the hydrolysis reaction determined before. The preliminary experiments indicated that the binding of chitinase required approximately 3 h; therefore, the binding reaction was performed for 3 h.
Assay for antifungal activity
The assay for antifungal activity was executed using 100 × 15-mm Petri dishes containing 10 mL of potato dextrose agar. At a distance of around 1 cm from the central disk (0.625 cm in diameter) were placed sterile blank paper disks of the same size. An aliquot (8 μL containing 140 or 210 μg) of chitinase in 0.01 mM Tris-HCl buffer (pH 7.2) was introduced onto a disk. The plates were incubated at 23°C for 72 h until mycelial growth from the central disk had enveloped peripheral disks containing the control (buffer) and had produced crescents of inhibition around disks containing samples with antifungal activity (Wang et al. 2009)
For a quantitative assay to determine the half maximal inhibitory concentration (IC50) of antifungal activity, P. aphanidermatum was taken as an example; the inhibition of hyphal growth of fungus by purified protein in series of concentration was observed (Wang et al. 2007). Three doses of the antifungal protein were added separately to three aliquots each containing 4 mL potato dextrose agar at 45°C, mixed rapidly, and poured into three separate small Petri dishes. After the agar had cooled down, the same small amount of mycelia was inoculated onto each plate. Buffer only without antifungal protein served as a negative control. After incubation at 27°C for 72 h, the radial growth rate of the mycelial colony under different sample concentration was measured, and the inhibition of fungal growth and the IC50 value were determined. Here, IC50 represented the concentration of purified protein that was required for 50% inhibition on fungal growth.
To further investigate its thermal stability in antifungal activity, the purified chitinase in Tris-HCl buffer (0.01 M, pH 7.2) was incubated at 20°C, 30°C, 40°C, 50°C, and 58°C for 10 min, respectively. After cooling the treated sample on ice for 10 min, the residual antifungal activities were measured as above.
Statistical analyses
All data are presented as means (SDs) of three independent experiments. Statistical analysis was done using Student’s t test. A value of P < 0.05 was considered statistically significant.
Results
Purification of the chitinase
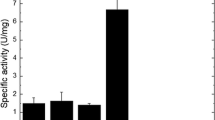
The crude extract of cranberry bean was first precipitated by ammonium sulfate, and the solution of the precipitate was applied to an open column of Affi-gel blue gel column previously equilibrated with 0.01 M Tris-HCl buffer (pH 7.2). Following removal of a large amount of unadsorbed proteins, the adsorbed fraction exhibiting chitinase activity was desorbed from the Affi-gel column with a linear NaCl concentration gradient (Fig. 1a). The active peak was pooled and dialyzed and chromatography on a CM-Sephadex C-50 column was carried out. After elution of a sizable quantity of unadsorbed materials, the column was eluted with a gradient of NaCl (0–0.5 M) in the same buffer to yield two peaks. The active peak CM1 was pooled, dialyzed, and subsequently concentrated (Fig. 1b). After that, gel filtration on a Sephadex G-75 column was carried out (Fig. 1c). The main peak designated Gm and displayed both chitinase and antifungal activities. The purified chitinase was shown by capillary reversed-phase high-performance liquid chromatography to be of high purity (data not shown). Its SDS-PAGE pattern was shown in Fig. 2, presenting a single band on the gel. The molecular weight of the chitinase obtained was estimated by SDS-PAGE to be 30,600 kDa. The protein yield and enzymatic activity at each purification step are presented in Table 1. There was an almost 122-fold increase in specific activity of the purified chitinase compared with that of the crude extract. The specific activity of the chitinase was estimated to be 68 U/mg.

a Fractionation of a solution of the (NH4)2SO4 precipitate extract on an Affi-gel blue gel column equilibrated with the binding buffer (0.01 M Tris-HCl buffer, pH 7.2). The gel was washed with the binding buffer and eluted with a linear gradient of 0 to 0.5 M NaCl in the same buffer. b Elution profile of adsorbed fraction from (a) on the CM-Sephadex column. The adsorbed fraction from the Affi-gel blue gel column was pooled, dialyzed and applied to the CM column in 0.01 M Tris-HCl buffer, pH 7.2. The column was then washed with the binding buffer. Adsorbed proteins were eluted with a linear gradient of NaCl from 0 to 0.5 M in the same buffer. c The adsorbed fraction CM1 with antifungal activity from CM-Sephadex column chromatography was pooled, concentrated and applied to a Sephadex G-75 column. Protein elution was carried out with 0.01 M Tris-HCl buffer, pH 7.2

SDS-polyacrylamide gel electrophoresis of cranberry bean chitinase. From left to right: Lane M molecular mass standards; lane DDT-, 18 μg purified chitinase from Sephadex G-75 under nonreducing conditions (without addition of dithiothreitol); lane DDT+, 18 μg purified chitinase under reducing conditions (with dithiothreitol added)
The purified enzyme exhibited a single band on SDS-PAGE under both non-reducing and reducing conditions, indicating the purified chitinase is a monomeric protein, as shown in Fig. 2. The isoelectric point of cranberry bean chitinase was determined to be 7.6 based on the results of isoelectric focusing electrophoresis (Fig. 3), which demonstrated that the newly reported chitinase was of basic property.

A profile of isoelectric focusing electrophoresis results
Chitinase activity as a function of pH and temperature was shown in Fig. 4. The optimum pH was 5.4, and the optimum temperature was 40°C to 55°C. The purified chitinase was stable below 58°C, but it was rapidly inactivated when incubated at temperature above 65°C. It was almost completely inactivated after incubation at a temperature above 85°C for 2 min (data not shown).

Effect of pH and temperature on activity of the purified chitinase. Activity at pH 5.4, and 50°C was used as standard
Effect of metal ions and EDTA on chitinase activity
In the presence of Na+, K+, Mg2+, Ca2+, Zn2+, Mn2+, Fe2+, and EDTA, the chitinase activity was either retained well or slightly increased. However, the activity was severely inhibited by Pb2+, Cu2+, and Hg2+. General characteristics of the purified chitinase were shown in Table 2. EDTA did not alter the activity of the enzyme, indicating that it does not require any metal ions for its activity.
Chitin-binding activity of purified chitinase
The double-reciprocal plots of chitin binding results were shown in Fig. 5. According to the method proposed by Gilkes et al. (1992), the relative equilibrium association constants (Kr)of chitinase toward regenerated chitin represents the binding activity of tested chitinase, and Kr was determined by the regression equation y = 1/Kr x+ b, based on the double-reciprocal plots. The regression equation for these experiments was estimated as y = 0.076 × +0.3086 (Fig. 5), the corresponding Kr was calculated to be 13.2 L·g−1.

Double-reciprocal plots of binding data for chitinase. The binding assay was performed by keeping the binding assay mixtures on ice for 3 h. [B] bound protein concentration; [F] free protein concentration
Antifungal activity
The antifungal activity of chitinase against four fungal species was illustrated in Fig. 6I-IV. Through crescents of inhibition around disks, it can be seen that the protein showed obvious antifungal activity toward B. cinerea (Fig. 6I), P. piricola (Fig. 6II), F. oxysporum (Fig. 6III) and P. aphanidermatum(Fig. 6IV). Also, the antifungal action showed concentration dependent, the crescents of inhibition around disks containing samples of 210 μg [(c) marked on the figure] were much more obvious than those with samples of 140 μg [(B) marked on the figure], as shown in Fig. 6. The IC50 value of the antifungal activity toward P. aphanidermatum was calculated to be 22.3 μM (Fig. 7). However, it showed no antifungal activity on fungi Mycosphaerella arachidicola. The results suggested the cranberry bean chitinase exhibit its own characteristic antifungal spectrum, which may contribute to the development of biological control of fungal pathogens typical of the particular species.

I–IV Inhibitory activity of cranberry bean chitinase against B. cinerea, P. piricola, F. oxysporum, and P. aphanidermatum. A 0.01 M Tris-HCl buffer, pH 7.2, b 140 μg cranberry bean chitinase, and c 210 μg cranberry bean chitinase

Determination of the IC50 value of the antifungal activity of cranberry bean chitinase. Each data point represents mean ± SD of triplicate inhibition radius of mycelia. The IC50 was calculated to be 22.3 μM
Interestingly, the purified chitinase retained almost 100% activity after treated by incubating at 20°C, 30°C, 40°C, 50°C and 58°C for 10 min, respectively. The residual antifungal activities of thermal stability determination demonstrated that the chitinase displayed a relatively strong thermostability in antifungal activity.
Discussions
The molecular weight for most legumious plant chitinases was reported within the range between 25–35 kDa (Ye and Ng 2002). Some chitinases have a molecular weight near 30 kDa, while others are well over 30 kDa even up to 45 kDa in molecular weight (Sahai and Manocha 1993; Radhajeyalakshmi et al. 2000). Two chitinases from chickpea and an endochitinase from bean leaves have a molecular weight near 35 kDa (Benhamou et al. 1993; Vogelsang and Barz 1993). The molecular weight of the newly purified enzyme, is similar to its counterparts from rice bean (Ye and Ng 2002), pinto beans (Ye and Ng 2003), peanuts (Wang et al. 2008a) and lima beans (Wang et al. 2008b).
Chitinases include acidic and basic in nature (Sahai and Manocha 1993). Among plant chitinases, there are various isoforms which have low homology and a different isoelectric point. Three chitinases designated PL Chi-A,-B,-C, isolated from Pineapple leaf (Taira et al. 2005), showed molecular weight of 25 kDa, 33 kDa, 39 kDa, and with isoelectric point of 4.4, 7.9, 4.6, respectively. Consequently, the individual chitinases from the same species often differ in their physical-chemical and biological properties.
The purified chitinase showed relatively higher optimum temperature when compared with its counterparts obtained by Boller et al. (1983), Yang and Luo (1998), and Wang et al. (2008a). Additionally, the optimum pH was somewhat different to the above mentioned chitinases reported by Boller et al. (1983) and Yang and Luo (1998). The former was at pH 6.5 and the latter was at pH 6.0. Moreover, the chitinases isoforms from gram-positive and gram-negative bacteria demonstrated extensive optimum pH and temperature, with optimum pH of 5.0-7.0, 4.5-6.5, 6.0, 8.0, respectively, and optimum temperature of 35-45°C, 40°C, 45°C, 50°C, 60°C, respectively (Konagaya et al. 2006). The fact that the individual chitinases are often different in their optimum pH and the optimum temperature represents the diversity of chitinases, even from the same species. Therefore, the thermostability of the purified enzyme was more higher than that of the chitinases reported by Boller et al. (1983), Yang and Luo (1998), and Wang et al. (2008a). The thermostable temperature of almost all of previously reported plant chitinases was less than 55°C, therefore the newly purified one was of a relatively heat-resistant property.
The effect of metal ions and EDTA on chitinase activity varies among different types. Hg2+ is a common inhibitor of most of the chitinases. The activity of a chitinase from Microbispora sp. V2 was just inhibited by Hg2+, in the presence of which only 10% activity was retained (Nawani et al. 2002). Among two chitinases were purified from the intestinal tract of the South American sea lion, one was inhibited with Cu2+, Fe2+, Hg2+ and Zn2+, while the other one was inhibited with Fe2+ (Konagaya et al. 2006). The lima bean chitinase was inhibited by Pb2+ and Hg2+ rather than Cu2+ (Wang et al. 2008b). The chitinase showed comparable chitin-binding activity when compared its Kr with those reported by Hashimoto et al. (2000) and Wang et al. (2008b). The result suggested that the catalytic domain is involved in the chitin-binding of the newly purified chitinase to a certain extent. The equation represented systematical linearity (R2 = 0.9931, Fig. 5), signifying that the model was a good fit to the behavior of chitin binding.
The protein showed antifungal activity toward a series of fungi (Fig. 6) when compared with the reported results of plant chitinase (Boller et al. 1983; Yang and Luo 1998; Wang et al. 2008a, b). Also, the cranberry bean chitinase exhibit its own characteristic antifungal spectrum, which may contribute to the development of biological control of fungal pathogens typical of the particular species. When it comes to the antifungal mechanism of chitinase, we could go back to its nature as one of the chitinolytic enzymes. Chitin, a β-(1,4)-linked polymer of N-acetyl D-glucosamine (GlcNAc), is a majorly structural polysaccharide in fungal cell walls. Chitinases hydrolyze the chitin located in fungal cell walls and demonstrate the antifungal activity, normally the hyphal morphological distortion along with stunted growth is observed for fungi with enzyme treatment. Also, the enzymatic activity of chitinase is measured with chitin as a substrate. It is therefore reasonable that the chitinase showed thermostable temperature up to 58°C in both enzymatic reaction and antifungal activity, which is definitely consistent in utilizing chitin as a specific reaction subject. A relatively thermostable enzyme newly obtained, demonstrating a potent inhibitory action toward fungal species, would certainly be significant for its utilization both in enzymology and in plant defense as well. The relatively heat-resistant enzyme obtained would possibly be helpful for elaborating strategies for constitutive expression of chitinase genes and their manipulation and general utilization in plant defense mechanisms through more recent techniques of genetic manipulation.
References
Benhamou N, Broglie K, Broglie R, Chet I (1993) Antifungal effect of bean endochitinase on Rhizoctonia solani: ultrastructural changes and cytochemical aspect of chitin breakdown. Canadian J Microbiol 39:318–328
Boller T, Gehri A, Mauch F, Vogeli U (1983) Chitinase in bean leaves: induction by ethylene,purification,properties,and possible function. Planta 157:22–31
Gilkes NR, Jervis E, Henrissat B, Tekant B, Miller RC, Warren RAJ, Kilburn DG (1992) The adsorption of a bacterial cellulase and its two isolated domains to crystalline cellulose. J Biol Chem 267:6743–6749
Gomes VM, Oliveira AEA, Xavier-Filho J (1996) A chitinase and α-1, 3-glucanase isolated from the seeds of cowpea (Vigna unguiculata (Walp)) inhibit growth of fungi and insect pests of the seed. J Sci Food Agric 72:86–90
Hashimoto M, Ikegami T, Seino S, Ohuchi N, Fukada H, Sugiyama J, Shirakawa M, Watanabe T (2000) Expression and Characterization of the Chitin-Binding Domain of Chitinase A1 from Bacillus circulans WL-12. J Bacteriol 182(11):3045–3054
Kabir KE, Hirowatari D, Watanabe K, Koga D (2006) Purification and characterization of a novel isozyme of chitinase from Bombyx mori. Bioscience, Biotechnol Biochem 70(1):252–262
Konagaya Y, Tsuchiya C, Sugita H (2006) Purification and characterization of chitinases from Clostridium sp. E-16 isolated from the intestinal tract of the South American sea lion (Otaria flavescens). Letters in Appl Microbiol 43:187–193
Laemmli UK, Favre M (1973) Gel electrophoresis of proteins. J Mol Biol 80:575–599
Lowry OH, Rosebrough NJ, Farr AL, Randall RJ (1951) Protein measurement with the Folin Phenol Reagent. J Biol Chem 193:265–275
Molano J, Duran A, Cabib E (1977) A rapid and sensitive assay for chitinase using tritiated chitin. Analytical Biochem 83(2):648–656
Nawani NN, Kapadnis BP, Das AD, Rao AS, Mahajan SK (2002) Purification and characterization of a thermophilic and acidophilic chitinase from Microbispora sp. V2. J Appl Microbiol 93:965–975
Nielsen MN, Sdpresen J (1999) Chitinolytic activity of Pseudomonas fluorescens isolates from barley and sugar beet rhizosphere. FEMS Microbiol Ecology 30:217–227
Radhajeyalakshmi R, Meena B, Thangavelu R, Deborah SD, Vidhyasekaran P, Velazhahan R (2000) 45-kDa chitinase purified from pearl millet (Pennisetum glaucum (L.) R. Br.) shows antifungal activity. J Plant Diseases Protection 107:605–616
Sahai AS, Manocha MS (1993) Chitinases of fungi and plants: their involvement in morphogenesis and host–parasite interaction. FEMS Microbiol 11:317–338
Shih CYT, Khan AA, Jia SF, Wu JL, Shih DS (2001) Purification, characterization and molecular cloning of a chitinase from the seeds of Benincasa hispida. Biosci Biotechnol Biochem 65(3):501–509
Taira T, Toma N, Ishihara M (2005) Purification, characterization and antifungal activity of chitinases from pineapple (Ananas comosus) leaf. Biosci Biotechnol Biochem 69(1):189–196
Usui T, Matsui H, Isobe K (1990) Enzymatic synthesis of useful chito-oligosaccharides utilizing transglycosylation by chitinolytic enzymes in a buffer containing ammonium sulfate. Carbohydrate Res 203:65–77
Vogelsang R, Barz W (1993) Purification, characterization and differential hormonal regulation of a β-1, 3-glucanase and two chitinases from chickpea (Cicer arietinum L.). Planta 189:60–69
Wang SY, Shao B, Rao PF, Lee YY, Ye XY (2007) Hypotin, a novel antipathogenic and antiproliferative protein from peanuts with sequence similarity to those of chitinase precursors. J Agric Food Chem 55:9792–9799
Wang SY, Shao B, Ye XY, Rao PF (2008a) Purification and characterization of a chitinase from peanut (Arachis hypogaea L.). J food biochem 32:32–45
Wang SY, Zhou JJ, Shao B, Lu YJ, Rao PF (2008b) A thermostable chitinase with chitin-binding activity from Phaseolus Limensis. J Food Sci 73(6):452–457
Wang SY, Ye XY, Rao PF (2009) Isolation and biochemical characterization of a novel leguminous defense peptide with antifungal and antiproliferative potency. Appl Microbiol Biotechnol 82(1):79–86
Yang HQ, Luo ZM (1998) A study of the purification and properties of chitinase from bean seeds. J Hunan Agric Uni (China) 24(3):194–198
Ye XY, Ng TB (2002) Delandin, a chitinase-like protein with antifungal, HIV-1 reverse transcriptase inhibitory and mitogenic activities from the ricebean (Delandia umbellata). Protein Expr Puri 24:524–529
Ye XY, Ng TB (2003) Isolation of Vulgin, a New Antifungal polypeptide with mitogenic activity from the pinto bean. J Peptide Sci 9:114–119
Acknowledgements
This study was supported by the Collaboration Funding of Queen’s University Belfast, Belfast, Northern Ireland, and the Fujian Science and Technology Foundation, Fujian, P. R.China (Funding number 2008N0102). The authors are grateful to Dr. Xiaoliang Shia in Bioprocessing engineering at University of Wisconsin, Madison, U.S.A, for his generous help.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Wang, S., Shao, B., Fu, H. et al. Isolation of a thermostable legume chitinase and study on the antifungal activity. Appl Microbiol Biotechnol 85, 313–321 (2009). https://doi.org/10.1007/s00253-009-2074-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-009-2074-9