
Abstract
A major disadvantage of cyclodextrin production is the limited cyclodextrin product specificity of cyclodextrin glycosyltransferase (CGTase). Here, we described mutations of Asp372 and Tyr89 at subsite −3 in the CGTase from Paenibacillus macerans strain JFB05-01. The results showed that Asp372 and Tyr89 played important roles in cyclodextrin product specificity of CGTase. The replacement of Asp372 by lysine and Tyr89 by aspartic acid, asparagine, lysine, and arginine resulted in a shift in specificity towards the production of α-cyclodextrin, which was most apparent for the mutants D372K and Y89R. Furthermore, the changes in cyclodextrin product specificity for the single mutants D372K and Y89R could be combined in the double mutant D372K/Y89R, which displayed a 1.5-fold increase in the production of α-cyclodextrin, with a concomitant 43% decrease in the production of β-cyclodextrin when compared to the wild-type CGTase. Thus, the D372K and Y89R single and double mutants were much more suitable for the industrial production of α-cyclodextrin than the wild-type enzyme. The enhanced α-cyclodextrin specificity of these mutants might be a result of stabilizing the bent conformation of the intermediate in the cyclization reaction.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cyclodextrins are cyclic, nonreducing oligoglucopyranose molecules linked via α(1,4)-glycosidic bonds mainly consisting of six, seven, or eight glucose residues (α-, β-, or γ-cyclodextrin, respectively; Chang et al. 1998). They can form inclusion complexes with various hydrophobic guest molecules (Saenger 1980). The advantageous changes of the guest molecular properties after the formation of inclusion complexes with cyclodextrins have led to many applications of cyclodextrins in the industries related to food, pharmaceuticals, etc. (Szente and Szejtli 2004; Del Valle 2004). Cyclodextrins are commonly produced by the enzymatic conversion of starch via an intramolecular transglycosylation reaction catalyzed by cyclodextrin glycosyltransferase (EC 2.4.1.19, CGTase). Apart from the cyclization reaction, CGTase also catalyzes three other reactions: coupling, disproportionation, and starch hydrolysis reactions (Li et al. 2007; van der Veen et al. 2000d).
All known wild-type CGTases produce a mixture of α-, β-, and γ-cyclodextrins when incubated with starch (Li et al. 2007; Schmid 1989). To isolate pure cyclodextrins from the reaction mixture, a series of additional steps are required, including selective crystallization of β-cyclodextrin (which is relatively poorly water-soluble) and selective complexation of α- or γ-cyclodextrins with organic solvents (Wind et al. 1998; van der Veen et al. 2000a). However, the additional procedures will increase significantly the costs of cyclodextrin production, especially for α- or γ-cyclodextrins. Moreover, the use of organic solvents also limits cyclodextrin applications involving human consumption (Pedersen et al. 1995; van der Veen et al. 2000a). Therefore, the availability of CGTase capable of producing an increased ratio of one particular type of cyclodextrin is highly desired (van der Veen et al. 2000a). This situation has strongly stimulated the constructions of mutant CGTases with improved cyclodextrin product specificity.
So far, large numbers of site-directed mutations in CGTases from different sources have been made (van der Veen et al. 2000b; Penninga et al. 1995; Fujiwara et al. 1992; Sin et al. 1994). Most of these mutations were based upon amino acid residues located in the active center cleft, which contains at least nine sugar-binding subsites with the catalytic site between subsites +1 and −1 (Strokopytov et al. 1996). Recently, some evidences supported that the amino acid residues contributing to the interactions with the glucoses bound at subsites −3 may be important for cyclodextrin product specificity of CGTase (van der Veen et al. 2000b, c; Wind et al. 1998; Kim et al. 1997).
The enzyme used in our studies, CGTase from Paenibacillus macerans, was most commonly used in the commercial production of α-cyclodextrin (Wind et al. 1998). Although it produces mainly α-cyclodextrin during the initial stage of starch conversion and is one of a few α-CGTases identified, the proportion of α-cyclodextrin in the total cyclodextrin products was even lower than that of β-cyclodextrin after prolonged incubation under conditions resembling industrial production nonsolvent process. Thus, mutants of this CGTase capable of producing an increased ratio of α-cyclodextrin are of high industrial interest for the production of α-cyclodextrin.
The amino acid residues at positions 372 and 89 (P. macerans CGTase numbering) are part of subsite −3 (Strokopytov et al. 1996). Moreover, it has been proved by X-ray studies of some CGTases that both the residues at the corresponding positions had the interactions with the glucose residue bound at subsite −3 (van der Veen et al. 2000c; Wind et al. 1998). Here, we thus chose Asp372 and Tyr89 in the CGTase from P. macerans strain JFB05-01 as targets for site-directed mutagenesis. The results showed that Asp372 and Tyr89 played important roles in cyclodextrin product specificity. The D372K and Y89R single and double mutants enhanced significantly the α-cyclodextrin specificity. The mechanisms for the effect of these mutations on cyclodextrin product specificity were also analyzed.
Materials and methods
Bacterial strains and plasmids
Escherichia coli JM109 [endA1 glnV44 thi-1 relA1 gyrA96 recA1 mcrB+ Δ(lac-proAB) e14- [F′ traD36 proAB+lacIqlacZΔM15] hsdR17(rK −mK +)] was used for recombinant DNA manipulations. The (mutant) CGTase proteins were produced with E. coli BL21 (DE3) [F−ompT gal dcm lon hsdSB(rB − mB −) λ(DE3 [lacI lacUV5-T7 gene 1 ind1 sam7 nin5])]. Plasmid cgt/pET-20b(+), in which the gene coding the mature wild-type CGTase from P. macerans strain JFB05-01 (CCTCC M203062) was placed downstream of pelB signal sequence and fused in frame to six histidine codons (Chen et al. 2008), was used for site-directed mutagenesis, sequencing, and expression of the (mutant) CGTase proteins.
DNA manipulations and sequencing
Restriction endonucleases were purchased from TaKaRa Shuzo (Otsu, Japan) and used according to the manufacturer’s instructions. DNA manipulations and calcium chloride transformation of E. coli strains were performed as described (Sambrook et al. 1989). DNA sequences were determined by cycle sequencing with an ABI PRISM BigDye primer cycle sequencing kit with AmpliTaq DNA polymerase (Perkin-Elmer, Foster City, CA, USA). Homology alignment was performed with the CLUSTAL W program (Thompson et al. 1994) using MacVector 6.5 software (Oxford Molecular Group).
Site-directed mutagenesis
Site-directed mutagenesis was performed by a one-step polymerase chain reaction (PCR) method using plasmid cgt/pET-20b(+) as template and a pair of complementary primers. The sequences of the mutagenic primers were shown in Table 1. The double mutant D372K/Y89R was made using plasmid cgt/pET-20b(+) with the D372K mutation as template and Y89R-For and Y89R-Rev as primers (Table 1) in the PCR reaction. The PCR products were treated with DpnI and used to transform chemically competent E. coli JM109 cells. The resulting (mutant) plasmids from these clones were used to transform chemically competent E. coli BL21 (DE3) cells. All the mutations were confirmed by DNA sequencing.
Production and purification of CGTase proteins
A single colony of E. coli BL21 (DE3) cells harboring (mutant) plasmid cgt/pET-20b(+) was inoculated into 10 ml Luria–Bertani medium containing 100 μg/ml ampicillin and grown at 37°C overnight. Overnight culture (1 ml) was then diluted into 100 ml of terrific broth containing 100 μg/ml ampicillin and incubated on a rotary shaker (200 rpm) at 30°C until the optical density at 600 nm (OD600) of the culture reached 0.6. Isopropyl β-d-1-thiogalactopyranoside was added to a final concentration of 0.01 mM to induce the expression of the target protein. Protein induction was performed at 25°C and continued for 90 h.
Culture supernatants were obtained by centrifugation at 10,000×g for 20 min at 4°C. The (mutant) CGTases in the culture supernatant were further purified by one-step affinity chromatography using a Ni-NTA agarose column (Qiagen, Chatsworth, CA, USA) according to the manufacturer’s instruction. The purified enzymes were divided into aliquots and stored at −80°C. Protein concentration was determined by the Bradford assay (Bradford 1976) using bovine serum albumin (Sigma-Aldrich, Milwaukee, WI, USA) as a standard.
Assay of CGTase
All enzyme assays were performed by incubating 0.1 ml of appropriately diluted enzyme with 0.9 ml of 3% (w/v) soluble starch in 50 mM phosphate buffer (pH 6.0) at 40°C for 10 min.
α-Cyclodextrin-forming activity was determined by the methyl orange method (Lejeune et al. 1989) with some modifications. The above reaction was terminated by the addition of 1.0 M HCl (1.0 ml), and then 1.0 ml of 0.1 mM methyl orange in 50 mM phosphate buffer (pH 6.0) was added. After the reaction mixture was incubated at 16°C for 20 min, the amount of α-cyclodextrin in the mixture was spectrophotometrically determined by measuring the absorbance at 505 nm. One unit of α-cyclodextrin-forming activity was defined as the amount of enzyme that was able to produce 1 μmol of α-cyclodextrin per minute.
β- and γ-cyclodextrin-forming activities were determined by the phenolphthalein (Makela et al. 1987) and bromocresol green (Kato and Horikoshi 1984) methods, respectively. One unit of each activity was defined as the amount of enzyme that was able to produce 1 μmol of the corresponding cyclodextrin per minute.
Stabilities of the (mutant) CGTases
The thermostabilities of the (mutant) CGTases were determined by incubating the purified enzymes in 50 mM phosphate buffer (pH 6.0) at 40°C. At various times, aliquots of the enzyme were removed and assayed for residual α-cyclization activity under the standard conditions.
HPLC product analysis
The formation of cyclodextrins under conditions resembling industrial production nonsolvent process was measured by incubating 5% (wet basis, w/v) soluble starch in 50 mM phosphate buffer (pH 6.0) with 0.2 U/ml (total cyclization activity) CGTase at 40°C for 40 h. Samples were taken at regular time intervals and boiled for 10 min. To eliminate contaminating oligosaccharides while leaving cyclodextrins intact, the glucoamylase (Novozymes, Bagsvaerd, Denmark) was added to the boiled sample at a final concentration of 1 U/ml, and then the mixture was incubated at 30°C for 2 h, followed by boiling for 10 min. The concentrations of α-, β-, and γ-cyclodextrins in the final sample were determined by HPLC, using a Lichrosorb NH2 column (Merck, Darmstadt, FRG) eluted with acetonitrile/water (65:45) at 1 ml/min. Products were detected by a Waters 410 refractive index detector (Waters Corp., Milford, MA, USA).
Structure modeling of the (mutant) CGTases
The theoretical structures of the wild-type and mutant CGTases were obtained by homology modeling from the SWISS-MODEL protein-modeling server (http://www.expasy.ch/swissmod/SWISS-MODEL.html; Arnold et al. 2006). The proposed complex structures of the wild-type and mutant CGTases with a maltohexaose or maltononaose were modeled by means of superpositioning of the above theoretical structure and the complex structure of Thermoanaerobacterium thermosulfurigenes strain EM1 CGTase with a maltohexaose inhibitor in the active site (PDB accession codes 1A47; Wind et al. 1998) or Bacillus circulans strain 251 CGTase E257Q/D229N with a maltononaose in the active site (PDB accession codes 1CXK; Uitdehaag et al. 1999), respectively, followed by least-square fitting of the Cα atoms.
Results
Expression and purification of the wild-type and mutant CGTases
The mutants were successfully constructed by site-directed mutagenesis via one-step PCR; all the mutations were verified by DNA sequencing. The wild-type and mutant CGTases were expressed in E. coli BL21 (DE3). There was no significant difference in the expression levels between the recombinant wild-type and the mutant CGTases, as 40–50 mg of the CGTase proteins per liter was produced in the flask cultures. Purified (mutant) CGTase proteins were obtained by one-step nickel affinity chromatography on Ni-NTA resin. Purity and molecular weight of the wild-type and mutant CGTase proteins were checked by SDS-polyacrylamide gel electrophoresis (data not shown). All proteins were purified to apparent homogeneity and displayed a molecular mass of approximately 76 kDa.
Cyclization activities of the wild-type and mutant CGTases
Different cyclodextrin-forming activities of the (mutant) CGTases were shown in Table 2. The D372K mutation resulted in a slight increase in α-cyclodextrin-forming activity but a twofold decrease in β-cyclodextrin-forming activity when compared to the wild-type CGTase. γ-Cyclodextrin-forming activity was not affected significantly by the mutation. In addition, the D372K mutant showed a slight decrease in the total cyclization activity.
The Y89L mutation had almost no effect on different cyclodextrin-forming activities, while the mutations Y89D and Y89N resulted in 15% and 11% increases in α-cyclodextrin-forming activity, respectively, and slight increases in β-cyclodextrin-forming activity when compared to the wild-type enzyme. The mutants Y89K and Y89R showed 16% and 18% increases in α-cyclodextrin-forming activity but 1.3- and 2.3-fold decreases in β-cyclodextrin-forming activity, respectively. The contribution of γ-cyclodextrin-forming activity in total cyclization activity was not affected significantly by these mutations at position 89. It was noted that the mutants at position 89, except for the Y89L mutant, showed 7–14% increases in the total cyclization activity.
Compared to the wild-type enzyme, the double mutant D372K/Y89R showed a 24% increase in α-cyclodextrin-forming activity but a threefold decrease in β-cyclodextrin-forming activity. In addition, the double mutant showed a 10% increase in the total cyclization activity.
Cyclodextrin product ratio of the wild-type and mutant CGTases under conditions resembling industrial production processes
The production of cyclodextrins in the reaction mixture containing a (mutant) CGTase protein (0.2 U/ml cyclization activity) and 5% (wet basis, w/v) soluble starch incubated at 40°C was analyzed in time. For the wild-type CGTase, at the initial stage of the reaction, α-cyclodextrin was the main product, while small amounts of β- and γ-cyclodextrin were produced. At the later stages, the proportion of α-cyclodextrin in the total cyclodextrin products decreased while the proportion of β-cyclodextrin increased (Fig. 1a). After 40 h of incubation, the wild-type enzyme produced a mixture of α-, β-, and γ-cyclodextrin at a ratio of 41.8:54.4:3.8 (Table 3). Compared to the wild-type enzyme, the D372K mutant produced more α-cyclodextrin at the expense of β-cyclodextrin, while the production of γ-cyclodextrin was not affected significantly (Fig. 1b). After 40 h of incubation, the amount of α-cyclodextrin in the reaction mixture was about 1.3-fold of that of the wild-type enzyme, while the amount of β-cyclodextrin decreased to 77% of that of the wild-type enzyme (Table 3).

Cyclodextrins formed during incubation of the (mutant) CGTases from P. macerans strain JFB05-01 (0.2 U/ml cyclization activity) with 5% (wet basis, w/v) soluble starch for 40 h at pH 6.0 and 40°C. Each value represents the mean of three independent measurements. Blue diamonds, α-cyclodextrin; pink squares, β-cyclodextrin; red triangles, γ-cyclodextrin. a Wild-type CGTase, b mutant D372K, c mutant Y89L, d mutant Y89D, e mutant Y89N, f mutant Y89K, g mutant Y89R, h mutant D372K/Y89R
The mutation Y89L had almost no effect on the production of cyclodextrins, so the Y89L mutant had similar cyclodextrin product ratio to the wild-type enzyme (Fig. 1c and Table 3). For the mutants Y89D and Y89N, the production of α-cyclodextrin slightly increased while the production of β-cyclodextrin slightly decreased when compared to that of the wild-type enzyme (Fig. 1d, e). After 40 h of incubation, almost equal amounts of α- and β-cyclodextrin were produced by the two mutants (Table 3). The mutants Y89K and Y89R had significant increases in the production of α-cyclodextrin, with concomitant significant decreases in the production of β-cyclodextrin (Fig. 1f, g). Eventually, the amounts of α-cyclodextrin in the reaction mixture were higher than those of β-cyclodextrin (Table 3). Especially for the Y89R mutant, the amount of α-cyclodextrin was about 1.4-fold of that of the wild-type enzyme, while the amount of β-cyclodextrin decreased to 70% of that of the wild-type enzyme (Table 3). In addition, the production of γ-cyclodextrin was not affected significantly by all the mutations at position 89.
The changes in cyclodextrin product ratio for the single mutants D372K and Y89R were combined in the double mutant D372K/Y89R (Fig. 1h). Eventually, the amount of α-cyclodextrin was about 1.5-fold of that of the wild-type enzyme, while the amount of β-cyclodextrin decreased to 57% of that of the wild-type enzyme, and no significant effect on the production of γ-cyclodextrin was observed. The double mutant had a final product ratio of 65:31.9:3.1 (Table 3).
Compared to the wild-type enzyme, the mutants Y89L, Y89R, and D372K/Y89R had 3–5% decreases in the conversions of starch into cyclodextrins, whereas for other mutants the conversions remained unaffected (Table 3).
Discussion
Previous reports had shown that many mutations at subsite −3 in the CGTases from different sources could change cyclodextrin product specificity (van der Veen et al. 2000b, c; Wind et al. 1998; Kim et al. 1997), suggesting that subsite −3 was a key site for product specificity of CGTase. In the present study, further experimental evidences for this were obtained by modifying Asp372 and Tyr89 at subsite −3 in the CGTase from P. macerans strain JFB05-01, using site-directed mutagenesis.
The effect of the mutations at subsite −3 on cyclodextrin product specificity was firstly reflected in the change of initially cyclodextrin product ratio. Among all four CGTase catalyzed reactions, only the effect of cyclization is apparent at the start of the reaction (van der Veen et al. 2000b). Thus, this ratio depends on different cyclodextrin-forming activities of CGTase and shows the preference of the CGTase for the production of the specific cyclodextrins (van der Veen et al. 2000c). In the present study, except for Y89L, other mutations of Asp372 and Tyr89 resulted in increased proportions of α-cyclodextrin-forming activity in total cyclization activity, thus causing a shift in the preference towards the production of α-cyclodextrin, which was most apparent for the D372K and Y89R single and double mutants (Table 2).
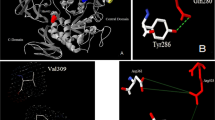
To explore the mechanisms for the effects of amino acid side chains at position 372 or 89 on cyclodextrin-forming activity of CGTase, we constructed theoretical structures of the wild-type and mutant CGTases and modeled complex structures of the above CGTases with a maltohexaose or maltononaose. An overlay of the −3 glucose residues in the maltohexaose and maltononaose conformations was shown in Fig. 2. In contrast to other subsites (not shown), at subsite −3, the maltohexaose-binding mode was radically different from the maltononaose-binding mode, the former being more bent towards the center (Tyr195) of the active site. The previous reports had revealed that the bent and straight conformations might represent (partly) specific intermediates in α- and β-cyclization reactions, respectively (van der Veen et al. 2000c; Wind et al. 1998). Thus, the underlying mechanisms for the observed shift of cyclization activity to α-cyclodextrin formation for the D372K mutant (Table 2) might be that: (a) The long side chain of lysine blocked the straight conformation, since the model of the mutant showed that the side chain was located too close to the −2 glucose residue (not shown) and −3 glucose residue (Fig. 2b) in this conformation. This suggested that the intermediate specific for the production of β-cyclodextrin was less frequently formed. (b) The Lys N-ζ atom could further stabilize the bent conformation by forming a hydrogen bond with the O3 atom of the −2 glucose residue and the O1 atom of the −3 glucose residue.

Models of the (mutant) CGTases from P. macerans strain JFB05-01 and superpositions of the −3 glucose residues in the maltohexaose (yellow) and maltononaose (purple) conformations. a Wild-type CGTase, b mutant D372K, c mutant Y89K, d mutant Y89R
For the mutations at position 89, the introduction of the hydrophilicity led to increases in α- and β-cyclodextrin-forming activities as shown by mutants Y89D and Y89N (Table 2), which was in agreement with the results of the Y89D mutation of the CGTase from B. circulans strain 251 (van der Veen et al. 2000c) and the Y89S mutation of the CGTase from alkalophilic Bacillus sp. strain I-5 (Kim et al. 1997). The possible reason was that these hydrophilic residues could stabilize the intermediates in α- and β-cyclization reactions. However, with the mutations of Tyr98 into Lys or Arg, the long and flexible side chains might clash with the straight maltononaose conformation, but be less hindering to the bent conformation (Fig. 2c, d). Moreover, both the mutations would also stabilize the bent conformation by adding hydrophilicity at subsite −3 and forming a hydrogen bond between residue 89 and the O6 atom of the −3 glucose residue. Thus, they resulted in significant increases in α-cyclodextrin-forming activity but decreases in β-cyclodextrin-forming activity.
The cyclization behavior of the double mutant D372K/Y89R (Table 2) was largely the sum of the single mutants. This additive behavior might be explained by that both the residues had synergistic effects on stimulating the maltohexaose conformation and hindering the maltononaose conformation.
The mutations of Asp372 and Tyr89 at subsite −3 also resulted in the change of cyclodextrin product ratio after reaching equilibrium. The final ratio is a combined result of all the four CGTase catalyzed reactions, as well as the solubilities of the diverse cyclodextrins (van der Veen et al. 2000c; Terada et al. 1997; Wind et al. 1998). With the incubation time prolonged, the degradation of cyclodextrins via the coupling reaction is not negligible. Meanwhile, α-, β-, and γ-cyclodextrin formation rates will gradually decrease due to product inhibition. Eventually, the formation and degradation of α-, β-, and γ-cyclodextrins may reach equilibrium. However, it was found that, for any (mutant) CGTase, β-cyclodextrin was continuously produced even after the production of α-cyclodextrin reached a plateau (Fig. 1), indicating that the formation and degradation of α-cyclodextrin reached equilibrium more quickly than those of β-cyclodextrin. Thus, the final ratio obtained after 40 h of incubation (Table 3) had obvious difference with the initial ratio expected from its cyclodextrin-forming activities (Table 2).
Generally, the mutations can alter not only cyclodextrin-forming activity but also other reaction activities and product inhibition (van der Veen et al. 2000c). In the present study, for the mutants, the altered other reaction activities, especially coupling activity, and product inhibition might also be the reasons for the change of cyclodextrin product ratio. Nevertheless, the changes of the final ratio for the mutants could be closely related to their altered cyclodextrin-forming activities; after 40 h of incubation, the mutant with higher proportion of α-cyclodextrin-forming activity in total cyclization activity (Table 2) still had an increased contribution of α-cyclodextrin at the expense of β-cyclodextrin when compared to the wild-type enzyme (Table 3), proving that the final ratio could be modified by changing the ratio in specific activities for the formation of the different cyclodextrins. This phenomenon was most pronounced for the D372K and Y89R single and double mutants.
In addition, although equal amounts of cyclization activity were used in our cases, at the final stages of the reaction, the amounts of starch converted into cyclodextrins for some mutants had slight decreases when compared to that of wild-type enzyme (Table 3). Stability tests revealed that these mutants with lower cyclodextrin production were companied by shorter half-life time at 40°C (Table 2), so the decreased total cyclodextrin production from starch was probably caused by the decreased stability of the enzyme.
In summary, Asp372 and Tyr89 at subsite −3 in the P. macerans strain JFB05-01 CGTase played important roles in cyclodextrin product specificity. The replacement of Asp372 by lysine and Tyr89 by arginine enhanced significantly the α-cyclodextrin specificity, while the double mutation D372K/Y89R could result in a larger shift in specificity towards the production of α-cyclodextrin than the single mutations. The enhanced α-cyclodextrin specificity of these mutants might be a result of stabilizing the bent conformation but hindering the straight conformation. These mutants were more suitable for the industrial production of α-cyclodextrin than the wild-type enzyme.
References
Arnold K, Bordoli L, Kopp J, Schwede T (2006) The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics 22:195–201
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Chang HY, Irwin PM, Nikolov ZL (1998) Effects of mutations in the starch-binding domain of Bacillus macerans cyclodextrin glycosyltransferase. J Biotechnol 65:191–202
Chen J, Wu J, Li ZF, Li B, Cheng C (2008) Cloning and expression of the gene encoding α-cyclodextrin glycosyltransferase. CN Patent 200810024162.3
Del Valle EMM (2004) Cyclodextrins and their uses: a review. Process Biochem 29:1033–1046
Fujiwara S, Kakihara H, Sakaguchi K, Imanaka T (1992) Analysis of mutations in cyclodextrin glucanotransferase from Bacillus stearothermophilus which affect cyclization characteristics and thermostability. J Bacteriol 174:7478–7481
Kato T, Horikoshi K (1984) Colorimetric determination of γ-cyclodextrin. Anal Chem 56:1738–1740
Kim YH, Bae KH, Kim TJ, Park KH, Lee HS, Byun SM (1997) Effect on product specificity of cyclodextrin glycosyltransferase by site-directed mutagenesis. Biochem Mol Biol Int 41:227–234
Lejeune A, Sakaguchi K, Imanaka T (1989) A spectrophotometric assay for the cyclization activity of cyclomaltohexaose (α-cyclodextrin) glucanotransferase. Anal Biochem 181:6–11
Li ZF, Wang M, Wang F, Gu ZB, Du GC, Wu J, Chen J (2007) γ-Cyclodextrin: a review on enzymatic production and applications. Appl Microbiol Biotechnol 77:245–255
Makela M, Korpela T, Laakso S (1987) Colorimetric determination of β-cyclodextrin: two assay modifications based on molecular complexation of phenolphtalein. J Biochem Biophys Meth 14:85–92
Pedersen S, Dijkhuizen L, Dijkstra BW, Jensen BF, Jorgensen ST (1995) A better enzyme for cyclodextrins. Chemtech 25:19–25
Penninga D, Strokopytov B, Rozeboom HJ, Lawson CL, Dijkstra BW, Bergsma J, Dijkhuizen L (1995) Site-directed mutations in tyrosine 195 of cyclodextrin glycosyltransferase from Bacillus circulans strain 251 affect activity and product specificity. Biochemistry 34:3368–3376
Saenger W (1980) Cyclodextrin inclusion compounds in research and industry. Angew Chem 19:344–362
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning, a laboratory manual, 2nd edn. Cold Spring Harbor Laboratory Press, New York
Schmid G (1989) Cyclodextrin glycosyltransferase production: yield enhancement by overexpression of cloned genes. Trends Biotechnol 7:244–248
Sin KA, Nakamura A, Masaki H, Matsuura Y, Uozumi T (1994) Replacement of an amino acid residue of cyclodextrin glucanotransferase of Bacillus ohbensis doubles the production of γ-cyclodextrin. J Biotechnol 32:283–288
Strokopytov B, Knegtel RM, Penninga D, Rozeboom HJ, Kalk KH, Dijkhuizen L, Dijkstra BW (1996) Structure of cyclodextrin glycosyltransferase complexed with a maltononaose inhibitor at 2.6 Angstrom resolution. Implications for product specificity. Biochemistry 35:4241–4249
Szente L, Szejtli J (2004) Cyclodextrins as food ingredients. Trends Food Sci Tech 15:137–142
Terada Y, Yanase M, Takata H, Takaha T, Okada S (1997) Cyclodextrins are not the major cyclic α-1,4-glucans produced by the initial action of cyclodextrin glucanotransferase on amylose. J Biol Chem 272:15729–15733
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680
Uitdehaag JC, Mosi R, Kalk KH, van der Veen BA, Dijkhuizen L, Withers SG, Dijkstra BW (1999) X-ray structures along the reaction pathway of cyclodextrin glycosyltransferase elucidate catalysis in the a-amylase family. Nat Struct Biol 6:432–436
van der Veen BA, Uitdehaag JC, Dijkstra BW, Dijkhuizen L (2000a) Engineering of cyclodextrin glycosyltransferase reaction and product specificity. Biochim Biophys Acta 1543:336–360
van der Veen BA, Uitdehaag JC, Dijkstra BW, Dijkhuizen L (2000b) The role of arginine 47 in the cyclization and coupling reactions of cyclodextrin glycosyltransferase from Bacillus circulans strain 251 implications for product inhibition and product specificity. Eur J Biochem 267:3432–3441
van der Veen BA, Uitdehaag JC, Penninga D, van Alebeek GJ, Smith LM, Dijkstra BW, Dijkhuizen L (2000c) Rational design of cyclodextrin glycosyltransferase from Bacillus circulans strain 251 to increase α-cyclodextrin production. J Mol Biol 296:1027–1038
van der Veen BA, van Alebeek GJ, Uitdehaag JC, Dijkstra BW, Dijkhuizen L (2000d) The three transglycosylation reactions catalyzed by cyclodextrin glycosyltransferase from Bacillus circulans (strain 251) proceed via different kinetic mechanisms. Eur J Biochem 267:658–665
Wind RD, Uitdehaag JC, Buitelaar RM, Dijkstra BW, Dijkhuizen L (1998) Engineering of cyclodextrin product specificity and pH optima of the thermostable cyclodextrin glycosyltransferase from Thermoanaerobacterium thermosulfurigenes EM1. J Biol Chem 273:5771–5779
Acknowledgments
This work was supported financially by the Program for Innovative Research Team of Jiangnan University, the Natural Science Foundation of Jiangsu Province (BK2007019), the Major State Basic Research Development Program of China (973 Program; 2007CB714036), the National Outstanding Youth Foundation of China (20625619), Research Program of the State Key Laboratory of Food Science and Technology (No. SKLF-MB-200802), the National High-tech Research and Development Program of China (863 Program; 2006AA10Z335), and the Graduate Student Creative Research Program of Jiangsu Province in 2008 (CX08B_127Z).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Li, Z., Zhang, J., Wang, M. et al. Mutations at subsite −3 in cyclodextrin glycosyltransferase from Paenibacillus macerans enhancing α-cyclodextrin specificity. Appl Microbiol Biotechnol 83, 483–490 (2009). https://doi.org/10.1007/s00253-009-1865-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-009-1865-3