
Abstract
Bacillus subtilis endo-β-1,4-glucanase (Cel5A) hydrolyzes cellulose by cleavage of the internal bonds in the glucose chains, producing new ends randomly. Using directed evolution techniques of error-prone polymerase chain reaction (PCR) and DNA shuffling, several Cel5A variants with improved catalytic activity had been screened from the mutant library, which contained 71,000 colonies. Compared with the wild-type enzyme, the variants (M44-11, S75 and S78) showed 2.03 to 2.68-fold increased activities toward sodium carboxymethyl cellulose (CMC), while the M44-11 also exhibited a wider pH tolerance and higher thermostability. Structural models of M44-11, S75, S78, and WT proteins revealed that most of the substitutions were not located in the strictly conserved regions, except the mutation V255A of S75, which was closed to the nucleophile Glu257 in the catalytic center of the enzyme. Moreover, V74A and D272G of M44-11, which were not located in the substrate binding sites and the catalytic center, might result in improved stability and catalytic activity. These results provided useful references for directed evolution of the enzymes that belonged to the glycoside hydrolase family 5 (GH5).
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Endo-β-1,4-glucanase (EC 3.2.1.4) randomly cleaved internal β-1,4-linkages in cellulose polymers and completely hydrolyzed cellulose to glucose by the synergistic action with the other types of cellulases, cellobiohydrolase (EC 3.2.1.91), and β-glucosidase (EC 3.2.1.21). The widely accepted mechanism for enzymatic cellulose hydrolysis was that endoglucanases hydrolyzed accessible intramolecular β-1,4-glucosidic bonds of cellulose chains randomly to produce new chain ends, cellobiohydrolases processively cleaved cellulose chains by removing the cellobiose unit from the nonreducing end, and β-glucosidases hydrolyzed cellobioses and oligosaccharides to glucoses, and these three hydrolysis processes occurred simultaneously (Kim et al. 1987; Han et al. 1995).
In the past 50 years, much effort had gone into the studies of cellulases as a potential means to obtain sustainable biobased products to replace depleting fossil fuels from an abundant, renewable energy resource, plant biomass. However, the high cost of cellulase production seemed to be a very important and difficult challenge in the cellulose bioconversion process (Kim et al. 1987). One way to increase enzyme volumetric productivity was to isolate hyperproducers and constitutive mutants with higher expression (Kim et al. 1987; Percival Zhang et al. 2006). Another way was to improve the necessary characteristics of cellulases for biorefineries, such as higher catalytic efficiency, increased stability at elevated temperature and at a certain pH by current genetic engineering biotechnology (Percival Zhang et al. 2006).
To this aim, directed evolution was employed to improve characteristics of enzymes and showed its power for protein engineering. Without detailed knowledge of the protein structure and accurate predictions on the active site or the binding pocket, this strategy could successfully generate a large library of random mutations by error-prone polymerase chain reaction (PCR) and DNA shuffling, followed by screening mutants for desired characteristics (Leisola and Turunen 2007). By this approach, many cellulases and other enzymes with the improved properties were obtained (Kim et al. 2000; Percival Zhang et al. 2006; Kim and Lei 2008). For examples, the hydrolysis rate of the Thermotoga neapolitana 1, 4-β-d-glucan glucohydrolase mutant was increased by 31% after error-prone PCR mutagenesis (McCarthy et al. 2004). A β-glycosidase mutant was found to display lactose hydrolysis rates 3.5- and 8.6-fold higher than the parent after DNA family shuffling (Kaper et al. 2002). Likewise, Wang et al. (2005) found that a Trichoderma reesei EG III mutant generated by error-prone PCR technique had an optimal pH of 5.4, corresponding to a basic pH shift of 0.6. Castle et al. (2004) also reported that a glyphosate N-acetyltransferase mutant with 10,000-fold increases in catalytic efficiency was obtained by 11 rounds of DNA shuffling.
In this study, we reported the use of directed evolution for improving the catalytic efficiency of endoglucanase from B. subtilis BME-15 (Cel5A). Since this approach used large libraries of variants, rapid and efficient methods were performed to screen and select the desired mutant by the halo-forming activity on the CMC plate. After two rounds of error-prone PCR and another round of DNA shuffling, seven mutants were obtained with 1.25- to 2.68-fold improved catalytic activities toward CMC, and one of them also exhibited improved pH tolerance and thermostability.
Materials and methods
Bacterial strains and plasmids
Escherichia coli DH5α was used for general cloning and construction of mutagenesis library. E. coli BL21-CodonPlus (DE3)-RIL strain was used as a host for protein production. B. subtilis BME-15 was isolated from soil sample of Shizi Mountain in Wuhan, China by screening for endoglucanase activity on CMC plate. It had been deposited in China Center for Type Culture Collection (CCTCC AB208216). The taxon of this strain was identified by comparison of the 16S rDNA sequence (GenBank accession number: FJ172349) with that in GenBank. Plasmid pGEX-6P-1 was used for preparation of mutant library and purification of Cel5A and its mutants.
Cloning and sequencing of the cel5A gene
The whole genomic DNA of B. subtilis BME-15 was purified and used to produce the whole 1,500 bp coding sequence of the cel5A gene by PCR. PCR program was performed with two primers (pEG-F1: CATGGATCCATGAAACGGTCAATCTCTA; pEG-R: CATCTCGAGCTAATTTGGTTCTGTTCCC; The BamHI and XhoI sites are underlined) and Pfu DNA polymerase (Fermentas) (PCR program: 5 min 94°C followed by 30 cycles of 30 s 94°C, 30 s 53°C, 90 s 72°C, and finally 7 min 72°C). The products were purified with the AxyPrep DNA purification kit (Axygen) and cloned into pGEX-6P-1 with BamHI and XhoI sites to construct the plasmid pGEX-cel5A, and then transformed into E. coli DH5α-competent cells by electroporation transformation method (Sambrook and Russell 2001).
Construction of the error-prone PCR mutant library
The mutant library was carried out as described by the protocol of DiversifyTM PCR Random Mutagenesis Kit (Clontech) with some modifications. Error-prone PCR was preformed using pGEX-cel5A as a template. The reaction mixture contained 0.5 μM primers (pEG-F1 and pEG-R), 0.2 mM dATP and dGTP, 1 mM dCTP and dTTP, 2 U TITANIUM Taq DNA Polymerase and Taq buffer containing 5 mM MgCl2 and 0.64 mM MnSO4 (PCR program: 30 s 94°C followed by 25 cycles of 30 s 94°C, 90 s 68°C, and finally 3 min 68°C). The products were digested with BamHI and XhoI and cloned into pGEX-6P-1, and then transformed into E. coli DH5α to obtain the mutant library.
Preparation of DNA shuffling mutant library
The mutant library was carried out as described by Zhao and Arnold (1997) with some modifications. The PCR products derived from the plasmids of different mutants by using Pfu DNA polymerase with two primers (pEG-F1 and pEG-R as described above) were purified and mixed equally. After digestion of about 2 μg of the DNA fragment with 0.10 U DNase I (10 U/μl, Fermentas) at 25°C for 5 min, 50 to 200 bp DNA fragments were saved on gel purification column (Axygen) and assembled by no primer PCR by using Pfu DNA polymerase (PCR program: 3 min 94°C followed by 50 cycles of 30 s 94°C, 30 s 48°C, 30 s + 5 s per cycle 72°C, and finally 7 min 72°C). A 50-fold dilution of this reaction was used as template in a final PCR reaction for the single product at the correct size with primers, pEG-F1 and pEG-R (5 min 94°C followed by 30 cycles of 30 s 94°C, 30 s 53°C, 90 s 72°C, and finally 7 min 72°C). The products were purified with the DNA purification kit and cloned into pGEX-6P-1 with BamHI and XhoI sites, and then transformed into E. coli DH5α for screening.
Selection and screening by Congo red staining method
Transformants of Cel5A mutant library were spread on LB plate containing 100 μg/ml ampicillin. After an incubation about 14 h at 37°C, the colonies were picked up and transferred on CMC plates (0.001% MgSO4, 0.005% KH2PO4, 0.001% CaCl2, 0.6% NaCl, 0.2% (NH4)2SO4, 0.2% K2HPO4, 0.1% yeast extract, 0.5% CMC and 1.5% agar). After another growth for 14 h at 37°C, the plates were stained with 0.1% Congo red (Amresco) for 10 min. The Congo red solution was poured off, and the plates were washed with 1 M NaCl for 5 min. The clones with larger halo-forming activities were analyzed and selected for sequencing (Kim et al. 2000; Yang et al. 2004).
Protein expression and purification
The mature genes, except for the portion that encodes the signal sequence, were amplified by PCR from mutants and cel5A using following two primers (pEG-F2: CGCGGATCCGCAGGGACAAAAACGCCAG; pEG-R: CATCTCGAGCTAATTTGGTTCTGTTCCC; The BamHI and XhoI sites are underlined), respectively. The cel5A genes were cloned into pGEX-6P-1 by using BamHI and XhoI sites to obtain pGEX-cel5A and the mutants. The accuracy of cloned sequences was confirmed by sequencing at Shanghai Sangon Biological Engineering Technology and Services (Shanghai, China).
To optimize expression of enzymes, the plasmids were transformed into E. coli BL21 (DE3)-RIL competent cells. The cells were grown in LB medium containing 100 μg/ml ampicillin at 37°C. When OD600 reached 0.6, IPTG was added into the medium at a final concentration of 0.2 mM. After an induction overnight at 18°C, cells were harvested and resuspended in 50 ml phosphate-buffered saline (PBS; 140.0 mM NaCl, 2.7 mM KCl, 10.0 mM Na2HPO4, 1.8 mM KH2PO4, pH 7.4) buffer, and disrupted by using French cell press. The cell lysate was centrifuged at 12,000×g for 30 min, and the supernatant was collected for GST-free affinity purification (Pharmacia).
The Glutathione (GSH)-Sepharose column (bed volume, 1 ml) was pre-equilibrated with 50 ml PBS buffer. The clear supernatant was directly packed into the GSH-Sepharose column with an initial elute speed of about 1 ml/min. The column was then washed with 200 ml PBS buffer to elute the unbound proteins. One milliliter of PBS buffer was added into 10 μl of 3C protease stock solution (10°U/μl, PreScission, Pharmacia), and the mixture was added to the column. After incubation for 16 h at 4°C, 1 ml PBS buffer was added to elute the purified protein (Cao et al. 2008). The quantification of the protein was determined with Bradford reagent (Sigma) by the method of Bradford (1976), using bovine serum albumin as a standard. The purity of the extracted proteins was analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE).
Enzyme assay
The enzyme assay consisted of 50 μl 0.5% CMC (Sigma) in 100 mM sodium acetate buffer (pH 5.0) and 50 μl diluted enzyme solution. After incubation at 50°C for 30 min, dinitrosalicylic acid reagent (100 μl) was added, and the mixture was heated in a boiling water bath for 5 min and then added 800 μl H2O. The absorbance was measured at 540 nm. One unit of enzyme activity was defined as the quantity of enzyme capable of releasing 1 μmol of glucose equivalent per min (glucose as standard). The effects of temperature and pH on the enzyme activity were performed at different temperatures (30–80°C) and at different pH values by using 0.2 M HAc-NaAc (pH 2.6–5.0), 0.2 M Na2HPO4–citric acid buffer (pH 4.0–8.0), 0.2 M Na2HPO4–NaH2PO4 (pH 6.0–pH 8.0), and 0.05 M glycine–NaOH (pH 9.0–12.0) buffers. Thermal stability studies were carried out by incubating the enzyme at different temperatures (30–90°C) for 1 h. Samples were withdrawn to determine residual enzyme activity. For pH stability, relative activity was determined after the enzyme had been incubated with different pH buffers (pH 2.6–pH 12) at 25°C for 2 h (Li et al. 2006).
To investigate the substrate specificity of the enzymes, the activities were determined replacing CMC by 0.5% barley glucan (Sigma), 1% avicel (Sigma), 1% filter paper (Whatman), 1% salicin (Fluka), 1% chitin (Sigma), 1% oat spelts xylan (Sigma), 1% birch wood xylan (Sigma), and 1% locust bean gum (Fluka) as substrates under the same condition.
Molecular modeling and analysis
Structural models of Cel5A (wild-type enzyme, WT) and mutant enzymes were based on the reported crystal structure of endoglucanase from Bacillus. agaradhaerens (Protein Data Bank code 1HF6) and family IIIA CBD domain from Clostridium cellulolyticum (Protein Data Bank code 1G43; Shimon et al. 2000; Varrot and Davies 2003). The hypothetical conformations of the proteins were predicted by Swiss-Model workspace and illustrated as ribbon diagrams using Swiss-Pdb viewer (Guex and Peitsch 1997; Schwede et al. 2003; Arnold et al. 2006).
Result
Cloning of cel5A gene
The endo-β-1,4-glucanase gene was PCR-cloned from B. subtilis BME-15 using the genomic DNA as the template. The nucleotide sequence was determined by sequencing and the open-reading frame of the gene contained 1,500 bp, which encoded a 499-residue polypeptide including a signal peptide of 29 residues. The overall sequence showed the identities with the cellulase gene from B. subtilis AH18 (98%, EF070194.1), cellulase gene from Bacillus amyloliquefaciens TB-2 (93%, EU022559.1), cel5A gene from B. agaradhaerens (69%, AF067428.1), celV1 gene from Pectobacterium carotovorum (68%, X79241.2), and celV gene from Erwinia carotovora (67%, X76000.1). This nucleotide sequence of cel5A data also had been deposited in GenBank database. (GenBank accession number: FJ172348).
Error-prone PCR and DNA shuffling mutagenesis
Random mutant library of cel5A generated by error-prone PCR was performed and screened by the Congo red staining method. From sequencing ten colonies, the PCR program yielded 6.4 bp substitutions per gene and corresponded to four amino acid substitutions per mutant. Fourteen clones which showed larger halo than the parent were selected from the resulting library with over 30,000 clones. Six best mutants were selected, and their plasmids were purified and used as template for the second cycle of random mutagenesis by error-prone PCR. Approximately 30,000 clones were screened from the second round random mutant library, 11 mutants with larger halo were selected for the further study.
In order to recombine the beneficial mutations generated by the error-prone PCR, 25 improved genes from the two round random mutagenesis were subjected to gene shuffling. About 12,000 clones were screened by the halo-forming activities, and 12 recombinants were obtained that had improved hydrolytic activities than the parents.
Expression and characterization of the improved enzymes
Seven clones with visually larger halo-forming activities obtained through error-prone PCR and DNA shuffling mutagenesis were characterized. All variants through the screening steps showed larger halos than the wild type (Fig. 1). Sequence analysis of the seven mutants showed that four to 12 amino acid positions were changed during the evolution procedure (Table 1). All of the total 35 amino acid substitutions, 15 were located in the glycoside hydrolase domain, and others were located in the linker and carbohydrate-binding domain (Lo et al. 1988; Park et al. 1993; Han et al. 1995).

Congo red staining of E. coli DH5α colonies displaying improved mutants on CMC plates. 1 E. coli DH5α/pGEX-6P; 2 E. coli DH5α/pGEX-cel5A; 3 E. coli DH5α/pGEX-m1; 4 E. coli DH5α/pGEX-m44; 5 E. coli DH5α/pGEX-m1-23; 6 E coli DH5α/pGEX-m44-11; 7 E. coli DH5α/pGEX-s40; 8 E. coli DH5α/pGEX-s75; 9 E. coli DH5α/ pGEX-s78 E. coli DH5α/pGEX-6P and other colonies were grown on CMC plate for 14 h at 37°C and followed by Congo red staining. In contrast, this assay showed that E. coli DH5α/pGEX-6P could barely produce halo on the CMC plate
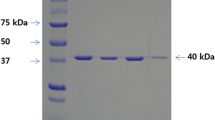
In order to characterize the seven mutants, the mature enzymes (from 30 aa to 499 aa) were produced in E. coli BL21-CodonPlus (DE3)-RIL with GST tag and purified by Glutathione Sepharose 4B as described above. The purified proteins were obtained and observed with SDS-PAGE (Fig. 2). The molecular weights of improved enzymes were estimated at 52.17 kDa, and no change compared with the parent enzyme. Using CMC as the substrate, all the mutant enzymes showed higher hydrolytic activities than the wild-type enzyme. M1 and M44 from the first round random mutation showed 1.25- to 1.56-fold increases in activity, while M1-23 and M44-11 from the second round random mutation and S40, S75, and S78 from the DNA shuffling showed 1.51- to 2.03-fold and 1.86- to 2.68-fold increased activities, respectively (Fig. 3). These results demonstrated that colony growth on the CMC plates, and the larger halo-forming were indeed due to the increased catalytic activities of the enzymes.

SDS-PAGE of the purified proteins. Lane 1, the standard protein markers; lane 2, Cel5A (the wild-type enzyme); lane 3-9, M1, M44, M1-23, M44-11, S40, S75 and S78

Enzyme activities of improved variants compared with the wild-type enzyme. WT was the wild-type enzyme (Cel5A); M1 and M44 were the protein obtained by the first round of random mutagenesis; M1-23 and M44-11were obtained through the second round of random mutagenesis derived from M1 and M44, respectively; S40, S75, and S78 were obtained by DNA Shuffling. Enzymatic reactions were performed for 30 min at pH 5.0 and 50°C. One unit of enzyme was defined as the quantity of enzyme capable of releasing 1 μmol of glucose equivalent per min. Error bars denoted SD from means
Substrate specificity
Different polysaccharides were used to test the specificity of the wild-type Cel5A and three selected mutants (Table 2). Among the tested substrates, all of the Cel5A and the mutants only exhibited the hydrolysis activity toward CMC and barley glucan, and activities against others were not observed.
Effects of temperature and pH on enzyme activity and stability
M44-11, S75, and S78 all had the same optimal activity at pH 5.0 and also showed similar pH stability profiles compared with the wild-type enzyme (Fig. 4a). Moreover, M44-11 had greater activity than others at pH 7.0 and maintained about 60% activity, while less 40% activities of the others was retained. All of the enzymes were stable over a wide pH range, and more than 70% activities were retained after incubating at 20°C for 2 h at pH ranging from 6.0 to 10.0 (Fig. 4b).

Effect of temperature and pH on enzyme activity and stability. a Effect of pH on enzyme activity. Enzyme activity was measured at 50°C and at the indicated pHs in 0.2 M Na2HPO4–Citric acid buffer (pH 4.0–8.0) with 0.5% CMC. The maximum activity observed was taken as 100%. b Effect of pH on enzyme stability. Enzymes were incubated at 20°C for 2 h at indicated pHs in various buffers. All the reactions were measured under the same condition of CMC activity assay. The activity without treatment was taken as 100%. Buffers used: 0.2 M HAc-NaAc (pH 2.6–5.0), 0.2 M Na2HPO4–NaH2PO4 (pH 6.0–8.0) and 0.05 M glycine–NaOH (pH 9.0–12.0). c Effect of temperature on enzyme activity. Enzymes were added to the reaction mixture (100 mM HAc-NaAc pH 5.0, CMC 0.5%) and the reaction was carried out at indicated temperatures. The maximum activity observed was taken as 100%. d Effect of temperature on enzyme stability. Enzymes were incubated for 1 h at indicated temperatures. Then samples were measured under the same conditions of CMC activity assay. The activity without treatment was taken as 100%. Error bars represent the SD of the mean calculated for three replicates. Open squares represent WT, open triangles represent M44-11, open circles represent S75, and open diamonds represent S78
The optimal temperature study showed that Cel5A and M44-11 exhibited maximum activity at 50°C, while S75 and S78 acted efficiently at 40°C and 60°C, respectively (Fig. 4c). In the temperature stability study, S75 and S78 were very stable after 1 h incubation at temperatures below 50°C, and their activity decrease accelerated above 50°C, while Cel5A and M44-11 were very stable below 55°C under the same condition (Fig. 4d). Moreover, M44-11 showed higher stability than others and retained more than 50% of its activity after incubation at 80°C for 1 h; compared with others, no enzyme activities were observed after incubation at 70°C for 1 h.
Protein modeling
Crystal structures of the enzymes were predicted and determined for two completely distinct structures at the tertiary level, the GH5 and the type 3 carbohydrate-binding module (CBM3; Fig. 5). The GH domain at the N-terminal was a catalytic module, consisted a total of 294 residues, from amino acids 36 to 329. This domain folded as a (β/α)8 barrel with two glutamates, Glu169 and Glu257, on strands IV and VII, acting catalytic acid/base and nucleophile, respectively (Ducros et al. 1995; Henrissat and Bairoch 1996; Gloster et al. 2004). Such structure was the typical module for clan GH-A glycoside hydrolase families (http://www.cazy.org/fam/acc_GH.html). The C-terminal domain consisted of 142 residues, from amino acid 354 to 495, was related to the function of binding to the cellulose surface and belonged to the CBM3, which folded into a β-sandwich fashion (Fig. 5). The structure of endoglucanase from B. agaradhaerens (Protein Data Bank code 1HF6) was used to construct GH domain models of both WT and mutant enzymes, and these models suggested the relative locations of the substituted residues in the tertiary level of the enzymes (Fig. 6a). Compared with the WT protein, the two mutations of M44-11 (K120E and D272G) were found in the different α-helix domains at the surface regions of the GH domain, and the other one (V74A) was located on the loop between the β-strand and the α-helix (Fig. 6b). While the three substitutions N39D, K120E, and S308P of S75, shared with S78, were positioned in different α-helix and loops, respectively. N175H and V255A of S75, situated on the opposite loops, were close to the catalytic acid/base (Glu169) and nucleophile (Glu257), respectively (Fig. 6c). In addition, the other mutations of the S78 (S248G, S283G, and R314G) were found on the surface regions of the protein far away from the catalytic center (Fig. 6d).

Modular and structure predicted for Cel5A. Cel5A three-dimensional structure predicted by Swiss-model and visualized in Swiss-Pdb viewer. The GH5 module, on the left, is folded as (β/α)8 barrel and discrete in relation to the right module, which represents the CBM3 folded in a β-sandwich fashion

Location of amino acid substitutions in the predicted GH5 modules. a catalytic sites (Glu169 and Glu257) and cellotriose located in Cel5A; b substitutions located in M44-11; c substitutions located in S75; d substitutions located in S78. The ribbon diagrams of the three-dimensional structure were generated using the Swiss-Pdb viewer
Discussion
In this study, we showed that directed evolution can be used to improve the activity of endoglucanase from B. subtilis BME-15. A library of variants was generated through two rounds of error-prone PCR and another round of DNA shuffling. To make it possible to screen large libraries, we used the Congo red staining method for selection of the halo-forming activity.
Seven mutant enzymes were purified without signal peptide and showed 1.25- to 2.68-fold increased activities toward CMC compared with the wild type. Three of them (M44-11, S75, and S78) were selected to be studied in detail including substrate specificity and the effect of temperature and pH. The results showed that S75 and S78 only obtained increased activities, while M44-11 exhibited a good stability at pH 10 and higher thermostability after incubation at 80°C for 1 h. The phenomenon that simultaneous improvements in thermostability and catalytic activity had been obtained were also found in other enzymes (Song and Rhee 2000; Kim and Lei 2008). Although the activities of the wild-type and mutant enzymes are lower than the other family 5 endoglucanases, such as Umcel5G from Bacillus cellulosilyticus (56.56 U/mg, P06565), EngE from Clostridium cellulovorans (106.6 U/mg, AAD39739), EG II from Trichoderma viride (49 U/mg, BAA36216), CelB from Streptomyces lividans (110 U/mg, AAB71950), and EglB from Aspergillus niger (8.6 U/mg, CAK45103), their results demonstrated the power of directed evolution used in protein engineering.
In order to understand the functions of the amino acid substitutions, the identified mutations in the selected mutants were distributed throughout the structure models of GH5 domain based on the crystal structures of endoglucanase from B. agaradhaerens. These three-dimensional structures suggested some strictly conserved residues such that Arg92, His131, Asn168, Glu169, His229, Tyr231 and Glu257 were positioned in a spatial arrangement and in close proximity to each other. They were all situated on the same side of the β-strand barrel and in the active-site cleft on the protein surface which were located on the loops interconnecting the β-strands. Moreover, Trp69 and Trp207 were opposite and parallel to each other at the entrance of the cleft, and participated in the binding of sugars through aromatic stacking interactions with the glucopyranosyl rings (Ducros et al. 1995; McCarthy et al. 2004). However, in this study, most of the mutations were located in loops and α-helix domains at the surface regions and were not found in those strictly conserved regions throughout GH5, except the substitution V255A of S75, which was very close to the nucleophile Glu257 in the catalytic center of the enzyme. This change might not be involved in hydrogen bonding with other residues, but presumably could result in the formation of a larger active-site pocket to increase catalytic activity. Similarly, the other substitutions might replace a hydrophobic residue and change the geometry of the immediate vicinity, presumably making new hydrogen bonds and tightening the turns of the short coil structure result in a repositioning of catalytic residues in the active site, to improve catalytic efficiency (McCarthy et al. 2004). Also, changes in backbone angles could appear to make catalytic center more accessible to the substrate. This observation that mutations outside the catalytic center or the binding sites resulted in increased catalytic activity was in agreement with the results obtained in other studies (van der Veen et al. 2004; Percival Zhang et al. 2006; Fan et al. 2007). Furthermore, most variants have the same substitution K120E in the α-helix domain indicating that this position could be important for improved activity and could be a significant target for saturation mutagenesis.
In comparison with other substitutions, the V74A and D272G probably resulted in most benefit to the pH tolerance and thermostability of M44-11. Since residue V74A introduced a smaller side chain in the loop between the β-strand and the α-helix, this substitution might change the conformations of the β-strands and eliminate structural hindrance to strengthen stabilizing interactions (Kim and Lei 2008). D272G, which is located in the α-helix domain at the protein surface, presumably introduced geometrical alterations of the helix and different hydrogen bondings with the adjacent residues, and then stabilized the protein surface through these local interactions. Moreover, by the sequence alignment of GH5 (pfam00150), Val74 was found in the similar positions of the endoglucanase from Bacillus cellulosilyticus (P19570), EngF from Clostridium cellulovorans (P94622), and endoglucanase from Pectobacterium atrosepticum (Q59394). Likewise, Asp272 was found in the relative positions of endoglucanase from Anaerocellum thermophilum (Q59154), EG-IV from Ruminococcus albus (Q07940), and endoglucanase from Actinomyces sp. 40 (O66064). Although it was unclear about the true functions of V74A and D272G, these positions might be important for improved stability of other endoglucanases in GH5.
Furthermore, as a part of the cellulase molecule, CBM could increase the enzyme concentration on the surface of the substrate and supply the catalytic domain with a more easily degradable substrate, thereby improving catalytic activity. Some studies also showed that CBM could affect the activity and stability of the catalytic domains through the interaction between different domains (Arai et al. 2003; Zhang et al. 2007). Therefore, the improved activity and stability of the mutants also could be attributed to the additive benefits and overall charge changes or folding in both two molecules of the whole enzyme produced by the individual amino acid substitutions. Actually, further studies were also currently under evaluation.
In summary, we had demonstrated that directed evolution could be used to improve the activity of the endoglucanase, which belonged to the GH5. With better catalytic efficiency and the higher thermostability, these variants might become more desirable and an economical source for transformation of cellulosics biomass to biofuels. These results of the evolution provided useful information for protein engineering on the enzymes of GH5 and hold great promise for the improvement of the significant component of the model cellulase system in the bioenergy production.
References
Arai T, Araki R, Tanaka A, Karita S, Kimura T, Sakka K, Ohmiya K (2003) Characterization of a cellulase containing a family 30 carbohydrate-binding module (CBM) derived from Clostridium thermocellum CelJ: Importance of the CBM to cellulose hydrolysis. J Bacteriol 185:504–512
Arnold K, Bordoli L, Kopp J, Schwede T (2006) The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics 22:195–201
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Cao S, Liu Z, Guo A, Li Y, Zhang C, Gaobing W, Chunfang F, Tan Y, Chen H (2008) Efficient production and characterization of Bacillus anthracis lethal factor and a novel inactive mutant rLFm-Y236F. Protein Expr Purif 59:25–30
Castle LA, Siehl DL, Gorton R, Patten PA, Chen YH, Bertain S, Cho HJ, Duck N, Wong J, Liu DL, Lassner MW (2004) Discovery and directed evolution of a glyphosate tolerance gene. Science 304:1151–1154
Ducros V, Czjzek M, Belaich A, Gaudin C, Fierobe HP, Belaich JP, Davies GJ, Haser R (1995) Crystal structure of the catalytic domain of a bacterial cellulase belonging to family 5. Structure 3:939–949
Fan YH, Fang WG, Xiao YH, Yang XY, Zhang YJ, Bidochka MJ, Pei Y (2007) Directed evolution for increased chitinase activity. Appl Microbiol Biotechnol 76:135–139
Gloster TM, Macdonald JM, Tarling CA, Stick RV, Withers SG, Davies GJ (2004) Structural, thermodynamic, and kinetic analyses of tetrahydrooxazine-derived inhibitors bound to beta-glucosidases. J Biol Chem 279:49236–49242
Guex N, Peitsch MC (1997) SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis 18:2714–2723
Han SJ, Yoo YJ, Kang HS (1995) Characterization of a bifunctional cellulase and its structural gene—the cel gene of Bacillus sp. D04 has exoglucanase and endoglucanase activity. J Biol Chem 270:26012–26019
Henrissat B, Bairoch A (1996) Updating the sequence-based classification of glycosyl hydrolases. Biochem J 316:695–696
Kaper T, Brouns SJJ, Geerling ACM, De Vos WM, Van der Oost J (2002) DNA family shuffling of hyperthermostable beta-glycosidases. Biochem J 368:461–470
Kim MS, Lei XG (2008) Enhancing thermostability of Escherichia coli phytase AppA2 by error-prone PCR. Appl Microbiol Biotechnol 79:69–75
Kim JM, Kong IS, Yu JH (1987) Molecular cloning of an endoglucanase gene from an alkalophilic Bacillus sp. and its expression in Escherichia coli. Appl Environ Microbiol 53:2656–2659
Kim YS, Jung HC, Pan JG (2000) Bacterial cell surface display of an enzyme library for selective screening of improved cellulase variants. Appl Environ Microbiol 66:788–793
Leisola M, Turunen O (2007) Protein engineering: opportunities and challenges. Appl Microbiol Biotechnol 75:1225–1232
Li Y-H, Ding M, Wang J, Xu G-J, Zhao F (2006) A novel thermoacidophilic endoglucanase, Ba-EGA, from a new cellulose-degrading bacterium, Bacillus sp. AC-1. Appl Microbiol Biotechnol 70:430–436
Lo AC, MacKay RM, Seligy VL, Willick GE (1988) Bacillus subtilis beta-1,4-endoglucanase products from intact and truncated genes are secreted into the extracellular medium by Escherichia coli. Appl Environ Microbiol 54:2287–2292
McCarthy JK, Uzelac A, Davis DF, Eveleigh DE (2004) Improved catalytic efficiency and active site modification of 1,4-beta-d-glucan glucohydrolase A from Thermotoga neapolitana by directed evolution. J Biol Chem 279:11495–11502
Park JS, Nakamura A, Horinouchi S, Beppu T (1993) Identification of the cellulose-binding domain of a Bacillus subtilis endoglucanase distinct from its catalytic domain. Biosci Biotech Bioch 57:260–264
Percival Zhang YH, Himmel ME, Mielenz JR (2006) Outlook for cellulase improvement: screening and selection strategies. Biotechnol Adv 24:452–481
Sambrook J, Russell DW (2001) Molecular cloning: a laboratory manual (3-volume set). Cold Spring Harbor Laboratory Press, Cold Spring Harbor New York
Schwede T, Kopp J, Guex N, Peitsch MC (2003) SWISS-MODEL: an automated protein homology-modeling server. Nucleic Acids Res 31:3381–3385
Shimon LJW, Pages S, Belaich A, Belaich J-P, Bayer EA, Lamed R, Shoham Y, Frolow F (2000) Structure of a family IIIa scaffoldin CBD from the cellulosome of Clostridium cellulolyticum at 2.2 ANG resolution. Acta Crystallogr D Biological Crystallography 56:1560–1568
Song JK, Rhee JS (2000) Simultaneous enhancement of thermostability and catalytic activity of phospholipase A1 by evolutionary molecular engineering. Appl and Environ Microbiol 66:890–894
van der Veen BA, Potocki-Veronese G, Albenne C, Joucla G, Monsan P, Remaud-Simeon M (2004) Combinatorial engineering to enhance amylosucrase performance: construction, selection, and screening of variant libraries for increased activity. Febs Lett 560:91–97
Varrot A, Davies GJ (2003) Direct experimental observation of the hydrogen-bonding network of a glycosidase along its reaction coordinate revealed by atomic resolution analyses of endoglucanase Cel5A. Acta Crystallogr D Biological Crystallography 59:447–452
Wang T, Liu X, Yu Q, Zhang X, Qu Y, Gao P, Wang T (2005) Directed evolution for engineering pH profile of endoglucanase III from Trichoderma reesei. Biomol Eng 22:89–94
Yang MJ, Jung SH, Shin ES, Kim J, Yun HD, Wong SL, Kim H (2004) Expression of a Bacillus subtilis endoglucanase in protease-deficient Bacillus subtilis strains. J Microbiol Biotechnol 14:430–434
Zhang S, Yin QY, Li YH, Ding M, Xu GJ, Zhao FK (2007) Molecular and biochemical characterization of Ba-EGA, a cellulase secreted by Bacillus sp. AC-1 from Ampullaria crosseans. Appl Microbiol Biotechnol 75:1327–1334
Zhao H, Arnold FH (1997) Optimization of DNA shuffling for high fidelity recombination. Nucleic Acids Res 25:1307–1308
Acknowledgment
We would like to thank Dr. Qifa Zhang for many valuable suggestions. This work was supported by grants from the National Natural Sciences Foundation of China (30770021 and 30570057) and the 111 Project (B07041).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Lin, L., Meng, X., Liu, P. et al. Improved catalytic efficiency of Endo-β-1,4-glucanase from Bacillus subtilis BME-15 by directed evolution. Appl Microbiol Biotechnol 82, 671–679 (2009). https://doi.org/10.1007/s00253-008-1789-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-008-1789-3